

Abstract
Optogenetics, genetically encoded engineering of proteins to respond to light, has enabled precise control of the timing and localization of protein activity in live cells and for specific cell types in animals. Light-sensitive ion channels have become well established tools in neurobiology, and a host of new methods has recently enabled the control of other diverse protein structures as well. This review focuses on approaches to switch proteins between physiologically relevant, naturally occurring conformations using light, accomplished by incorporating light-responsive engineered domains that sterically and allosterically control the active site.
Graphical Abstract
Introduction
Nature has evolved mechanisms to precisely regulate the level of protein activity in a wide variety of biological processes. These mechanisms include post-translational modifications and interaction events that control localization and catalytic or binding activity. Chemogenetic and optogenetic manipulations have been used to interrogate how a protein’s function depends on transient localization events and associated activation kinetics. Here we focus on approaches to control protein conformation. They have provided new avenues to create protein switches that adopt physiologically relevant activation states [1,2], and can retain a full set of protein activities and regulation.
Proteins undergo a spectrum of conformational changes from their initial folding to their degradation and disposal. These changes, especially the ones regulating protein activity, include changes in the relative orientation of domains and intra-domain order-disorder transitions that may include both side chain and backbone movements (Figure 1). At one extreme of the conformational change spectrum, intrinsically disordered proteins become structurally ordered upon binding to partner proteins. At the other end of the spectrum, structurally-ordered protein domains that undergo little or no intra-domain structural changes are modulated through steric blocking, e.g by auto-inhibitory domains (AID) [3,4] (Figure 1). For the many proteins between these two extremes (e.g., kinases, phosphatases, methyltransferases, deacetylases, transmembrane proteins, proteases [5]), structural order-disorder transitions occur locally in the vicinity of active sites to regulate protein activity. It should be noted that changes in the active site induced at an allosteric site can be experimentally hard to detect, as there are enzymes that are reported to undergo allostery through dynamics without conformational change [6]. All these shifts in the structural state of proteins are the hallmark of biochemical regulation. To create physiologically relevant optogenetic tools, it is desirable to exploit nature’s structural mechanisms, switching proteins between different naturally occurring states through control of active site accessibility and allosterically-regulated conformational change. Here we discuss the structural engineering strategies that leverage these mechanisms in living cells.
Figure 1. The conformational regulation of proteins and their engineered analogs.
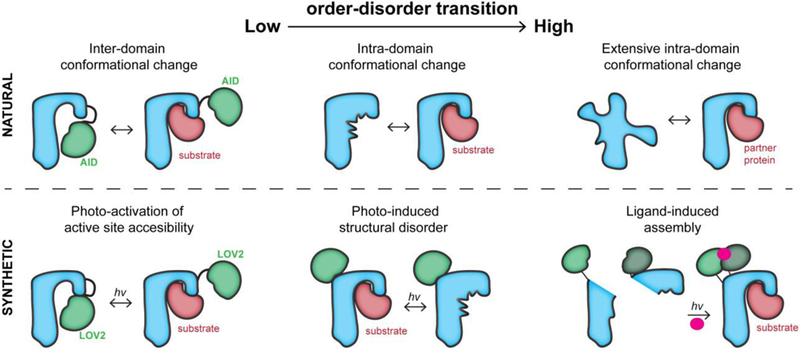
Top, left: An auto-inhibitory domain (AID) blocks the active site in the inactive state. Upon post-translational modification or protein binding, the AID undocks from the active site. Top, center: The active site undergoes a disorder-order transition during activation, enabling binding. Top, right: An intrinsically disordered protein becomes ordered upon binding to a partner protein. Bottom, left: The light-sensitive domain LOV2 blocks the active site. Upon irradiation, it undocks from the active site. Bottom, center: The LOV2 domain inserted into a loop of the host protein undergoes a light-induced conformational change. This distorts the active site allosterically. Bottom, right; The split halves of a protein reassemble upon binding a small molecule that enables dimerization. The small molecule is activated through cleavage of a light-sensitive protecting group.
Optogenetic steric control of active site accessibility
In addition to their catalytic or active domains, many signaling proteins also carry separate regulatory domains that control localization and/or activity. Optogenetic manipulation of protein localization has been used to control signaling [7–10], usually with strategies that use two proteins that associate or disassociate upon irradiation. The target protein is often mislocalized in the dark, and then released and/or targeted to a region where it is biologically active [10–12]. However, these strategies often require using a constitutively active protein analog, causing residual off-state signal, or limiting localization to only some of the subcellular positions where a protein normally acts. “Conformation-based” control approaches are valuable because they enable the protein to travel to all relevant locations in the cell, in addition to switching between physiologically relevant states. They can also provide greater spatio-temporal resolution, as they do not need to diffuse between a mislocalized and targeted distribution [1,2,13].
Based on the conformational selection model [14], all protein conformations are occupied, but in different proportions. A molecular event such as protein binding, ligand binding or post-translational modification redistributes the conformational states. A high-degree of structural disorder enables intrinsically disordered proteins to bind multiple targets with high specificity, as the unfavorable entropic contribution upon binding uncouples the affinity from specificity, resulting in high specificity without high affinity. Alternately, an auto-inhibitory domain (AID) or a separate protein can sterically block an active site. This block can be removed by a post-translational modification or protein binding to the AID, resulting in a shift of the equilibrium to a state in which the active sites of most conformational ensemble members are accessible. This feature was first exploited to engineer a photo-activatable Rac1 (PA-Rac1) by fusing a light sensitive light-oxygen-voltage 2 (LOV2) domain from plant phototropin to the N-terminus of Rac1 [1]. Because LOV2 is structurally ordered in the dark state, it bound to a portion of Rac1 and sterically blocked the Rac1 active site, like an AID. Upon blue light absorption, LOV2 partially melted and undocked from the active site, resulting in Rac1 activation (inter-domain conformational change in Figure 1). This photo-controlled switching was reversible and repeatable, enabling control of actin polymerization within living cells, and directed movement of cells within live animals. PA-Rac1 was used to show that localized Rac1 activation was sufficient to induce cell polarization and migration [1,15,16], and elucidated the causal link between the learning-induced potentiated dendritic spines and motor memory in vivo The use of LOV2 as a light-controlled AID has enabled optogenetic control of diverse proteins, including potassium channels [18], Caspase [19], the formin mDia [20], and others [12].
The photo-activatable green fluorescent protein DRONPA has also been used as a light-gated AID [13]. DRONPA and its mutants tetramerize or dimerize and become fluorescent upon irradiation at circa 400 nm. With 500 nm light, fluorescence switches off and the protein complexes fall apart. DRONPA fused to target proteins can multimerize and thereby block the active site. For both the LOV2-based AID and DRONPA, inhibition required precise placement of the photosensitive protein. DRONPA was engineered so that it could be inserted in the middle of protein sequences rather than only at the C-terminus [21].
Optogenetic allosteric control of structural order
Disorder is present everywhere and proteins are no exception. In the protein data bank, only one fourth of protein structures have complete structures because there are missing electron density maps from less ordered regions [22]. The degree and extent of disorder varies for each protein; some are completely disordered (intrinsically disordered proteins), whereas rigid proteins have almost no detectible disorder. Sequence-based prediction algorithms indicate that almost half of the eukaryotic proteins have disordered regions consisting of at least thirty amino acids [23]. Because higher organisms bear more disordered protein segments, it is tempting to speculate that the signaling regulation of complex biological systems incorporates more mechanisms based on protein disorder.
In contrast to the striking transformations of intrinsically disordered proteins, enzymes and other proteins undergo more subtle order/disorder transitions that nonetheless profoundly affect their activity. In kinases, the glycine-rich loop (G-loop), C-helix, and activation loop (A-loop) undergo disorder to order transitions that enhance activity: in the inactive state, the G-loop and the A-loop are close together and collapsed on the substrate binding site. Upon phosphorylation, the A-loop moves away from the G-loop, freeing the site for substrate binding. Although the helix in the A-loop becomes an unstructured loop in the active state, it is held away from the substrate binding site by the conserved DFG site (which coordinates Mg2+/ATP) and a salt bridge between a C-helix glutamate and a catalytic lysine.
We tested whether it is possible to artificially induce order-disorder transitions in the active site of kinases using light or ligand-responsive “sensory domains” inserted at an allosteric site [2,24,25]. In one application, the LOV2 domain was inserted in a surface exposed loop that is allosterically coupled to the ATP binding site of Src kinase [2]. Because the termini of LOV2 are close together (within 15 angstroms) in the dark state, insertion of LOV2 into small exposed surface loops could be accomplished without unfolding the kinase. When LOV2 absorbs blue light, its termini become disordered and perturb the connected kinase domain, resulting in kinase inactivation (intra-domain conformational change in Figure 1). The activity of this photo-inhibitable Src (PI-Src) is fully rescued when the light is removed. Importantly, the extrinsically induced conformational change of PI-Src kinase is similar to the natural conformational change of wild type Src (Figure 2a). The native state of inactive (closed) Src kinase is stabilized by a C-terminal phosphorylated tyrosine (Y527) which binds to the Src SH2 domain. A cascade of modifications including binding of the SH3 domain to another protein and dephosphorylation of Y527 results in opening, phosphorylation of the activation loop and activation. Molecular modeling and molecular dynamics simulations revealed that the structure of PI-Src in the dark closely resembles that of native Src in the active state, whereas PI-Src in the lit state was similar to native inactive Src. Current work, focused on understanding why a structural perturbation at a distant allosteric site shifts Src between its native active and inactive states, can guide engineering of other physiologically relevant transitions. LOV2 in the dark fluctuates between the folded dark state and the partially unfolded lit state with a free energy difference of ΔGdark= 2.4 kcal/mol, whereas blue light absorption induces a conformational change with a free energy difference of ΔGdark= −1.4 kcal/mol [26]. Therefore, a total of 3.8 kcal/mol energy becomes available to distort the attached protein. Considering that the stability of most proteins is on the order of 5 to 15 kcal/mol, this may be just enough to drop PI-Src into the native energy minimum that is the inactive state (Figure 2a).
Figure 2. Allosteric control of the active site.
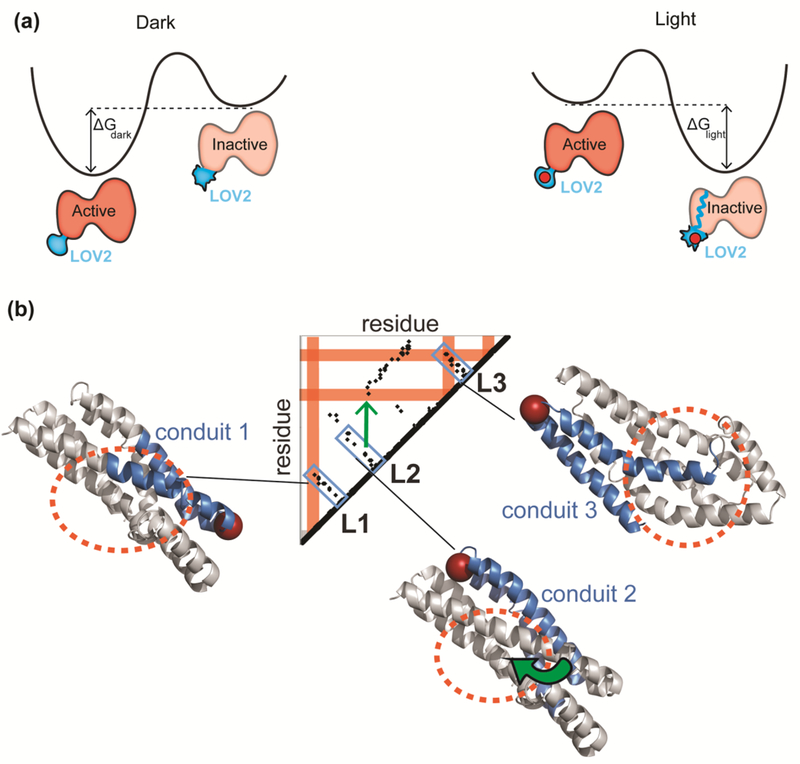
(a) Left: The lit/dark state equilibrium of the LOV2 domain is affected by irradiation [2,26]. In the dark, the active state (Jα helix wound) is favored. When LOV2 is irradiated, a covalent bond formed to the flavin shifts the equilibrium, favoring the inactive state (Jα unwound). The maximum energy that could be derived from this conformational shift in the LOV2 domain alone is 3.8 kcal/mol [26], and protein stabilities range between roughly 5 and 15 kcal/mol [36]. (b) A contact map showing pairwise contacts in the DH domain. The orange circles show the active site of the protein. The blue portions are “conduits” connecting the allosteric insertion sites (shown with red spheres) to the active site. The conduits can be seen as close proximity residues on the contact map. Allosteric effects can propagate through a secondary structure element that connects the loop to the active site (L1, L3) or the loop can communicate with the active site by passing through two secondary structure elements that affect one another at a point of close contact (green arrow).
Like Src kinase, Rac1 GTPase is a switch protein, transitioning between an active and an inactive state. For Rac1, we also identified a surface loop allosterically connected to the active site and there inserted the LOV2 domain [2]. A light-induced conformational change between native states appeared to occur in photo-inhibitable Rac1 (PI-Rac1). The crystal structures of Rac1 and dark-state PI-Rac1 are almost identical. Moreover, the two proteins have very similar activities in cells. As with PI-Src, molecular dynamics simulations indicated that active site regions of PI-Rac1 and wild type Rac1 (switch 1 and 2) undergo similar conformational changes. (Figure 2a).
Is it possible to perturb a domain that does not naturally undergo a transition between two states, and does not have a catalytic activity? We tested this by controlling the catalytic DH domains of guanine exchange factors that are regulated by auto-inhibitory domains (AID). The AID sterically blocks the DH active sites through interaction of broad binding surfaces. Molecular dynamics simulations and hydrogen-deuterium exchange coupled to mass spectrometry showed that the DH interaction surfaces of PI-ITSN1 were perturbed upon irradiation, and inhibition of the DH domain could therefore be controlled by light. Extrinsic disorder was also able to control rigid AID domains that do not undergo intra-domain transitions (e.g. the CH domain of Vav2 [2]) and anti-CRISPR proteins [27]. Synthetic induction of these conformational changes suggests that regulatory order-disorder transitions may occur in Nature, perhaps controlled by pH, temperature, oxidative stress, light, mechanical force, membrane interaction, or ligand binding [23].
From the standpoint of protein engineering, extrinsic allosteric control of disorder is a flexible approach, because the responsive domain can be inserted away from the active site and because the domain does not need to be at the target’s termini. To find LOV2 insertion sites for allosteric control, we developed a workflow that begins with the identification of non-conserved, surface exposed loops in a specific length range (so-called tight loops (Figure 2b)). Allosteric coupling between these loops and the active site can be detected using a pairwise distance contact map, or using motion correlations computed through molecular dynamics simulations [2]. In a contact map, points indicate close contacts between two residues, often because they reside within interacting structural units (e.g. between parallel interacting helices (Figure 2b)). A series of close contacts running through the interior of a protein can connect a surface loop to the active site; this was found to predict an allosteric connection. A perturbation produced at the allosteric loop through disordering a sensory can induce disorder in a distant active site. Making an optimum switch that mimics the activity of both the on and off states typically requires optimization of the linkers connecting the sensory domain to the host protein [28].
Control of reassembly of split proteins
A potential problem with single chain conformational switches is undesired off-state activity (‘leakiness’). This background activity can be reduced by introducing structural perturbation specifically into the off state of the target protein. In fact, a protein can be split into two inactive halves, which are reassembled by chemogenetic or optogenetic dimerizers to produce the intact protein [11,29]. To control protein activity through splitting one must find split sites that reassemble only when triggered, and that do not cause irreversible protein misfolding. We recently developed an algorithm named split proteins reassembled by ligands or light (SPELL) that uses an engineered small domain to generate chemogenetic and optogenetic split proteins with reduced background activity and effective split sites [30]. For each potential split site, one computes the “split energy”, the loss of interaction energy between the two split halves. Iterative computation of the split energy for each residue provides a split energy profile that reveals hidden subdomains within which split sites should be avoided.
Using the split energy profile helps pinpoint promising split sites, but the half proteins may undergo spontaneous assembly. To address this, we used a truncated version of the FKBP12 domain with termini brought close together for insertion into tight loops (insertable FKBP, or iFKBP, with termini 7 angstroms apart). When it is free of its ligands rapamycin and FRB, this domain is more disordered and destabilizes the protein half it is attached to. When the small molecule rapamycin is added, the protein half bearing iFKBP can form a ternary complex with rapamycin and with the FRB that is fused to the other half protein [24,25,31–33]. Because the half protein linked to iFKBP is destabilized it is less likely to spontaneously assemble until it interacts with the FRB fused to the other half protein. This interaction both induces assembly and stabilizes the assembled protein. This approach was successfully applied to proteins with varied topologies and sizes, and we have generated rapamycin analogs that bear a photoreactive protecting group so they can only bind iFKBP after irradiation [30].
Conclusions
The optogenetic tools described above are based on three different conformational switches: (1) a light-sensitive domain positioned where it sterically inhibits the target protein until it is irradiated (2) an inserted light-sensing domain that allosterically distorts the target protein’s active site upon irradiation, and (3) light-induced reassembly of protein halves. For both steric inhibition and split proteins, inactive forms can act as dominant negative analogs, unless mutations can be introduced to eliminate all interactions other than those with desired downstream targets. If full regulation of the activated protein is desired this approach is not feasible, but often one simply wants to control specific activities of the targeted protein. Careful regulation of expression level (e.g. with inducible expression) is required when dark state activity is high, or when small differences in protein activity produce biological effects. Light-induced allosteric effects (approach 2) have the potential to avoid these problems because they can be used to make intact protein analogs with active sites or regulatory sites far from the site where the light-sensitive domain was inserted. However, as with other engineered proteins, the activity of the photo-inhibitable analog in the dark should be close to that of inactive wild type protein.
The LOV2 domain is photo-activated in under a second. The rate at which LOV2 returns to the dark state can be adjusted with mutations, to provide a t1/2 of 1.7 seconds to over 10 minutes at 37 °C [10,34]. Fast mutants are valuable for dynamic processes happening in seconds, while mutants enabling slow switching can be useful for processes with long time courses, such as gene expression or animal studies, because prolonged activation can be produced simply by brief irradiation every few minutes. As with other ligand-mediated dimerizers [11,29], the SPELL approach is largely irreversible due to the subnanomolar affinity of rapamycin and the iFKBP/FRB pair. Differences in expression level of the split halves and their mislocalization can also decrease the efficiency of split proteins [35].
Genetic perturbations such as gene knock-out and mRNA knock-down have produced groundbreaking discoveries, but examining rapid and dynamic processes requires faster perturbation techniques that do not lead to compensatory changes. Small molecule inhibitors or activators are useful for acute perturbation, but their target specificity is a major concern. Ideally, the techniques described above can have both absolute specificity and activation within seconds. The unprecedented spatial and temporal control provided by chemogenetics and optogenetics can pave the way to explore a world of cell regulation occurring on seconds and at least micron scales.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, Hahn KM: A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009, 461:104–0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dagliyan O, Tarnawski M, Chu PH, Shirvanyants D, Schlichting I, Dokholyan NV, Hahn KM: Engineering extrinsic disorder to control protein activity in living cells. Science 2016, 354:1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trudeau T, Nassar R, Cumberworth A, Wong ET, Woollard G, Gsponer J: Structure and intrinsic disorder in protein autoinhibition. Structure 2013, 21:332–341. [DOI] [PubMed] [Google Scholar]
- 4.Pufall MA, Graves BJ: Autoinhibitory domains: modular effectors of cellular regulation. Annu Rev Cell Dev Biol 2002, 18:421–462. [DOI] [PubMed] [Google Scholar]
- 5.Darling AL, Uversky VN: Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front Genet 2018, 9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nussinov R, Tsai CJ: Allostery without a conformational change? Revisiting the paradigm. Curr Opin Struct Biol 2015, 30:17–24. [DOI] [PubMed] [Google Scholar]
- 7.Levskaya A, Weiner OD, Lim WA, Voigt CA: Spatiotemporal control of cell signaling using a light-switchable protein interaction. Nature 2009, 461:997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL: Rapid blue-light-mediated induction of protein interactions in living cells. Nature Methods 2010, 7:973–U948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J, Heo WD: Reversible protein inactivation by optogenetic trapping in cells. Nat Methods 2014, 11:633–636. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, Kuhlman B, Liu R, Danuser G, Hahn KM: LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods 2016, 10.1038/nmeth.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q, Tucker CL: Engineering genetically-encoded tools for optogenetic control of protein activity. Curr Opin Chem Biol 2017, 40:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weitzman M, Hahn KM: Optogenetic approaches to cell migration and beyond. Curr Opin Cell Biol 2014, 30:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou XX, Chung HK, Lam AJ, Lin MZ: Optical Control of Protein Activity by Fluorescent Protein Domains. Science 2012, 338:810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Changeux JP, Edelstein S: Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol Rep 2011, 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang XB, He L, Wu YI, Hahn KM, Montell DJ: Light-mediated activation reveals a key role for Rac in collective guidance of cell movement in vivo. Nature Cell Biology 2010, 12:591–U154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dietz DM, Sun H, Lobo MK, Cahill ME, Chadwick B, Gao V, Koo JW, Mazei-Robison MS, Dias C, Maze I, et al. : Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci 2012, 15:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi-Takagi A, Yagishita S, Nakamura M, Shirai F, Wu YI, Loshbaugh AL, Kuhlman B, Hahn KM, Kasai H: Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 2015, 525:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cosentino C, Alberio L, Gazzarrini S, Aquila M, Romano E, Cermenati S, Zuccolini P, Petersen J, Beltrame M, Van Etten JL, et al. : Optogenetics. Engineering of a light- gated potassium channel. Science 2015, 348:707–710. [DOI] [PubMed] [Google Scholar]
- 19.Mills E, Chen X, Pham E, Wong S, Truong K: Engineering a Photoactivated Caspase-7 for Rapid induction of Apoptosis. Acs Synthetic Biology 2012, 1:75–82. [DOI] [PubMed] [Google Scholar]
- 20.Baarlink C, Wang HC, Grosse R: Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 2013, 340:864–867. [DOI] [PubMed] [Google Scholar]
- 21.Zhou XX, Fan LZ, Li P, Shen K, Lin MZ: Optical control of cell signaling by single-chain photoswitchable kinases. Science 2017, 355:836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bardwell JC, Jakob U: Conditional disorder in chaperone action. Trends Biochem Sci 2012, 37:517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakob U, Kriwacki R, Uversky VN: Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function. Chem Rev 2014, 114:6779–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dagliyan O, Karginov AV, Yagishita S, Gale ME, Wang H, DerMardirossian C, Wells CM, Dokholyan NV, Kasai H, Hahn KM: Engineering Pak1 Allosteric Switches. ACS Synth Biol 2017, 6:1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dagliyan O, Shirvanyants D, Karginov AV, Ding F, Fee L, Chandrasekaran SN, Freisinger CM, Smolen GA, Huttenlocher A, Hahn KM, et al. : Rational design of a ligand-controlled protein conformational switch. Proc Natl Acad Sci U S A 2013, 110:6800–6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao X, Rosen MK, Gardner KH: Estimation of the available free energy in a LOV2-J alpha photoswitch. Nat Chem Biol 2008, 4:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bubeck F, Hoffmann MD, Harteveld Z, Aschenbrenner S, Bietz A, Waldhauer MC, Borner K, Fakhiri J, Schmelas C, Dietz L, et al. : Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat Methods 2018, 15:924–927. [DOI] [PubMed] [Google Scholar]
- 28.Ohlendorf R, Schumacher CH, Richter F, Moglich A: Library-Aided Probing of Linker Determinants in Hybrid Photoreceptors. ACS Synth Biol 2016, 5:1117–1126. [DOI] [PubMed] [Google Scholar]
- 29.Shekhawat SS, Ghosh I: Split-protein systems: beyond binary protein-protein interactions. Curr Opin Chem Biol 2011, 15:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dagliyan O, Krokhotin A, Ozkan-Dagliyan I, Deiters A, Der CJ, Hahn KM, Dokholyan NV: Computational design of chemogenetic and optogenetic split proteins. Nat Commun 2018, 9:4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM: Engineered allosteric activation of kinases in living cells. Nature Biotechnology 2010, 28:743–U1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu PH, Tsygankov D, Berginski ME, Dagliyan O, Gomez SM, Elston TC, Karginov AV, Hahn KM: Engineered kinase activation reveals unique morphodynamic phenotypes and associated trafficking for Src family isoforms. Proc Natl Acad Sci US A 2014, 111:12420–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karginov AV, Tsygankov D, Berginski M, Chu PH, Trudeau ED, Yi JJ, Gomez S, Elston TC, Hahn KM: Dissecting motility signaling through activation of specific Src-effector complexes. Nat Chem Biol 2014, 10:286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zayner JP, Sosnick TR: Factors that control the chemistry of the LOV domain photocycle. PLoS One 2014, 9:e87074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler T, Moglich A: Photoreceptor engineering. Front Mol Biosci 2015, 2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fersht A, Fersht UA: Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding: W. H. Freeman; 1999. [Google Scholar]