

Abstract
Rationale.
Mitragyna speciosa (kratom) may hold promise as both an analgesic and treatment for opioid use disorder. Mitragynine, its primary alkaloid constituent, is an opioid receptor ligand. However, the extent to which the in vivo effects of mitragynine are mediated by opioid receptors, or whether mitragynine interacts with other opioid agonists, is not fully established.
Objectives.
The effects of mitragynine and the prototypical opioid agonist morphine were compared for their capacity to decrease operant responding for food delivery, and to increase response latency to a thermal stimulus.
Methods.
Male and female Sprague-Dawley rats responded under a multiple cycle fixed ratio 10 schedule of food delivery and were tested on a hot plate (52°C) immediately after each cycle. Morphine and mitragynine were administered alone, in combination with each other, and in combination with the opioid antagonist naltrexone.
Results.
Morphine and mitragynine dose-dependently decreased schedule-controlled responding; the ED50 values were 7.3 and 31.5 mg/kg, respectively. Both drugs increased thermal antinociception (the ED50 value for morphine was 18.3). Further, doses of naltrexone that antagonized morphine did not antagonize mitragynine. Mitragynine (17.8 mg/kg) did not alter the rate-decreasing or antinociceptive effects of morphine.
Conclusions.
The antinociceptive effects of mitragynine and morphine occur at doses larger than those that disrupt learned behavior. Opioid receptors do not appear to mediate the disruptive effects of mitragynine on learned behavior. Mitragynine had lesser antinociceptive effects than morphine, and these did not appear to be mediated by opioid receptors. The pharmacology of mitragynine includes a substantial non-opioid mechanism.
Keywords: pain, kratom, analgesia, hot plate, opioid, rodent
Introduction
The plant Mitragyna speciosa is native to the Southeast Asian countries of Malaysia and Thailand, where it is commonly grown by locals; its leaves are brewed as a tea for ceremonial and recreational purposes (Gong et al., 2012; Hassan et al., 2013). Additionally, field workers in these countries chew the leaves to boost mood, and increase energy (Jansen & Prast, 1988). As far back as 1836 the leaves of Mitragynia speciosa, also referred to as kratom, have been reported to have analgesic properties and opioid-like effects (Swogger et al., 2015; Boyer et al., 2008; Jansen & Prast, 1988; Burkill & Haniff, 1930). Amid the current United States opioid epidemic, these reports have led to considerable consumer interest in kratom. However, use of kratom in the United States as a dietary supplement has been associated with serious adverse reactions including seizures and in isolated incidences, death, which in 2016 caused the United States Drug Enforcement Administration (DEA) to briefly consider placing two kratom-related alkaloid constituents (mitragynine and 7-hydroxymitragynine) under schedule I of the Controlled Substances Act (US Drug Enforcement Agency, 2016). Yet, only limited information about kratom’s pharmacology currently exists, which has led to the inability to make a scientifically sound decision on its therapeutic versus adverse effects (Henningfield et al., 2018). Thus, there is a need to gather pharmacological information on the in vivo effects of kratom and its constituent alkaloids.
Kratom contains over 30 unique alkaloids, including mitragynine and 7-hydroxymitragynine (Hassan et al., 2013; Gogineni et al., 2014). Mitragynine comprises ~60% of the alkaloid fraction of kratom, is thought to be one of kratom’s main psychoactive components (Prozialeck et al., 2012), and has been characterized in vitro to bind and act as an agonist at mu- and kappa-opioid receptors, as well as at alpha2 adrenergic receptors (Boyer et al., 2008; Kruegel et al., 2016; Varadi et al., 2016; Yue et al., 2018). Specifically, mitragynine has a Ki value of 502 ± 19.4 for mu-opioid receptors, 1200 ± 79.7 for kappa-opioid receptors and 7910 ± 1140 for delta-opioid receptors (Yue et al., 2018). In comparison, morphine has a Ki value of 2.33 ± 0.261 for mu-opiod receptors, 82.4 ± 3.92 for kappa-opioid receptors and 105 ± 11.5 for delta-opioid receptors (Yue et al., 2018). Previous studies have shown that mitragynine can produce antinociceptive effects in rodent assays of thermal nociception. Specifically, it has been found that mitragynine produces antinociceptive effects in the hot plate assay, in both rats and mice, which are antagonized by both naltrexone and naltrindole, and suggests an opioid-related analgesic effect profile (Shamima et al., 2012; Carpenter et al., 2016). Mitragynine has been trained as a discriminative stimulus in the drug discrimination procedure, with morphine producing full substitution in mitragynine-trained rats and conversely, with mitragynine producing substitution in morphine-trained rats (Harun et al., 2015). Recently, it was found that i.v. mitragynine was not self-administered when it was substituted for morphine (Hemby et al., 2018) or methamphetamine in rats (Yue et al., 2018). Collectively, these results suggest that mitragynine may be useful as a medication for opioid abuse. However, more evidence is needed on the preclinical effects of mitragynine, specifically with regard to the interactions of mitragynine with opioid receptors and with opioid drugs in behavioral assays that are sensitive to the pharmacological effects of opioids.
Here we examined the effects of mitragynine and morphine alone and in combination in two different behavioral assays that are sensitive to both opioids and non-opioids: schedule-controlled responding and antinociception. We specifically examined schedule-controlled responding as it has been previously utilized to examine the neuropharmacology of various classes of opioids (Harris, 1980). Accordingly, one goal of the present study was to examine the extent to which the effects of mitragynine are mediated by opioid receptors. This was accomplished by combining the opioid antagonist naltrexone with both mitragynine and the well-characterized opioid agonist morphine. A second goal was to examine whether mitragynine can modify the effects of morphine. An opioid receptor antagonist was reported to block the antinociceptive effects of mitragynine (Shamima et al., 2012), whereas mitragynine was reported to decrease opioid self-administration (Hemby et al., 2018), and in particular to produce a downward shift in the self-administration dose-response curve of heroin (Yue et al., 2018). Mitragynine therefore appears to produce opioid agonist activity in vivo, although its potential to exert opioid antagonist activity, as well as non-opioid receptor mediated effects, cannot be discounted. We hypothesized that the addition of an ineffective dose of mitragynine would alter the analgesic and operant rate-decreasing effects of morphine. In addition to establishing the relative potency of mitragynine and morphine administered i.p., the onset and duration of effects of both drugs were compared.
Methods
Animals
Adult male (n=8) and female (n=8) Sprague Dawley rats (245-281 gram upon arrival, Jackson Laboratory, Charleston, SC) were singly housed in a temperature-(20–22 °C), humidity-(55 ± 10 %), and light-controlled (12 hour light/dark; lights on at 0600 hours) facility that was approved by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Water was available ad libitum. Rats were food restricted to 90% of their free-feeding body weight, with access to food (Dustless Precision Pellets Grain-Based Rodent Diet, Bio-Serv, Frenchtown, NJ) 30 min following daily experimental sessions, as well as to food available during experimental sessions as described below.
Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council, 2011). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Drugs
(−)-Morphine sulfate pentahydrate (morphine) and (−)-naltrexone hydrochloride (naltrexone) were obtained from the National Institute on Drug Abuse Research Technology Branch (Rockville, MD) and Sigma-Aldrich (St. Louis, MO). Mitragynine was isolated as follows: The alkaloid enriched fraction was obtained by usual acid-base extraction of the ethanol (190 proof) extract from the leaves of M. speciosa (1000 g, purchased from Pure Land Ethnobotanicals) using a protocol originally described by Ponglux et al. (1994) and Ali et al. (2014). The alkaloid fraction (5 g) was fractionated using vacuum silica gel (5” x 4.2”) column chromatography with gradients increasing the polarity from hexanes to ethyl acetate (EtOAc). The fraction was eluted with hexanes/EtOAc (7:3) and was subjected to successive medium pressure chromatography columns using a combiflash instrument in isocratic mobile phase hexanes-EtOAc (8:2) to yield mitragynine (2.4 g). Mitragynine as (−)-mitragynine hydrochloride salt was subjected to spectroscopic analysis and found to contain > 99% purity Figure 1. The hydrochloride salt of mitragynine was prepared and dissolved in a vehicle consisting of 20% Tween 80 and 80% sterile saline (0.9 % NaCl). Morphine and naltrexone were also dissolved in sterile saline containing 20% Tween 80. Drugs and vehicle were administered intraperitoneally (i.p.). Doses of mitragynine higher than 56 mg/kg produced lethal effects in a small pilot study (not shown), thus the top of the dose range of mitragynine in these studies was 56 mg/kg.
Figure 1.

Purity analysis of mitragynine. A. 1H NMR spectra of mitragynine (Methanol-d/4). Each signal shows the presence of a proton in mitragynine HC1 and the total signals observed in the resultant spectra are consistent with those for mitragynine in previously published data. B. HPLC-PDA chromatogram of mitragynine at a wavelength of 222 nm showing ≥99% purity. C. Parent Ion scan (MS1) of mitragynine using high resolution mass spectrometer (HR-MS) ≥99% purity.
Schedule-Controlled Responding
Apparatus
Operant conditioning chambers were 25 cm long, 31 cm high and 25 cm wide, as commercially supplied by Med Associates (Fairfax, VT). Each chamber was equipped with two centrally-mounted, 5 cm-long levers located 9 cm from the chamber floor and 3 cm from either wall. When operated, a pellet dispenser delivered a 45-mg food pellet (soy-free PJAI, Test Diet, St. Louis, MO) into a pellet trough, which was centrally mounted between the two levers. Located above each lever was a stimulus light. Houselights were centrally mounted on the ceiling. All chambers were equipped with a fan, which supplied ventilation and white noise.
Operant Procedures
Rats were trained to lever press seven days a week, as previously described (McMahon & Cunningham, 2001), but modified for food reinforcement. Initially rats were placed in operant conditioning chambers for 30-90 min, under a fixed ratio (FR)1 schedule, where one press on the lever designated by illumination of the light above the lever resulted in delivery of one 45-mg pellet; the second lever was inactive. Rats could receive a maximum of 50 pellets; once response rate (responses per s) during each of three consecutive sessions deviated by no more than +/−20% of the 3-day running average, the FR was increased in the following increments: FR3, FR6, FR10. Responding under the FR10 schedule continued until response rate was stable, defined as three consecutive sessions that deviated by no more than +/−20% of the 3-day running average. Thereafter, sessions were divided into consecutive, discrete, 20-min cycles. Each cycle began with a 15-min timeout during which the stimulus light was not illuminated and responses on the lever had no programmed consequence. The timeout was followed by a 5-min period during which food pellets were available under the FR10 schedule. Each session consisted of up to six cycles. Completion of 10 responses resulted in delivery of a food pellet; a maximum of 10 pellets could be delivered per cycle. Rats received vehicle or drug injections within the first min of a given cycle. Drug tests were initiated once responding stabilized, i.e., responding during each of three consecutive sessions deviated by no more than +/−20% of the 3-day running average calculated for all cycles in all sessions.
Hot plate Assay
Antinociceptive testing was performed in the hot plate test, as previously described (Ignatowska-Jankowska et al., 2015), but modified for rats. Rats were placed on a heated (52°C) enclosed Hot Plate Analgesia Meter (Columbus Instruments, Columbus, OH) and latency to jump or lick/shake the back paws was determined. If there was no response within 60 s, the rat was removed from the apparatus.
Experimental Design
Immediately following one 20-min cycle in the operant conditioning procedure, rats were tested in the hot plate assay. After hot plate testing rats were injected with vehicle or a drug dose and returned to the operant conditioning chamber for testing in the next cycle. As shown in Supplemental Figure 2, exposure to the hot plate did not modify operant response rates, and vice versa, through six consecutive 20-min cycles. For the time course study of morphine and mitragynine, the length of time between testing was adjusted to correspond with the times indicated. For all studies, a within-subject design was used for both operant and hot plate measurements. After each drug test session, rats received a minimum 48-hour washout period before receiving the next drug. During these washout periods, rats were given daily sessions consisting of 3-6 cycles to lever press for food, without being exposed to the hot plate apparatus.
Data analysis
A within-subject design was used to test dose and dose combinations and the order of the experimental test days was non-systematic. For each test day, doses where systematically and cumulatively increased. A daily baseline was collected for use in the analysis of hot plate data. Hot plate data were converted to percent maximum possible effect (% MPE) with the following equation: ([(experimental test value - daily baseline value) / (60 sec - daily baseline value)] ×100) and plotted versus log dose values. Lever-press response rates were expressed as a percentage of control for each rat, defined as the mean response rate from the previous three non-drug sessions, with each session defined as the individual cycles for that session averaged together. In each experiment, male and female data were analyzed together in all data analyses. The dose-effect curves of morphine were determined both initially, as well as at the end of all experiments, and were compared statistically. No tolerance was found in the dose effect curve of morphine to suppress rates of responding, as determined by a repeated-measures 2-way ANOVA [interaction between treatment and time: (F5,156=0.800; P < 0.551)], (Supplemental Figure 1A). However, tolerance had developed to the analgesic effects of morphine [significant interaction between treatment and time: F5,156=4.77; P < 0.001)], (Supplemental Figure 1B). As experiments were completed in a randomized order, for all experiments, the initial dose response of morphine is utilized for graphing and analysis. When the mean effect of a drug to produce a reduction in schedule-controlled responding, or an increase in thermal antinociception was greater than 50% MPE, the ED50 values and corresponding 95% confidence limits were calculated using linear regression, where slopes were allowed to vary, according to Tallarida (2000). If the mean effect of a drug did not produce a 50% or greater effect, an ED50 value was not generated. Potency ratios and 95% CL were calculated as the ratio of ED50 values calculated from the dose-response curves, and a potency ratio not including 1 within the 95% CL indicated a statistically significant difference in potency. For the comparison of morphine and mitragynine potency ratios, as both compounds are in a salted form, with different free-base/salt molecular weight ratios (0.75 for morphine and 0.92 for mitragynine), potency ratios were calculated in terms of their respective free-base molecular weight. In mitragynine thermal analgesia studies, ED50 and potency ratio values could not be calculated, thus a two-way repeated measures ANOVA was used to compare effects. All dose response data were analyzed using a one-way repeated measures analysis of variance (ANOVA) and all time course data were analyzed using a two-way repeated measures ANOVA. A Bonferroni comparison was used for post-hoc analysis following a significant ANOVA (P < 0.05). SigmaPlot version 14.0 (Systat Software Inc., San Jose, CA) was used in all statistical analyses. All data are expressed as the mean +/− SEM.
Results
Effects of morphine and mitragynine on schedule-controlled responding and latency to respond to thermal stimuli.
Figure 2 shows the effects of vehicle, morphine, and mitragynine on the rate of fixed ratio responding and latency to respond in the hot plate test. Repeated injections of vehicle did not alter response rates (Supplemental Figure 2A, the range of absolute rates of responding was 0.88-0.94 responses/s) or nociceptive latency in the hot plate assay (Supplemental Figure 2B, 15.2-16.9 s). Thus, stable baselines were obtained across the six cycles in both schedule-controlled responding, as well as nociceptive latency. Morphine dose-dependently decreased response rates F5,75= 38.9; P < 0.001 (Figure 2A) and increased thermal latency in the hot plate assay F5,75= 34.1; P < 0.001 (Figure 2B). Doses of 3.2 and 5.6 mg/kg had no effect on either operant behavior or hot plate latency. Meanwhile the dose of 10 mg/kg morphine significantly decreased operant behavior but did not alter thermal latency, and the doses of 17.8 and 32 mg/kg significantly decreased operant responding and increased thermal latency. The ED50 value of morphine to produce reductions in operant responding was 7.3 (95% confidence limits: 6.4 - 8.4) mg/kg. The ED50 value of morphine to produce thermal antinociception was 18.3 (95% confidence limits: 15.1 −22.2) mg/kg. Operant responding was 2.5-fold more sensitive to the effects of morphine than was thermal nociception (potency ratio: 2.5 (95% confidence limits: 2.4 - 2.6).
Figure 2.
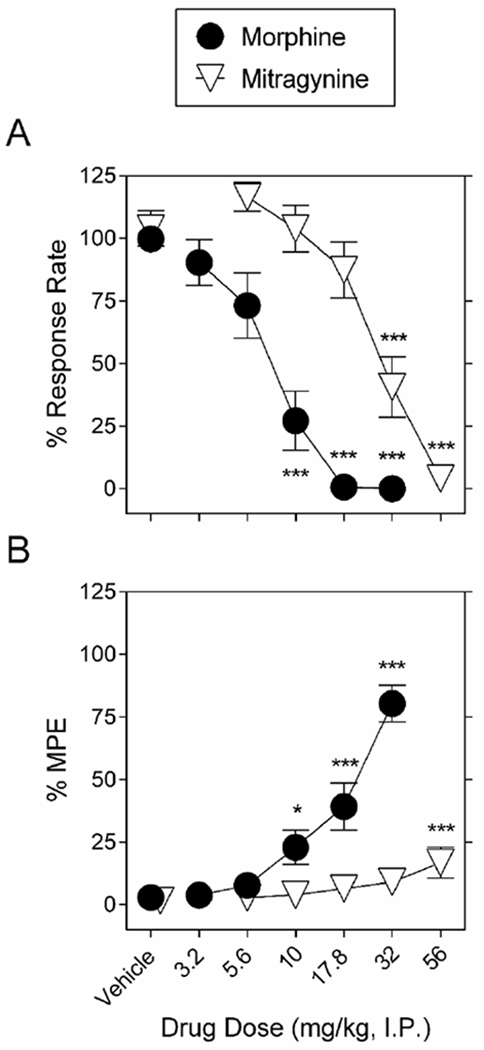
Morphine and mitragynine dose-dependently decrease schedule-controlled behavior and increase thermal response latency. A. Morphine (filled circles) and mitragynine (downward-facing open triangles) dose-dependently decrease schedule-controlled behavior. Ordinates: food-maintained responding expressed as a percentage of control. B. Morphine (filled circles) and mitragynine (downward-facing open triangles) dose-dependently increase thermal response latency. Ordinates: thermal antinociception expressed as a percentage of maximum possible effect (MPE). Abscissae for both A, B: drug dose (mg/kg, I.P.), log scale. Each point represents the mean ± S.E.M. of data points for sixteen (eight males and eight females) subjects. *P < 0.05, ***P < 0.001 vs. vehicle.
Figure 2 also shows the effects of mitragynine on operant responding and hot plate latency. Mitragynine dose-dependently decreased operant responding F5,75=33.9; P < 0.001 (Figure 2A) and increased thermal latency in the hot plate assay F5,75=4.37; P = 0.002 (Figure 2B). Mitragynine doses of 5.6, 10 and 17.8 mg/kg had no effect on either operant responding or hot plate latency. The dose of 32 mg/kg mitragynine significantly decreased operant behavior but did not alter thermal latency, and the dose of 56 mg/kg significantly decreased operant responding and increased thermal response latency. The ED50 value of mitragynine to decrease operant response rate was 31.5 (95% confidence limits: 20.4 - 48.6) mg/kg. Potency ratio analysis confirmed that morphine was more potent than mitragynine to decrease operant responding (5.3 (95% confidence limits: 3.9 - 7.1)). A t-test comparing 32 mg/kg morphine and 56 mg/kg mitragynine indicated that the magnitude of thermal latency after morphine was greater than that for mitragynine (P < 0.001). Because a dose 100 mg/kg of mitragynine was lethal, further studies were conducted with doses equal to or smaller than 56 mg/kg.
Time course of morphine and mitragynine on schedule-controlled responding and latency to respond to thermal stimuli.
The following doses were chosen to determine the time course of effects: the lowest dose of morphine that significantly decreased schedule-controlled responding and increased thermal latency (17.8 mg/kg); and 56 mg/kg mitragynine, which altered both schedule-controlled responding and thermal latency. The time course for the effects of 17.8 mg/kg morphine as well as 56 mg/kg mitragynine on operant responding and latency to respond to thermal sensitivity are shown in Figure 3. At 15-120 min post-injection, both morphine and mitragynine significantly decreased operant responding [significant interaction between treatment and time: (F5,40=3.33; P < 0.05) and (F5,70=4.65; P < 0.05), respectively] whereas thermal nociceptive latency was only increased at 30-60 min post-injection for both morphine [significant interaction between treatment and time: (F5,40=3.61; P < 0.05)] and mitragynine [significant effects of treatment (F1,14=5.16; P < 0.05) and time: (F5,70=4.44; P < 0.05)], (Figure 3). That is, the onset of action of both morphine and mitragynine to decrease operant responding was less than or equal to 15 min. The effects of morphine on response rate, but not hot plate latency, were significant at 120 min, and the effects of mitragynine on response rate, but not hot plate latency were significant at 120-240 min (Figure 3).
Figure 3.
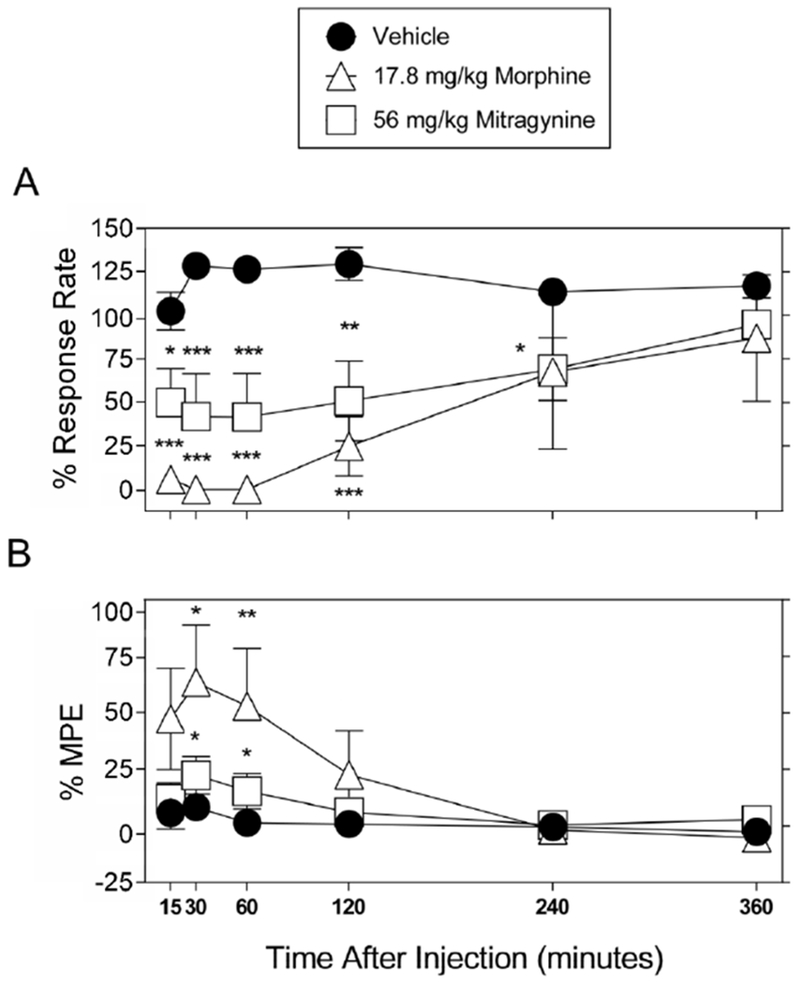
Effects of morphine and mitragynine on schedule-controlled responding and thermal nociception as a function of time. A. Time course of morphine (17.8 mg/kg, upward-facing open triangles), mitragynine (56 mg/kg, open squares), or vehicle (filled circles) effects on schedule-controlled responding. Ordinates: Food-maintained responding expressed as a percentage of control. B. Time course of morphine (17.8 mg/kg, upward-facing open triangles), mitragynine (56 mg/kg, open squares), or vehicle (filled circles) effects on thermal nociception. Ordinates: thermal antinociception expressed as a percentage of maximum possible effect (MPE). Abscissae: time after injection; Each point represents the mean ± S.E.M. of data points for eight (vehicle and 56 mg/kg mitragynine, four males and four females), or five (17.8 mg/kg morphine, three males and two females) subjects. *P < 0.05, **P < 0.01, and *** P < 0.001 vs. vehicle injection at each time point (two-way repeated-measures ANOVA).
Effects of 0.032 and 1 mg/kg naltrexone on morphine and mitragynine-induced changes in operant responding and latency to respond to thermal stimuli.
By themselves, neither 0.032 nor 1 mg/kg naltrexone administered during the first cycle followed by vehicle in subsequent cycles, altered either rates of responding (Supplemental Figure 2A) or nociceptive latency in the hot plate assay (Supplemental Figure 2B). Thus, stable baselines were obtained across the six cycles for both schedule-controlled responding and nociceptive latency. Morphine in the presence of either 0.032 or 1 mg/kg naltrexone still significantly reduced schedule-controlled responding (F5,75= 32.5; P < 0.001, and F5,55=20.4; p<0.001, respectively) and produced antinociception (F5,75= 68.1; P < 0.001, and F5,55=3.48; p=0.008, respectively). However, as indicated by a rightward shift in dose-response functions (Figure 4 panels A and B), both 0.032 and 1 mg/kg naltrexone antagonized the effects of morphine on operant responding and latency to respond to a thermal stimulus. After either 0.032 or 1 mg/kg naltrexone, doses of 10 and/or 17.8 mg/kg morphine no longer significantly modified operant response rate or hot plate latency; doses of morphine equal to or larger than 32 mg/kg were required to decrease operant responding and to increase thermal latency (Figure 4 panels A and B). In the presence of 0.032 mg/kg naltrexone, the ED50 values of morphine to produce reductions in operant responding and to increase thermal latency were 28.0 (17.7 - 44.2) and 55.9 (42.8 - 73.2) mg/kg, respectively. In the presence of 1 mg/kg naltrexone, the ED50 value of morphine to produce reductions in operant responding was 32.2 (17.3 - 59.9) mg/kg. A potency ratio analysis confirmed significant antagonism of morphine by 0.032 mg/kg naltrexone, as evidenced by a 3.8 (95% confidence limits: 2.8 - 5.3) -fold rightward shift in the morphine dose-response function to decrease operant responding and a 3.1 (95% confidence limits: 2.8 - 3.3) - fold rightward shift in the morphine dose-response curve to increase latency to a thermal stimulus. Meanwhile, a potency ratio analysis confirmed significant antagonism of morphine by 1 mg/kg naltrexone, as evidenced by a 4.4 (95% confidence limits: 2.7 - 7.1) -fold rightward shift in the morphine dose-response function to decrease operant responding.
Figure 4.
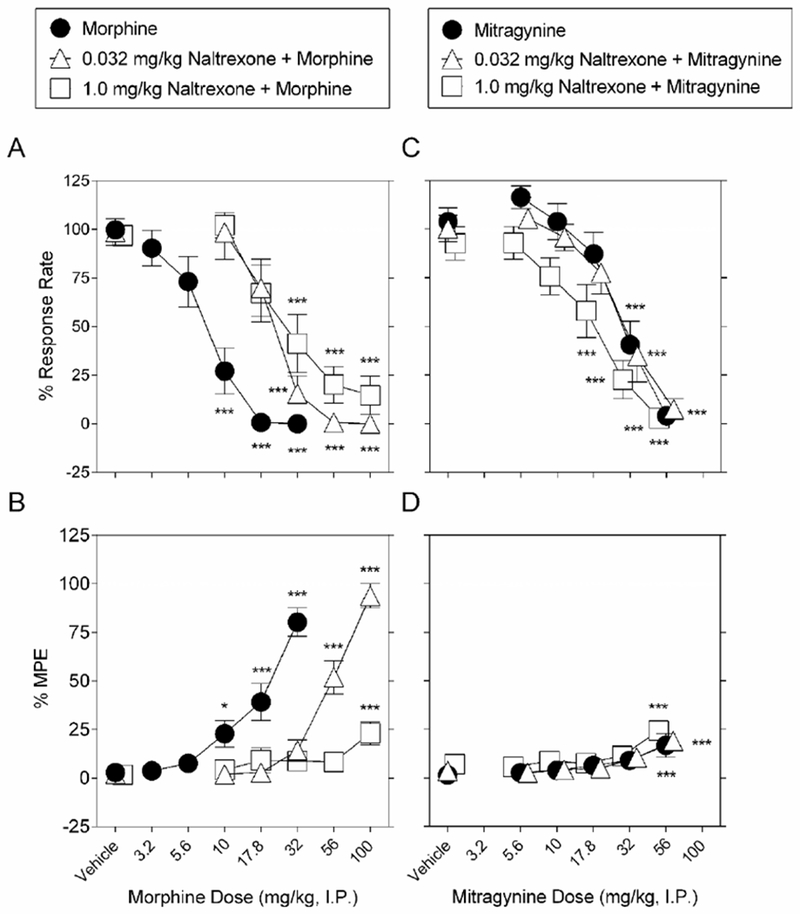
Effects of pretreatment with naltrexone on morphine-induced changes in schedule-controlled responses and thermal nociception. Pretreatment with either 0.032 mg/kg naltrexone (upward-facing open triangles) or 1 mg/kg naltrexone (open squares) antagonizes the dose-response effect of morphine with vehicle pretreatment (filled circles), to dose-dependently A. decrease schedule-controlled behavior, and B., increase thermal latency. Meanwhile, pretreatment with either 0.032 mg/kg naltrexone (upward-facing open triangles) or 1 mg/kg naltrexone (open squares) does not antagonize the dose-response effect of mitragynine with vehicle pretreatment (filled circles), to dose-dependently C. decrease schedule-controlled behavior, and D., increase thermal latency. Abscissae: drug dose (mg/kg, I.P.), log scale. Ordinates: A, C: Percent control rates of food-maintained responding. B, D: Percent maximum possible effect (MPE). Each point represents the mean ± S.E.M. of data points for 16 subjects (eight males and eight female for morphine alone and in combination with 0.032 mg/kg naltrexone and mitragynine alone and in combination with 0.032 mg/kg naltrexone), 12 (six males and six female for morphine in combination with 1.0 mg/kg naltrexone), and 13 (seven males and six female for mitragynine in combination with 1.0 mg/kg naltrexone). **P < 0.01, and ***P < 0.001 vs. data during the first cycle (i.e., either vehicle or naltrexone). The morphine control is re-plotted from Figure 2.
In the presence of either 0.032 or 1 mg/kg naltrexone, mitragynine still significantly reduced schedule-controlled responding (F5,75= 29.9; P < 0.001, and F5,60=21.2; P < 0.001, respectively) and produced antinociception (F5,75=7.04; P < 0.001, and F5,60=6.23; P < 0.001, respectively). In the presence of 0.032, 1 mg/kg naltrexone, the ED50 values of mitragynine to produce reductions in operant responding were 32.0 (21.0 - 49.0) and 18.1 (12.0 - 27.3) mg/kg, respectively. Figure 4 shows that neither 0.032 nor 1 mg/kg naltrexone altered the effects of mitragynine on operant responding, with a potency ratio analysis confirming no change in effect (1.0 (95% confidence limits: 1.0 - 1.0) and (0.57 (95% confidence limits: 0.2 - 1.2), respectively). Thermal antinociception after a dose of 56 mg/kg mitragynine did not differ in the presence versus the absence of either 0.032 or 1 mg/kg naltrexone pretreatment.
Effects of 17.8 mg/kg mitragynine on morphine-induced changes in operant responding and latency to respond to a thermal stimulus.
By itself 17.8 mg/kg mitragynine did not alter operant responding (Figure 5A) or thermal response latency in the hot plate assay (Figure 5B). We hypothesized that if mitragynine acts as an opioid agonist, and dependent on the degree of competitive binding at specific opioid receptors, we would see either augmented or disrupted morphine effects if animals were pretreated with a subthreshold dose of mitragynine. When administered with mitragynine pretreatment, morphine still produced decreases in operant responding (F5,35= 9.0; P < 0.0001). Mitragynine did not significantly alter the morphine dose-response function for producing decreases in operant responding. The ED50 value of morphine determined in the presence of 17.8 mg/kg mitragynine to produce reductions in operant responding was 7.2 (95% confidence limits: 4.6 - 11.3) mg/kg, and the potency ratio calculated versus the morphine control was 1.0 (95% confidence limits: 0.7 - 1.3). When administered with mitragynine pretreatment, morphine still produced antinociception (F5,35= 6.2; P < 0.001). Pretreatment with 17.8 mg/kg mitragynine did not significantly alter the dose-response function for morphine to produce thermal antinociception. The ED50 value of morphine determined in the presence of 17.8 mg/kg mitragynine to produce increases in thermal latency was 18.5 (95% confidence limits: 10.3 - 33.0) mg/kg, and the potency ratio calculated versus the morphine control was 1.0 (95% confidence limits: 0.7 - 1.5).
Figure 5.
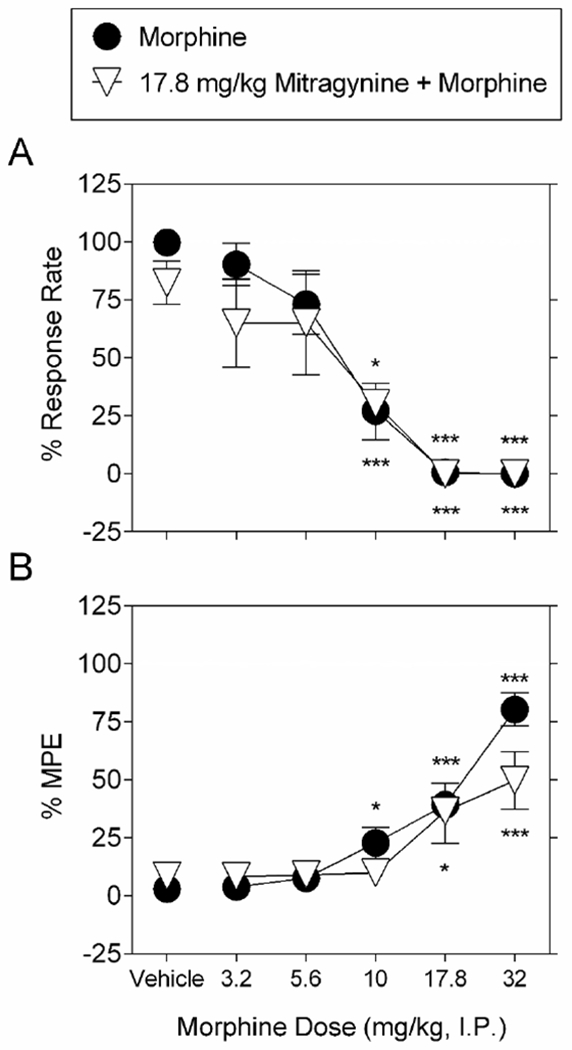
Effects of pretreatment with 17.8 mg/kg mitragynine on morphine-induced changes on schedule-controlled behavior and nociception. Pretreatment with 17.8 mg/kg mitragynine (upward-facing open triangles) does not alter the dose-response effect of morphine with vehicle pretreatment (filled circles), to dose-dependently A. decrease schedule-controlled behavior, and B., increase thermal latency. Ordinates: A: Percent control rates of food-maintained responding. B: Percent maximum possible effect (MPE). Abscissae: drug dose (mg/kg, I.P.), log scale. Each point represents the mean ± S.E.M. of data points for 16 subjects (eight males and eight female for morphine alone) and seven (four males and three female for morphine in combination with 17.8 mg/kg mitragynine). The morphine control is re-plotted from Figure 2.
Discussion
Here we show the potency of morphine to decrease schedule-controlled responding is 5.0-fold higher in comparison to mitragynine’s potency in the same assay, and morphine was more potent than mitragynine to produce antinociception in the hot plate assay. Consistent with previous literature (Goldberg et al., 1981; Shamima et al., 2012), naltrexone significantly antagonized the effects of morphine in both assays. However, here we report that naltrexone does not significantly antagonize the effects of mitragynine to decrease schedule-controlled responding. Only the dose of 56 mg/kg mitragynine produced significant antinociception. We considered the possibility that mitragynine might antagonize the effects of morphine, especially under conditions where mitragynine appeared to exert lesser effects than morphine (i.e., antinociception). However, mitragynine did not significantly modify the operant rate-decreasing or antinociceptive effects of morphine. These findings do not support a role for opioid receptors as the primary mechanism of action for at least some of the behavioral effects of mitragynine.
The present study sought to maximize data collection by not only using animals repeatedly, but also by measuring the rate-decreasing and antinociceptive effects of drugs successively in the same animals. When using the same subjects for more than one behavioral assay, the testing and timing of exposure to conditions from one behavioral assay (i.e., rat paw exposure to noxious thermal stimulus) could potentially impact behavior in a second assay (i.e., rat lever pressing with paws that had previously been exposed to noxious thermal stimulus). Thus, these studies also examined the combination of repeated sequential testing of the hot plate assay for antinociception with schedule-controlled responding in the same experimental subjects. In the present study, examination of these behaviors was found to be feasible when the two assays are performed in conjunction with one another. In particular, the rate of lever pressing was not impacted by exposure to the hot plate as evidenced by stable lever-pressing for food in the presence and absence of hot plate exposure.
Morphine and mitragynine decreased operant behavior at doses smaller than those that produced antinociception. In agreement with previous literature (Shamima et al., 2012; Harun et al., 2015; Carpenter et al., 2016), here we show that mitragynine was less potent than morphine. Mitragynine, at the dose of 56 mg/kg, produced significant thermal antinociception in the hot plate test, but the magnitude of the effect was significantly less than that produced by morphine. Doses of mitragynine higher than 56 mg/kg were not studied in any animals because higher doses of mitragynine were lethal. Although we expand upon previous studies reporting that mitragynine produces hot plate antinociception in rats (Carpenter et al., 2016) and mice (Shamima et al., 2012), this effect is relatively small in magnitude and appears to be limited by toxicity. Indeed, toxic effects of mitragynine have been observed in rats (Sabetghadam et al., 2013) and mice (Matsumoto et al., 1996). In previous studies the largest effect of mitragynine produced less than a 10 s change from vehicle treatment, and while no lethal effect of mitragynine was reported, neither study attempted to achieve a greater antinociceptive latency with higher mitragynine doses (Shamima et al., 2012; Carpenter et al., 2016). This less than 10 s change in thermal response latency reported previously is seemingly replicated with the 56 mg/kg dose of mitragynine tested here insofar as the latency in the current study was less than maximal (i.e., 60 s).
We report that two different doses of naltrexone that antagonize morphine’s effects on schedule-controlled responding and thermal response latency do not significantly alter mitragynine’s effects. Previous in vitro studies reported that mitragynine binds to and acts as an agonist at mu- and kappa-opioid receptors (Kruegel et al., 2016; Varadi et al., 2016; Yue et al., 2018). In previous studies measuring effects on operant behavior, mitragynine did not alter operant responding up to the largest doses studied (Harun et al., 2015; Hemby et al., 2018; Yue et al., 2018). It is notable that here we do not see antagonism of mitragynine’s effects with 1 mg/kg naltrexone, as this is a dose of naltrexone that has been found to antagonize delta- and kappa-opioid receptor effects in schedule-controlled responding (Harris, 1980). Here we conclude that opioid receptors likely do not mediate mitragynine’s ability to decrease operant responding or to increase latency to respond in the hot plate test, at least under conditions of the current study. Previous studies have reported that opioid receptor antagonists block mitragynine induced-antinociception (Shamima et al., 2012; Carpenter et al., 2016). Additionally, naltrexone was reported to block the interoceptive effects of mitragynine (Harun et al., 2015). Although disruptions in schedule-controlled responding can be a relatively non-selective measure of a drug’s pharmacology, the rate-decreasing effects of morphine were antagonized by the lower dose of naltrexone. Any involvement of mu-opioid receptors in the effects of mitragynine should therefore have been evidenced by similar antagonism, and any involvement of delta- and kappa-opioid receptors should have been antagonized with the higher dose of naltrexone. The failure of naltrexone to antagonize some of the effects of mitragynine points to the involvement of non-opioid receptors. Indeed, mitragynine has affinity for the adenosine A2A receptor, as it inhibited 65.66% of adenosine A2A receptor radioligand binding (Boyer et al., 2008). Although reported elsewhere, we did not examine the possibility that adenosine A2A receptors may mediate the antinociceptive (Matsumoto et al., 1996) or rate-decreasing effects of mitragynine observed herein.
Given the widespread use of kratom, the presence of mitragynine as the primary alkaloid constituent of kratom, and the potential for users to combine kratom with other opioid agonists, this study examined the effects of mitragynine in combination with morphine. Mitragynine did not antagonize the rate-decreasing effects of morphine. Mitragynine was reported in two previous studies to decrease i.v. self-administration of morphine (Hemby et al., 2018;) and heroin (Yue et al., 2018). The inverted, U-shaped dose-response function for heroin self-administration was shifted downward (Yue et al., 2018). If mitragynine had been acting to competitively antagonize heroin, a rightward shift of both the ascending and descending limbs of the heroin self-administration dose-response function would have been expected. The results of the current study suggest that any reduction of opioid self-administration produced by mitragynine results from its opioid agonist actions or from non-opioid receptor actions. Additionally, it was recently found that mitragynine is predominantly metabolized via the cytochrome P450 enzyme CYP3A4, with minor contributions from CYP2D6 and CYP2C9 Kamble et al., 2018). These enzymes play critical roles in the metabolism of opioids. Thus, one intriguing possibility is that mitragynine might alter metabolism of morphine. Further, additional studies are needed to assess involvement of non-opioid receptors to the effects of mitragynine, e.g., alpha2 adrenergic receptors where mitragynine has been reported to bind with low micromolar affinity (Boyer et al., 2008; Kruegel et al., 2016; Varadi et al., 2016; Yue et al., 2018).
In summary, the effects of mitragynine to markedly decrease schedule-controlled responding and to produce significant, although relatively small, decreases in thermal latency did not appear to be mediated by opioid receptors. In contrast, the same effects of morphine were mediated by opioid receptors. These data strongly suggest that mitragynine, the predominant alkaloid in kratom, has a pharmacological mechanism that differs from that of prototypical opioids. The current data do not exclude the possibility that opioid receptors mediate the effects of mitragynine, as suggested by the results of other studies and as would be predicted by the binding affinity of mitragynine at opioid receptors. Nevertheless, there appears to be a non-opioid pharmacology of mitragynine that could prove to be exceptionally important to the ongoing debate into the relative benefits and risks of human kratom consumption.
Supplementary Material
Acknowledgements
We humbly dedicate this work in memory of our friend and colleague, Dr. Bonnie A. Avery. She had an invaluable influence on many students and colleagues, and her contributions in the areas of pharmacokinetics, analytical chemistry, and drug metabolism will remain a driving force in better understanding novel, potential drug candidates as well as complex natural product mixtures. Her research interests also spanned into the complex pharmacophore of Mitragyna speciosa. We will always be grateful for her unparalleled support and guidance as well as the inspiration to live by the qualities and ideals she promoted through her boundless courage, hard work, dedication, and loyalty. She will be dearly missed.
The authors would like to thank Dr. Hannah Harris, Ms. Lisa Wilson, and Ms. Ariana Brice for their technical assistance.
Grant acknowledgements: Research was supported by NIH grants DA25267 and DA48353.
Abbreviations:
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- CL
confidence limits
- DEA
Drug Enforcement Administration
- EtOAc
ethyl acetate
- FR
fixed ratio
- MPE
maximum possible effect
- NMR
nuclear magnetic resonance
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Ali Z, Demiray H, Khan IA (2014) Isolation, characterization, and NMR spectroscopic data of indole and oxindole alkaloids from Mitragyna speciosa. Tetrahedron Letters 55: 369–372. [Google Scholar]
- Boyer EW, Babu KM, Adkins JE, McCurdy CR, Halpern JH (2008) Self-treatment of opioid withdrawal using kratom (Mitragynia speciosa korth). Addiction 103: 1048–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkill IH, Haniff M (1930) Malay Village Medicine. The Gardens’ Bulletin Straits Settlements 6, 165–207. [Google Scholar]
- Carpenter JM, Criddle CA, Craig HK, Ali Z, Zhang Z, Khan IA, Sufka KJ (2016) Comparative effects of Mitragyna speciosa extract, mitragynine, and opioid agonists on thermal nociception in rats. Fitoterapia 109: 87–90 [DOI] [PubMed] [Google Scholar]
- Goldberg SR, Morse WH, Goldberg DM (1981) Acute and chronic effects of naltrexone and naloxone on schedule-controlled behavior of squirrel monkeys and pigeons. J Pharmacol Exp Ther 216: 500–9 [PubMed] [Google Scholar]
- Gong F, Gu HP, Xu QT, Kang WY (2012) Genus Mitragyna: ethnomedicinal uses and pharmacological studies. Phytopharm 3:263–272. [Google Scholar]
- Gogineni V, Leon F, Avery BA, McCurdy CR, Cutler SJ (2014) Phytochemistry of mitragyna speciosa In Kratom and other mitragynines: The chemistry and pharmacology of opioids from a non-opium source, Ed Raffa RB CRC press [Google Scholar]
- Harris RA (1980) Interactions between narcotic agonists, partial agonists and antagonists evaluated by schedule-controlled behavior. J Pharmacol Exp Ther 213: 497–503 [PubMed] [Google Scholar]
- Harun N, Hassan Z, Navaratnam V, Mansor SM, Shoaib M (2015) Discriminative stimulus properties of mitragynine (kratom) in rats. Psychopharmacology (Berl) 232: 2227–38 [DOI] [PubMed] [Google Scholar]
- Hassan Z, Muzaimi M, Navaratnam V, Yusoff NH, Suhaimi FW, Vadivelu R, Vicknasingam BK, Amato D, von Horsten S, Ismail NI, Jayabalan N, Hazim AI, Mansor SM, Muller CP (2013) From Kratom to mitragynine and its derivatives: physiological and behavioural effects related to use, abuse, and addiction. Neurosci Biobehav Rev 37: 138–51 [DOI] [PubMed] [Google Scholar]
- Hemby SE, McIntosh S, Leon F, Cutler SJ, McCurdy CR (2018) Abuse liability and therapeutic potential of the Mitragyna speciosa (kratom) alkaloids mitragynine and 7-hydroxymitragynine. Addict Biol [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Fant RV, Wang DW (2018) The abuse potential of kratom according the 8 factors of the controlled substances act: implications for regulation and research. Psychopharmacology (Berl) 235: 573–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B, Wilkerson JL, Mustafa M, Abdullah R, Niphakis M, Wiley JL, et al. (2015). Selective Monoacylglycerol Lipase Inhibitors: Antinociceptive versus Cannabimimetic Effects in Mice. J Pharmacol Exp Ther 353:424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen KL, Prast CJ (1988) Ethnopharmacology of kratom and the Mitragyna alkaloids. J Ethnopharmacol 23: 115–9 [DOI] [PubMed] [Google Scholar]
- Kamble SH, Sharma A, King TI, Leon F, McCurdy CR, Avery BA (2018) Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom). Xenobiotica: 1–31 [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel AC, Gassaway MM, Kapoor A, Varadi A, Majumdar S, Filizola M, Javitch JA, Sames D (2016) Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J Am Chem Soc 138: 6754–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai S, Aimi N, Watanabe H (1996) Central antinociceptive effects of mitragynine in mice: contribution of descending noradrenergic and serotonergic systems. Eur J Pharmacol 317: 75–81 [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon LR, Cunningham KA (2001) Antagonism of 5-hydroxytryptamine(2a) receptors attenuates the behavioral effects of cocaine in rats. J Pharmacol Exp Ther 297: 357–363. [PubMed] [Google Scholar]
- National Research Council (2011). Guide for the Care and Use of Laboratory Animals. National Academies Press: Washington, DC. [Google Scholar]
- Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai S (1994) A New Indole Alkaloid, 7 alpha-Hydroxy-7H-mitragynine, from Mitragyna speciosa in Thailand. Planta Med 60: 580–1 [DOI] [PubMed] [Google Scholar]
- Prozialeck WC, Jivan JK, Andurkar SV (2012) Pharmacology of kratom: an emerging botanical agent with stimulant, analgesic and opioid-like effects. J Am Osteopath Assoc 112: 792–9 [PubMed] [Google Scholar]
- Sabetghadam A, Ramanathan S, Sasidharan S, Mansor SM (2013) Subchronic exposure to mitragynine, the principal alkaloid of Mitragyna speciosa, in rats. J Ethnopharmacol 146: 815–23 [DOI] [PubMed] [Google Scholar]
- Shamima AR, Fakurazi S, Hidayat MT, Hairuszah I, Moklas MA, Arulselvan P (2012) Antinociceptive action of isolated mitragynine from Mitragyna Speciosa through activation of opioid receptor system. Int J Mol Sci 13: 11427–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swogger MT, Hart E, Erowid F, Trabold N, Yee K, Parkhurst KA, Priddy BM, Walsh Z (2015) Experiences of kratom users: a qualitative analysis. J Psychoactive Drugs 5: 360–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida RJ (2000) Drug synergism and dose-effect data analysis. Chapman Hall/CRC Press, Boca Raton [Google Scholar]
- US Drug Enforcement Administration (2016) Schedules of controlled substances: placement of mitragynine and 7-Hydroxymitragynine into schedule I. Federal Register. https://www.federalregister.gov/documents/2016/08/3½016-20803/schedules-of-controlledsubstances-temporary-placement-of-mitragynine-and-7-hydroxymitragynine-into. Accessed 24 August 2018
- Varadi A, Marrone GF, Palmer TC, Narayan A, Szabo MR, Le Rouzic V, Grinnell SG, Subrath JJ, Warner E, Kalra S, Hunkele A, Pagirsky J, Eans SO, Medina JM, Xu J, Pan YX, Borics A, Pasternak GW, McLaughlin JP, Majumdar S (2016) Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit beta-Arrestin-2. J Med Chem 59: 8381–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue K, Kopajtic TA, Katz JL (2018) Abuse liability of mitragynine assessed with a self-administration procedure in rats. Psychopharmacology (Berl) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.