

Key Points
Memory reactivation to priming Ags induces real-time immune resetting in sepsis.
Sepsis response shifts from pathogen-driven to one driven by memory reactivation.
Resetting in sepsis increases survival by a new hypoinflammatory T cell response.
Visual Abstract
Abstract
Cure of severe infections, sepsis, and septic shock with antimicrobial drugs is a challenge because morbidity and mortality in these conditions are essentially caused by improper immune response. We have tested the hypothesis that repeated reactivation of established memory to pathogens may reset unfavorable immune responses. We have chosen for this purpose a highly stringent mouse model of polymicrobial sepsis by cecum ligation and puncture. Five weeks after priming with a diverse Ag pool, high-grade sepsis was induced in C57BL/6j mice that was lethal in 24 h if left untreated. Antimicrobial drug (imipenem) alone rescued 9.7% of the animals from death, but >5-fold higher cure rate could be achieved by combining imipenem and two rechallenges with the Ag pool (p < 0.0001). Antigenic stimulation fine-tuned the immune response in sepsis by contracting the total CD3+ T cell compartment in the spleen and disengaging the hyperactivation state in the memory T subsets, most notably CD8+ T cells, while preserving the recovery of naive subsets. Quantitative proteomics/lipidomics analyses revealed that the combined treatment reverted the molecular signature of sepsis for cytokine storm, and deregulated inflammatory reaction and proapoptotic environment, as well as the lysophosphatidylcholine/phosphatidylcholine ratio. Our results showed the feasibility of resetting uncontrolled hyperinflammatory reactions into ordered hypoinflammatory responses by memory reactivation, thereby reducing morbidity and mortality in antibiotic-treated sepsis. This beneficial effect was not dependent on the generation of a pathogen-driven immune response itself but rather on the reactivation of memory to a diverse Ag pool that modulates the ongoing response.
Introduction
One of the most enduring paradigms in medicine is that cure of infectious diseases is essentially achieved by antimicrobial drugs, which act as toxic selective agents capable of destroying and/or blocking pathogens in minimal concentrations and with limited damage to the host. As a class of drugs, they are employed monotherapeutically with the intent to resolve the disease because they are considered potentially curative (1–5). In severe cases of infections, sepsis, and septic shock, antimicrobials tend to have broad spectrum and are used early, in higher doses, and often as a combination of different kinds of antibiotics, avoiding microbial resistance. This universal approach is complemented by life support measures and eventually helped by preventive vaccines (6–14). Current research in this field is focused on the discovery of new antimicrobial agents, prevention of microbial resistance, modulation of proinflammatory and immune suppressive microenvironments, and development of prophylactic vaccines (7, 8, 14–24). Little else beyond these boundaries has been contemplated.
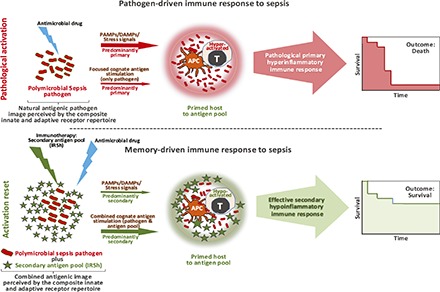
We propose in this article a new interpretation as to how antimicrobial drugs work in vivo. In our view, antimicrobials act as equilibrium shifters of the host–pathogen interaction, by impairing the pathogen viability/proliferation/action, and ultimately favoring its clearance by the host’s immune system. Accordingly, the use of any drug in vivo that possesses intrinsic antimicrobial effect would facilitate the work of the immune system in pathogen clearance in the context of a proper immune response, shifting the balance toward cure.
Paradoxically, morbidity and mortality associated with infectious disease are often caused by improper immune response (6–8, 13, 17–19, 21–23). In the case of sepsis, an antimicrobial drug acting as an equilibrium shifter is likely to have reduced chances to work fully because it may allow an ineffective/pathologic immune response to proceed without substantially changing its nature. For this reason, the challenge to cure extreme cases of infection might be successfully met only by the replacement of the faulty immune response in real time. We have attempted to achieve the latter goal by using as a rationale the following three observations, resulting from the earlier work of several other investigators: 1) the innate primary immune response is capable of localizing the pathogen in a given restricted anatomic territory but is ineffective to deal with a large systemic burden of microorganisms because of its natural proinflammatory profile (6–8, 13, 17, 18, 22, 23, 25, 26); 2) the secondary adaptive immune response is better equipped to systemically handle large numbers of invader pathogens because of its low inflammatory bias and enhanced pathogen clearance potency. The most convincing evidence to date that secondary immunity deals effectively with large numbers of pathogens was provided by Edward Jenner’s classical work and the more recent eradication of smallpox (27–42); and 3) the secondary adaptive immune response is the dominant one when it occurs simultaneously to primary adaptive responses, because memory lymphocytes can reset innate and adaptive cells to be more effective, and could induce the primary immune response to shift to a dominantly low inflammatory pattern (43–50).
We propose to change the ongoing pathological immune response by altering the way the immune system perceives the pathogen and the associated stress and danger signals. The pathogen “image” at the molecular level results from the engagement of the composite repertoire of adaptive and innate immune receptors, including pattern recognition receptors, expressed in lymphocytes and innate immune cells with memory and naive phenotypes (25, 26, 32, 51–67). We hypothesized that a new molecular image could be created by overlapping the one induced by primary pathogen activation with that generated by a diverse pool of newly reactivated secondary memory Ags derived from multiple pathogens—from now on referred to as Immune Response Shifter (IRSh). In this scenario, instead of only one or a few pathogens, the immune system would perceive multiple and diverse aggressors for which there are large numbers of memory precursors. A strong secondary activation and memory response would be elicited, and would take over the overall activity of the immune system, affecting the way it responds to the primary etiologic agent(s) of the ongoing disease by providing a new hypoinflammatory context that is more compatible with disease resolution.
We have tested the possibility to change inefficient and pathological immune responses by repeated stimulation with the IRSh secondary antigenic pool during ongoing infection in a highly stringent murine model of polymicrobial sepsis produced by cecum ligation and puncture. We show that systemic and repeated reactivation of established memory to diverse and mostly unrelated pathogens resets unfavorable immune responses in vivo, thereby reducing morbidity and mortality in antibiotic-treated high-grade sepsis.
Materials and Methods
In vivo experiments
Four- to five-week-old, pathogen-free, female C57BL/6j mice were obtained from the Multidisciplinary Center for Biological Investigation on Laboratory Animal Science at the State University of Campinas (UNICAMP). The project was approved by the UNICAMP Animal Experimentation Ethics Committee (Protocol no. 3486-1/2014) and was conducted according to guidelines set by the Brazilian National Council for Animal Experimentation. The mice had free access to water and feed and were kept in a controlled environment at 22 ± 2°C, with 60–80% humidity in 12-h-long light/dark cycles.
Prick test
The prick test was applied in two mice hemilaterally to analyze the immunization specificity and efficiency of IRSh and an unrelated Ag control (KLH) on day 35, with the use of 2.2 μg of IRSh and 2.5 μg of KLH in 50 μl of buffer solution per injection site, respectively. IRSh was injected into the left dorsal side and KLH was injected into the right dorsal side of the animals; two to three tests were conducted per mouse. Hair was removed from the application points 1 d before the test. Results were recorded during 7 d. The animals were euthanized after 3 wk of the end of the test. No abnormalities were observed.
Immunization
To mimic the immunological memory present in most humans against common pathogens, mice underwent a primary immunization with the IRSh Ag pool 5 wk before sepsis induction (Supplemental Table I). All Ags were obtained from Imunocenter (Rio de Janeiro, Brazil). Freund’s complete adjuvant (FCA)—inactivated mycobacteria (usually Mycobacterium tuberculosis) in 85% paraffinic oil and 15% mineral oil (v/v) emulsion—was purchased from Sigma-Aldrich. Administration of the Ag mixture was performed intradermally with 4.44 μg in 100 μl buffer plus 40 μl FCA. Half of the dose (70 μl) was injected into either side of the tail base. Immunization was performed under anesthesia with ketamine/xylazine (20/12.5 mg/kg). Three intradermal boosts were made under anesthesia with ketamine/xylazine (20 and 15 mg/kg) on days 3, 7, and 10, without any adjuvant, in multiple injection points (20 points, 40 μl/point), distributed over the ventral and dorsal surfaces of the animal (0.0444 μg/point, total dose per boost: 0.888 μg) according to the diagram shown in Supplemental Fig. 1A. Hair was removed from the application points 1 d before the first immunization.
Test for preexisting memory for IRSh Ags
Before beginning the sepsis experiments, we have discarded the possibility of any preexisting memory for the IRSh Ag pool in the experimental animals. For this purpose, two batches of 16 untreated C57BL/6j mice were first immunized as described above and observed for 2 mo for the development of any spontaneous skin reaction at the injection sites. Hair was removed from the injection points 1 d before immunization; these results have been recorded during this 2-mo period and were negative for all animals, showing no preformed memory (data not shown).
Experimental design
Experiments were planned for batches of 32 animals that were randomized immediately after surgery and divided in a blocked format into four experimental groups with six to eight mice: 1) sepsis control (administration of saline only); 2) antibiotic plus saline; 3) IRSh plus saline; and 4) IRSh plus antibiotic.
Protocol exclusion criteria
The major criterion for protocol exclusion was any skin reaction to the IRSh antigenic pool that could reveal preexisting memory to immunizing Ags from the first FCA + IRSh application, including the three consecutive boosts on days 3, 7, and 10 after priming, until the day of surgery. No mouse met this criterion during any phase of the experimental protocol. Additional criteria were any disease signs; skin lesions; and complications such as bleeding, wound dehiscence, and death due to anesthesia or surgery.
Sepsis induction
Animals were submitted to laparotomy with 1-cm-long abdominal incision for cecum exposure and nonobstructive ligation just below the ileocecal valve. Thereafter, the cecum was punctured twice with an 18-gauge needle, followed by gentle compression to ensure some feces extrusion. After perforation of the cecum, the intestines were reinserted into the abdominal cavity, and the muscles and skin were sutured (68). Following the surgical procedure, the animals received 1 ml prewarmed saline s.c. for fluid replacement (69); then, they received i.m. 2.0 mg/kg meloxicam as an analgesic drug and were kept in a warm environment until recovery from anesthesia (30–60 min) (70). Meloxicam was injected after surgery once a day for three consecutive days. All surgical procedures were conducted by a highly skilled surgeon (V.D.N.)
After cecum ligation/puncture, the animals were observed daily for 7 d and scored according to Manley et al. (71) to evaluate the signs consistent with murine sepsis, such as lethargy, piloerection, tremors, periorbital exudation, respiratory distress, and diarrhea. Each sign was evaluated by its absence/presence, not by its severity, and the maximum score was 6. A score >1 was considered as indicative of sepsis. Animals with a score ≥3 after 24 h of cecum ligation/perforation were euthanized by cervical dislocation and their blood was collected for phenotypic analysis of surface markers. Serum was frozen at −80°C for further analysis by mass spectrometry (MS).
Animals were euthanized after 7 d, and biological samples, such as the spleen and lymph nodes, were collected, snap frozen, and kept at −80°C for further use (68).
Therapeutic vaccination
To attempt immune resetting systemically, therapeutic vaccination with the IRSh pool was performed in randomized mice immediately after surgery and before administration of antimicrobial therapy in 24 points (20 points in dorsal and ventral sides of the animal plus four additional points in the limbs, 40 μl/point, total dose: 1.066 μg, Supplemental Fig. 2). The first therapeutic vaccination was performed during surgery anesthesia (ketamine/xylazine, 20 and 12.5 mg/kg), and repeated in the surviving animals 4 d after in the same conditions (days 0 and 4). Hair was removed from the application points 1 d before the surgery intervention. The mice that were randomized not to receive therapeutic vaccination were also anesthetized as described above but received only saline injections (40 μl/point).
Antimicrobial treatment
After therapeutic vaccination, the mice that were randomized to receive antimicrobial treatment were injected with imipenem s.c. 25 mg/kg in 1.0 ml and thereafter in 12 h intervals, until the end of the experiment on day 7, as described elsewhere (72). The animals that were randomized for not receiving the antimicrobial drug were treated with 1.0 ml of saline solution twice a day. The antibiotic imipenem is commercially available in combination with cilastatin, an enzyme that blocks the degradation of imipenem by dehydropeptidases. The term imipenem refers throughout this text to the combination of imipenem + cilastatin.
Phenotypic characterization
The spleens were obtained from sacrificed mice and mechanically disrupted in petri dishes to release the splenocytes. Mononuclear cells were isolated from the total splenocyte population by gradient centrifugation with Ficoll–Hypaque (Sigma-Aldrich, St. Louis, MO) and incubated with fluorochrome-conjugated mAbs for 20 min (eBioscience, San Diego, CA and Miltenyi Biotec, Bergisch Gladbach, Germany). After fixation with 0.5% paraformaldehyde (Labsynth, São Paulo, Brazil), they were analyzed with a FACSCanto flow cytometer (Becton Dickinson, Mountain View, CA). Anti-mouse Abs against the following surface markers were used: CD3e (PerCP-Vio 700; Miltenyi Biotec); CD4 (allophycocyanin, clone GK1.5; eBioscience); CD4 (allophycocyanin-Cy7; Miltenyi Biotec); CD8 (allophycocyanin; Miltenyi Biotec); CD62L (FITC, clone MEL-14; BioLegend); CD69 (FITC; Miltenyi Biotec); and CD44 (PE; Miltenyi Biotec). All the Abs were validated for use in flow cytometry. Additional data are available on the manufacturers’ Web sites. The gating strategy was as follows: the starting cell population was gated on the lymphocyte window defined by side-scattered light area/forward-scattered light area analysis made with the FACSDiva software, version 6.1.3, with acquisition of 10,000 events per sample. The positivity of the samples was inferred by comparison with isotype negative controls. Three isotype controls were used in our experiments (IgG1, IgG2a, and IgG3).
Protein quantitative analyses by MS
Sample digestion.
Thirty microliters of serum were diluted in 90 μl of 50 mM ammonium bicarbonate (NH4HCO3, pH 8.5). Flow-throughs containing the protein solution had their buffer exchanged and were concentrated with 50 mM NH4HCO3 (pH 8.5), with Vivaspin 6 cartridges (Sartorius, Gottingen, Germany). To an aliquot of each extract containing 50 μg of total protein, 25 μl of 0.2% v/v RapiGEST SF solution (73) was added and heated (80°C, 15 min). Afterwards, 2.5 μl of 100 mM DL-DTT (Sigma-Aldrich, Merck) (prepared in 50 mM NH4HCO3, pH 8.5) was added to each sample, incubated (30 min, 60°C), and centrifuged (14.000 × g, 10 min, 4°C). Then, 2.5 μl of 300 mM iodoacetamide (prepared in 50 mM NH4HCO3, pH 8.5) was added to the samples and kept at 20°C in the dark for 30 min. Samples were centrifuged in a microcentrifuge for 30 s, and 20 μl of trypsin (Promega, Madison, WI) (prepared in NH4HCO3, 50 mM, pH 8.5) was added at a ratio of 1:100 w/w trypsin/protein and left at 37°C for 16 h in a ThermoMixer C (Eppendorf, Hamburg, Germany). After digestion, 10 μl of 5% trifluoroacetic acid (Pierce, Rockford, IL) was added to stop the proteolysis, incubated (90 min, 37°C) for RapiGEST hydrolysis, and centrifuged (14.000 × g, 90 min, 4°C). The supernatants were placed into total recovery vials (Waters, Milford, MA) containing 5 μl of 1 N ammonium hydroxide for two-dimensional–nano-ultraperformance liquid chromatography (UPLC)–UDMSE proteomic analysis (74).
MS acquisition.
Prior to injection, each sample was acquired as “scouting runs” to access the total ion counts, and a stoichiometric normalization between conditions was conducted (75). Peptides were injected into a 2D-RP/RP ACQUITY UPLC M-Class System (Waters, Milford, MA), tandem to a Synapt G2-Si mass spectrometer (Waters, Manchester, U.K.). We selected this two-dimensional system to accomplish high peak capacity separations conjoined with the use of 5-μm and <2-μm particles, in both first-dimension and second-dimension analytical columns, respectively (76). Peptides (0.5 μg) were loaded into an ACQUITY M-Class BEH C18 Column (130 Å, 5 μm, 300 μm × 50 mm; Waters) for the first-dimension chromatography, which was performed through 10 discontinuous steps of acetonitrile (8.7, 11.4, 13.2, 14.7, 16.0, 17.4, 18.9, 20.7, 23.4, and 50%) at high pH fractionation (pH 10) for 10 min at a flow rate of 2 μl/min. After each step, peptide loads were carried to second-dimension separation in a ACQUITY M-Class UPLC HSS T3 Column (1.8 μm, 75 μm × 150 mm; Waters). Peptides trapped in the second-dimension were eluted with an acetonitrile gradient from 7 to 40% (v/v) for 29 min at a flow rate of 0.5 μl/min, directly into the nanoflow tandem mass spectrometer (77). The mass spectrometer data acquisition was performed in MSE multiplexed mode with ultradefinition ion mobility (UDMSE), switching low (trap traveling-wave T wave) and high collision energy. In the elevated collision energy, quasi m/z–specific collision energies were applied to the different drift time bins that were used to fragment precursor ions prior to orthogonal acceleration time-of-flight analysis and applied to the transfer T wave, collision-induced dissociation cell, filled with argon gas (78). Ions were acquired between m/z 50 and 2000, scanning time of 0.5 s, cone voltage of 30 V, capillary voltage of 2.7 kV, and source offset voltage of 30 V. The mass spectrometer operated in resolution mode with an m/z resolving power of at least 25,000 full width of the peak at half its maximum height, using ion mobility with a cross-section resolving power of at least 40 Ω/ΔΩ (79). Tandem MS (MS/MS) analyses were performed by nanoelectrospray ionization in positive ion mode using a NanoLock Spray ionization source (Waters, Manchester, U.K.). The lock mass channel was sampled every 30 s with a [M + 2H]2+ = 785.84261 [Glu1]-fibrinopeptide B (human Glu-Fib; Waters). The mass spectrometer was calibrated with an MS/MS spectrum of 100 fmol/μl Glu-Fib solution that was delivered through the reference sprayer of the NanoLockSpray ion source. The radio frequency offset (MS profile) was adjusted such that the nanoUPLC-UDMSE data were effectively acquired from m/z 400 to 2000, which ensured that any masses observed in the high energy spectra with less than m/z 400 arose from dissociations at the collision cell (80).
Label-free protein quantitation.
Proteins were identified using dedicated algorithms and searching against the UniProt database, version 2015_08. For proper spectra processing and database searching conditions, we used Progenesis QI for Proteomics software package (Nonlinear Dynamics, Newcastle, U.K.) with Apex3D, Peptide3D, and ion accounting informatics (Waters, Manchester, U.K.) (81). The parameters considered to identify peptides were the software defaults. Briefly, for database searching, tryptic peptides with only one missed cleavage were allowed, with carbamidomethylation as fixed modification and oxidation of methionine as variable modification. For peptide and protein assignments, the following filters were used: minimum of three out of three replicates, mass error (normal distribution) <20 parts per million (ppm), and coefficient of variation (CV; maximum absolute CV of 0.3). Also, as a default parameter, we have applied protein grouping, which combines the conserved domains from proteins whose peptides are subset of other proteins. The final, confirmed list of proteins only considered those identified by at least one unique peptide if it had been matched at least three times in a given condition. The databank used was reversed “on the fly” during the database queries and appended to the original database to assess the false-positive identification rate (82). Relative quantification was determined from the absolute intensities with the use of ion accounting Hi3 (Top3)–based quantitation method, as described (83). The obtained proteins were organized by the Progenesis QI for Proteomics software algorithm into a statistically significant list corresponding to increased and decreased regulation ratios between the different groups with a max fold change of log2 ± 1.6 and ANOVA (p < 0.05). Normalization was performed automatically by the software using default parameters (77). All the information about peptides and proteins identified in each replicate is available as Supplemental Table II, which contains the complete protein and peptide list, accession codes, protein description, ion accounting, scores, and mass error distribution (ppm), as indicated in the text.
Systems biology in silico analysis
Protein networks and canonical pathways associated with differentially expressed proteins were mapped using Ingenuity Pathway Analysis (Ingenuity Systems, Qiagen, Redwood, CA, www.ingenuity.com). This algorithm uses curated connectivity information extracted from the literature to determine the interaction network among the canonical pathways involved in the differentially expressed proteins identified (84). Significant biological functions are based on Fisher test, and multiple correlation hypothesis is performed on the basis of the Benjamini–Hochberg approach at 1% false discovery rate threshold (85).
Lipid analyses by MS
Sample preparation.
Three replicate plasma samples (200 μl each) were precipitated by the addition of three volumes of isopropyl alcohol precooled to −20°C. Samples were mixed by vortex for 1 min after 10-min incubation at room temperature and stored overnight at −20°C for further protein precipitation. After centrifugation at 14.000 × g for 20 min, the supernatant was collected (600 μl) and stored at −80°C until MS analysis. Samples were diluted to adjust the water content at 50% and analyzed by UPLC-MSE.
MS acquisition.
The data were collected in an ACQUITY i-Class UPLC (Waters) equipped with an ACQUITY UPLC HSS T3 1.8 μm 2.1 × 100 mm column tandem to a Synapt G2-S HDMS mass spectrometer. The mobile phases were 1) solvent A: acetonitrile/water (60:40) with 10 mM ammonium formate and 0.1% formic acid and 2) solvent B: isopropanol/acetonitrile (90:10, v/v) with 10 mM ammonium formate and 0.1% v/v formic acid. Chromatography runs were obtained over 10 min at a flow rate of 0.5 ml/min at 55°C. The initial solvent B gradient was programmed as follows: 0.0–1.0 min from 40% B to 50% B, 1.0−6.0 min to 60% B, 6.0−8.5 min to 98% B, 8.5 to 9.0 min kept on 98% B. Finally, the gradient was equilibrated down to 40% from 9.0 to 9.1 min and maintained until 10.0 min for the next run (sample manager, 10 μl injection). MS data were acquired with the instrument control software MassLynx 4.1v. The mass spectrometer was tuned in ESI(+) with the following parameters: positive electrospray ionization (ESI+) with capillary voltage set to 1.0 kV, cone voltage 30 V, source temperature 120°C, desolvation temperature: 450°C, desolvation gas (N2): 700 L/h, and MSE acquisition mode with an m/z range from 100 to 2000. Low collision energy to allow precursor ions to be detected was 2 eV and the multiplex MS/MS alternated high collision energy was ramped from 25 to 30 eV. Leucine-enkephalin (Waters), C28H37N5O7, [M + H]+ = 556.2771 was used as lock mass reference at concentration of 0.2 ng/L with a flow rate of 10 μl/min.
Data processing.
Data were processed with default parameters for peak detection, multivariate analysis and identification, using Progenesis QI 2.2v (Nonlinear Dynamics, Newcastle, U.K.). The detected features were identified using precursor mass error ≤10 ppm, fragment tolerance ≤10 ppm, and the LIPID MAPS Lipdomics Gateway database (Cardiff, U.K.). (https://www.lipidmaps.org). The remaining output lists were concatenated into a single sheet that is available as Supplemental Table II.
Statistical analyses
Statistical analysis was performed using GraphPad Prism software V 8.1.1 (GraphPad).
The survival curves were compared using the Kaplan–Meier method, with differences determined by the log-rank test and reported a two-tail p value. The curves were also individually analyzed by the Bonferroni method, and if a p value was lower than the Bonferroni-corrected threshold, the comparison was considered to be statistically significant using a family-wise 5% significance level.
The spleen weight data were analyzed by two-way ANOVA and Holm–Sidak test. The cytometric data statistical significance was determined by ordinary one-way ANOVA followed by Tukey multiple comparisons test. Outlier analysis for cytometric data validation was conducted with Grubbs test, which compared F values calculated with F tab, and rejected the F values larger than 1.938, considering n = 7 and α = 0.05.
Mean expected lifetime was analyzed by two-way ANOVA, and the statistical significance was determined by using the Tukey multiple comparisons test. The p values <0.05 were considered statistically significant.
Results
Memory induction
One batch of 33 C57BL/6j mice was immunized with IRSh—a pool of nine-well–defined Ags that are used in the classical skin test for delayed type hypersensitivity and against which most humans have preformed immune memory (86). The antigenic sources and immunizing protocol are described in detail in Materials and Methods. No mice showed signs of preformed memory during the entire induction experiment as indicated by the lack of any skin reaction. The efficacy of this protocol in inducing specific memory was tested in a pulse-chase experiment set up to monitor spleen CD3+ T cell phenotype over 8 wk after the last antigenic boost (Fig. 1A, 1B). There was statistically significant mobilization of all memory subpopulations of helper CD4+ T cells [Fig. 1A, p < 0.0001, n = 3 per point, F (3, 66) = 108.2], as well as cytotoxic CD8+ T cells [Fig. 1B, p < 0.0001, n = 3 per point, F (3, 66) = 3293]. As indicated in the encircled area in Fig. 1C, there was a parallel percentage increase of CD4+ and CD8+ lymphocytes displaying memory or central memory phenotypes that peaked at day 35 [p < 0.0001, n = 3 per point, F (3, 66) = 360.6].
FIGURE 1.

(A) Memory induction. Memory phenotype in total CD3+ T cells and their subsets. Comparison of the CD3+ T cell phenotype relative representation over 63 d after the last boost of the immunization protocol is presented. Day 0 on the graph corresponds to the mean of three mice tested before immunization. Thereafter, three additional animals were sacrificed, and their spleens were analyzed individually for the relevant T cell phenotype on days 3, 7, 14, 21, 28, 35, 42, 49, 56, and 63. The p values were calculated by two-way ANOVA. Holm–Sidak multiple comparison test was used for comparison between days 0 and 35 (CD4+ T cells: p < 0.0001; M-CD4+ T cells: p < 0.0001; CM-CD4+ T cells: p < 0.0001; and EM-CD4+ T cells: p < 0.0001). (B) Memory phenotype in total CD8+ T cells and their subsets. Comparison of the CD8+ T cell phenotype relative representation over 63 d after the last boost of the immunization protocol is presented. Three mice per point were sacrificed for analysis following the same schedule described in (A). Day 0 on the graph corresponds to the mice tested before immunization. The p values were calculated by two-way ANOVA. Holm–Sidak multiple comparison test was used for comparison between days 0 and 35 (CD8+ T cells: p = 0.0019; M-CD8+ T cells: p < 0.0001; CM-CD8+ T cells: p = 0.0174; and EM-CD8+ T cells: p = 0.8276). (C) Comparison of memory and central memory phenotypes in CD4+ versus CD8+ T cells. Day 0 on the graph corresponds to the mice tested before immunization. Three mice per point were sacrificed for analysis following the same schedule described in (A). The p values were calculated by two-way ANOVA. Holm–Sidak multiple comparison test was used for comparison between days 0 and 35 (M-CD4+ T cells: p < 0.0001; CM-CD4+ T cells: p < 0.0001; M-CD8+ T cells: p < 0.0001; and CM-CD8+ T cells: p = 0.0045). (D and F) Prick test for IRSh applied hemilaterally. (E and G) Prick test for KLH control Ag applied hemilaterally. Each point in panels (A)–(C) represents the mean ± SE of independent analyses of the spleens of three mice. CM, central memory; EM, effector memory; M, memory.
The immunization specificity and efficiency was corroborated by the results of a Prick test conducted with IRSh and an unrelated Ag control (KLH) on day 35. Fig. 1D and 1F show that IRSh rechallenge was able to promote a positive hypersensivity reaction 3 d later, as opposed to KLH boost that failed to do so (Fig. 1E, 1G). These tests were performed hemilaterally in each animal (Fig. 1D–G). Also, prior to immunization, two batches of 16 nonimmunized C57BL/6j mice were tested for preexisting memory. No preformed memory was detected in this previous test. The antigenic sources and immunizing protocol are described in detail in Materials and Methods.
Altogether, the above findings support the efficacy of the immunization protocol and also suggest the fifth week after the last boost as the optimal window for memory protection.
Combined immunological and antimicrobial treatments (IRSh plus imipenem) increase survival in a lethal polymicrobial sepsis experimental model
We have induced experimental polymicrobial sepsis by cecum ligation and puncture after 5 wk of the last immunization with a protocol that induces high-grade sepsis and is lethal in almost 80% of the untreated animals in 24 h (68). We have tested four different experimental conditions with six to eight animals per group. The experiment was conducted four independent times in a blocked format. Kaplan–Meier statistical analysis showed that they were comparable and could be analyzed as a single block. Fig. 2A shows the composed survival curves of the following groups: 1) IRSh plus imipenem (n = 29); 2) imipenem alone (n = 31); 3) IRSh alone (n = 27); and 4) saline control (n = 30).
FIGURE 2.

IRSh impact on survival and immune response to sepsis. (A) Survival curve. The survival of animals in the four experimental groups after cecum ligation/perforation was plotted in a Kaplan–Meier curve. The log-rank (Mantel–Cox test) revealed a p value < 0.0001 (n = 113). Dotted lines indicate the 95% confidence interval for each curve. (B) Mean expected lifetime. Statistical significance was calculated by two-way ANOVA followed by Tukey multiple comparisons test for IRSh and saline groups. The means with 95% confidence interval are indicated. ****p < 0.0001. (C) Spleen weight and immune response in murine sepsis. Two-way ANOVA followed by Holm–Sidak test were used to show correlation between spleen weight and survival (p < 0.0001). Saline (control): 30 dead animals; imipenem: 28 dead and 3 survivors; IRSh: 27 dead; and IRSh plus imipenem: 13 dead and 15 survivors. The means with 95% confidence interval are indicated. ****p < 0.0001.
The treatment with IRSh plus imipenem had the best effect, increasing the survival rate obtained by imipenem alone over five times (from 9.7 to 51.7%). Most importantly, the improvement was highly significant, with no overlap of confidence intervals (p < 0.0001, n = 113). These results suggest the superiority of the combined IRSh plus imipenem treatment over the traditional antimicrobial monotherapeutic approach in our model of lethal experimental sepsis in C57BL/6j mice. The treatment efficiency had the following order: IRSh plus imipenem > imipenem > IRSh > saline.
IRSh and imipenem have a positive synergic effect on the mean expected lifetime in experimental sepsis
The mean expected lifetime achieved by IRSh plus imipenem treatment (n = 29) was higher than that of the other three groups raising to 114 h from 54, 27, and 26 h in imipenem only (n = 31), IRSh only (n = 27), and saline (n = 30) groups, respectively [p < 0.0001 for each comparison, n = 113, F (3, 113) = 31.20, Supplemental Fig. 1C]. Imipenem alone has also increased the mean expected lifetime over the saline control from 26 to 54 h [p = 0.0332, n = 113 mice, F (3, 113) = 31.20] and over the IRSh alone group from 27 to 54 h [p = 0.0566, n = 113 mice, F (3, 113) = 31.20, Supplemental Fig. 1C]. Fig. 2B indicates that there was a synergic interaction between IRSh and imipenem [p < 0.0001, n = 113 mice F (1, 113) = 17.00], which has doubled the mean expected lifetime achieved by imipenem alone from 54 to 114 h. ANOVA test has also revealed a significant impact on survival for IRSh alone [p < 0.0001, n = 113, F (3, 113) = 18.63] and for imipenem alone [p < 0.0001, n = 113 mice, F (3, 113) = 65.14]. Thus, although both IRSh and antibiotic increased survival when used alone, their synergic interaction was more efficient in rescuing animals from death.
Spleen weight as a proxy of immune response in lethal experimental sepsis
Spleen weight is one of the best immune response indicators in murine sepsis models (87). Accordingly, spleen weight gain was the only parameter to correlate with survival in our experiments [Fig. 2C, p < 0.0001, n = 55 mice, F (1, 55) = 66.95]. Fifty-three percent of the animals that received IRSh plus imipenem survived and had a more effective immune response as inferred from the spleen weight analysis (Fig. 2C). Spontaneous immune responses could also be observed in a small fraction of mice (9.7%) that managed to survive on imipenem alone over the 7-d follow-up period. The rarity of spontaneous responses may explain the low cure rate induced by antimicrobial drugs in lethal polymicrobial sepsis. Thus, an effective immune response—induced or spontaneous—is necessary for survival to be achieved with antimicrobials in this model. The spleen weight results are in plain accordance with those obtained with the survival curves.
Antimicrobial treatment alone acts as equilibrium shifter inducing an ineffective T cell proliferation that correlates to death
Sepsis was induced as previously described in 29 animals that were allocated into four treatment groups of seven to eight mice. This experiment was one out of the four pooled that compose the survival curves presented in Fig. 2A. Fig. 3 shows that the use of imipenem alone promotes an increase in the percentage of CD3+ T cells in the spleen: 29.3% [n = 24, F (3, 24) = 8.745] as compared with 10.4% in the IRSh plus imipenem group [p = 0.0007, n = 24, F (3, 24) = 8.745], 11.6% in saline group [p = 0.0015, n = 24, F (3, 24) = 8.745], and 15% in the IRSh only group [p = 0.0107, n = 24, F (3, 24) = 8.745]. The only surviving animal in the antibiotic group had 6.8% CD3+ T cells in the spleen, similar to those that survived in the IRSh + imipenem group (Supplemental Fig. 1E), and also had the same spleen weight gain (Fig. 2C). This pathogen-driven expansion of the CD3+ T cell compartment negatively correlates with survival because it only occurred in nonsurviving animals treated with antibiotic alone. However, the uncontrolled lymphocyte expansion that results from the equilibrium shift effect of imipenem is held in check by the IRSh stimulation, or more rarely by a spontaneous proper immune response, so that animals treated with IRSh plus imipenem are more likely to survive (Fig. 3).
FIGURE 3.

CD3+ T cell population expansion in sepsis. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey multiple comparisons test. The means with 95% confidence interval are indicated. *p < 0.05, **p < 0.01, ***p < 0.001.
Imipenem alone promotes CD3+ splenocyte expansion in sepsis but does not alter the hyperactivation status of proinflammatory T cells that are characteristic of sepsis
Although imipenem treatment in sepsis shifts the host–pathogen balance in favor of the host increasing survival as compared with saline-injected mice (Fig. 2A), this effect is accompanied by an uncontrolled and ineffective T cell immune response in those mice that die. The relative proportion of the three major CD3+ T cell subsets (CD4+, CD8+, and CD4−CD8−) that accumulate in the spleen is similar to that found in other treatment groups with two exceptions: the CD3+CD4+ subset was higher in the IRSh only group as compared with the imipenem only group [55.8% versus 37.7%, p = 0.0443, n = 24 mice, F (3, 24) = 4.028], and the CD3+CD8+ subset was higher in the imipenem only group as compared with the saline control group [28.7% versus 20.2%, p = 0.0474, n = 24 mice, F (3, 24) = 3.305, Supplemental Fig. 1D]. Nevertheless, there are important differences as regards the activation status of these cells. Fig. 4 shows that splenocytes from imipenem-treated mice tend to be as hyperactivated as those from untreated control animals that follow the natural course of sepsis. The CD69+ subpopulation represented 51–71% of total T cells regardless of the use of antibiotic, except for CD3+CD8+CD69+ T cells from antibiotic-treated animals that amounted to 37.2%, but this difference had no statistical significance. The lymphocyte activation status as measured by the CD69 activation marker was even higher in the antibiotic-only versus the saline-only groups in CD4+ T lymphocytes with memory [p = 0.0107, n = 23, F (3, 23) = 21.21] and effector memory [p = 0.0144, n = 22, F (3, 22) = 19.92] phenotypes (Fig. 4).
FIGURE 4.

Flow cytometric evaluation of T cell subsets in four experimental groups. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey multiple comparisons test. The means with 95% confidence interval are indicated. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
IRSh fine-tuning disengages the hyperactivation status of most spleen T cell populations and their subsets
The IRSh antigenic stimulation was capable of fine-tuning the immune response in sepsis not only by contracting the total CD3+ T cell compartment in the spleen but also by reducing the proinflammatory hyperactivation of all classical T cell populations (Fig. 4). Thus, cell activation was reduced in the IRSh plus imipenem group as indicated by the lower positivity for the CD69 surface marker in flow cytometry. CD69 values dropped 22–62% in all tested subpopulations, except CD3+CD8+ effector memory T cells, which were in too small numbers to be accurately tested (Table I). The difference in T cell activation status between imipenem only versus IRSh plus imipenem groups was most evident in the CD3+CD8+ total population [62.4% drop in CD69 positivity, p = 0.0085, n = 24, F (3, 24) = 11.50] and the CD3+CD8+ central memory subpopulation [62.3% drop of CD69 positivity, p = 0.0006, n = 24, F (3, 24) = 9.901] (Table I). These activation results are also in agreement with the survival curves.
Table I. Activation phenotype profile in the four treatment groups of experimental sepsis.
| Cell Populations | IRSh + Imipenem | Saline + Imipenem | IRSh | Saline |
|---|---|---|---|---|
| CD3+CD69+ | 29.69 (15.42–43.95) | 60.67 (45.89–75.45) | 38.37 (33.96–42.78) | 55.17 (39.34–71.00) |
| (p = 0.0006) F (3, 23) = 11.81 | ||||
| CD3+CD44+CD69+ | 32.24 (28.42–36.07) | 65.43 (55.59–75.27) | 48.10 (37.03–59.17) | 63.07 (51.79–74.36) |
| (p < 0.0001) F (3, 24) = 18.29 | ||||
| CD3+CD4+CD69+ | 38.23 (30.37–46.09) | 53.34 (46.53–60.15) | 39.39 (35.32–43.46) | 52.47 (45.70–59.24) |
| (p = 0.0012) F (3, 24) = 10.94 | ||||
| CD3+CD4+CD44+CD69+ | 39.94 (32.79–47.09) | 59.10 (32.79–85.41) | 44.67 (38.16–51.18) | 55.29 (46.05–64.53) |
| (p < 0.0001) F (3, 23) = 21.21 | ||||
| CD3+CD4+CD44+CD62L+CD69+ | 43.29 (36.23–50.35) | 70.87 (59.57–82.17) | 49.23 (34.20–64.26) | 60.06 (52.54–67.58) |
| (p = 0.0004) F (3, 24) = 9.031 | ||||
| CD3+CD4+CD44+CD62LnegCD69+ | 47.37 (40.26–54.48) | 60.74 (33.93–87.55) | 40.76 (22.65–58.87) | 59.20 (49.86–68.54) |
| (p < 0.0001) F (3, 22) = 19.32 | ||||
| CD3+CD8+CD69+ | 13.97 (8.88–19.06) | 26.94 (20.28–33.60) | 51.11 (30.54–71.68) | |
| (p = 0.0085) F (3, 24) = 11.50 | ||||
| CD3+CD8+CD44+CD69+ | 25.57 (19.25–31.89) | 62.70 (46.52–78.88) | 44.26 (26.05–62.47) | 63.73 (43.73–83.73) |
| (p = 0.0012) F (3, 24) = 8818 | ||||
| CD3+CD8+CD44+CD62L+CD69+ | 23.60 (17.95–29.25) | 62.66 (46.85–78.47) | 43.07 (24.89–61.25) | 63.21 (43.40–83.02) |
| (p = 0.0006) F (3, 24) = 9.901 | ||||
| CD3+CD4negCD8negCD69+ | 26.90 (23.15–30.65) | 57.77 (48.17–67.37) | 38.13 (23.91–52.35) | 61.30 (45.62–76.98) |
| (p = 0.0003) F (3, 24) = 13.46 | ||||
| CD3+CD4negCD8negCD44+CD69+ | 23.50 (19.85–27.15) | 54.59 (41.71–67.47) | 44.66 (26.43–62.89) | 59.04 (42.67–75.41) |
| (p = 0.0018) F (3, 24) = 8.990 | ||||
| CD3+CD4negCD8negCD44+CD62L+CD69+ | 25.70 (21.27–30.13) | 60.60 (48.67–72.53) | 51.67 (31.15–72.19) | 65.03 (49.70–80.36) |
| (p = 0.0007) F (3, 24) = 10.60 | ||||
| CD3+CD4negCD8negCD44+CD62LnegCD69+ | 24.93 (18.90–30.96) | 55.39 (48.47–62.31) | 45.84 (28.53–63.15) | 61.81 (43.46–80.16) |
| (p = 0.0015) F (3, 24) = 10.08 | ||||
Results are means with 95% confidence intervals. The p value (IRSh + imipenem versus saline + imipenem) was calculated by ANOVA followed by Tukey multiple comparison posttest.
Altogether, our data indicate that an “equilibrium shifter” of the host–pathogen interaction, such as the imipenem antibiotic, does not hold complete curative power because it does not change the course of the pathological immune response. It requires the immune resetting provided by the IRSh antigenic stimulation to do so, or a rare spontaneously effective immune response.
IRSh fine-tuning of the immune response in sepsis increases recovery and reduces activation of naive CD4+ and CD8+ T cell subsets
Immune suppression in human and murine sepsis is attributed in part to the lack of quantitative and qualitative recovery of CD4+ naive T cells (23). Fig. 5A shows higher recovery of naive CD4+ T cells in the IRSh plus imipenem group: 72.7% compared with 42.8, 51.3, and 55.8% in the saline [p = 0.0008, n = 24, F (3, 24) = 7.130], IRSh only [p = 0.0182, n = 24, F (3, 24) = 7.130], and imipenem only [p = 0.0779, n = 24, F (3, 24) = 7.130] treatment groups, respectively. Fig. 5B illustrates the recovery of naive CD8+ T cells, which were induced equally well by treatment with imipenem alone [73.7% versus 44.9% in saline group, p = 0.0012, n = 23, F (3, 23) = 8.655] and IRSh plus imipenem [72.6% versus 44.9% in saline group, p = 0.0011, n = 23, F (3, 23) = 8.655]. The naive CD8+ T cell phenotype accounted for 63.3% in the IRSh only group versus 44.9% in the saline control [p = 0.0371, n = 23, F (3, 23) = 8.655].
FIGURE 5.

Naive recovery and activation status of CD4+ and CD8+ T cell populations. (A) Naive CD4+ T cell recovery. (B) Naive CD8+ T cell recovery. (C) Naive CD4+ T cell activation. (D) Naive CD8+ T cell activation. Statistical analysis was performed by ordinary one-way ANOVA followed by Tukey multiple comparisons test. The means with 95% confidence interval are indicated. *p < 0.05, **p < 0.01, ***p < 0.001.
As for the activation status of naive cells, there was a reduction of the CD69+ subpopulation in the group that received IRSh alone (Fig. 5C), but neither the antibiotic alone nor its combination with IRSh stimulation affected the CD4+ T cell compartment. The most impressive impact of the IRSh antigenic modulation was evident on naive CD8+ T cells that retained the CD69+ activated phenotype. The naive CD8+/CD69+ lymphocytes shrunk to 6.9% in the spleens from mice treated with IRSh plus imipenem as compared with 31% in those from the saline group [p = 0.0010, n = 24, F (3, 24) = 7.246]. It is noteworthy that the number of activated naive CD8+ splenocytes found in mice treated with imipenem alone was also significantly higher than that present in animals exposed to the IRSh plus imipenem treatment [24.1% versus 6.9%, p = 0.0203, n = 24, F (3, 24) = 7.246, Fig. 5D].
Thus, the accumulated data suggest that the IRSh antigenic stimulation coupled to the antimicrobial drug decreases expansion of total CD3+ T cells in sepsis, but this effect does not occur at the expense of the recovery of naive CD4+ and CD8+ subsets. The hyperactivation state is disengaged throughout all tested CD3+ memory populations and subpopulations, as well as in the naive CD4+ and CD8+ subsets, most notably in CD8+ T cells.
The proteome/interactome profile of the peripheral blood obtained from mice treated with the combination of IRSh plus antibiotic is very different from that of untreated sepsis
We have carried out proteomic/lipidomic analyses to better understand the immune mechanisms that underlie the integrated action of IRSh stimulation and use of antibiotics in promoting survival in sepsis. For this purpose, we have compared surviving animals on day 7 after treatment with IRSh plus imipenem with mice in the saline control group. Sepsis was induced as described in Materials and Methods in C57BL/6j mice, which were randomly assigned to either the experimental group (IRSh plus imipenem, n = 54) or the control group (saline, n = 7). The survival was 65% in 7 d for the experimental group and <24 h for five out of seven control animals (Supplemental Fig. 1F), reproducing the same pattern of high-grade sepsis described in Fig. 2A and in reference (68) for controls. The serum of the surviving animals was pooled and used to set up the techniques for proteomic and lipidomic studies. Similarly, the serum of the dying mice in the control group that had to be euthanized were pooled for analyses.
Protein expression profiles in survival and nonsurviving conditions are shown in the volcano plot depicted in Fig. 6A. In total, 121 differentially expressed proteins were identified (Supplemental Table II). Fig. 7A shows the results of an in silico study of systems biology performed with the Ingenuity Pathway Analysis software. Forty proteins were differentially downregulated in survival, including proinflammatory mediators, molecules that promote cytokine storm, feedback regulatory proteins, and proapoptotic factors. Fig. 7B depicts the interactome from 81 proteins that were differentially upregulated in survival, being associated to processes that are as diverse as intracellular detoxification, muscle metabolism improvement, cell regeneration, and secretion of some cytokines possibly connected to recovery and survival. As indicated in Fig. 6B and Supplemental Fig. 2B, the proteins uniquely expressed in survival groups induce a selective expression of TNFSRF1A, and to a lesser extent TNFSRF1B, that probably neutralize the overproduction of TNF-α, one of the key cytokines in the pathogenesis of sepsis. Also, they induce a selective expression of factors related to maintenance and renewal of the lymphoid system as well as cytokines implicated in regulation of inflammation and of regenerative settings.
FIGURE 6.

Differential protein expression profile ratio in survival and death outcomes. Protein expression profile ration between survival and nonsurvival conditions: a volcano plot was obtained after applying the filters (CV < 0.3; maximum fold change: log2 = ±1.6 and p < 0.05, n = 56 for survival and n = 7 for nonsurvival) to understand the level of significance and magnitude of changes observed in differentially expressed proteins between each of the two conditions (A). Differences with ANOVA. A p value < 0.05 and log2 ≥ ±1.6 were considered significant. In this comparison set, the most significantly up- and downregulated proteins are shown in green and red, respectively. The comparison between survival and nonsurvival samples identified 17 exclusive proteins for either condition: 7 proteins for survival and 10 for nonsurvivor groups (B).
FIGURE 7.

Interactome of proteins differentially expressed in survival versus nonsurvival outcomes. The interactome from 40 proteins downregulated in survival and upregulated in nonsurvival is shown (A). The interactome from 81 proteins upregulated in survival and downregulated in nonsurvival is shown (B).
The opposite profile of high and low protein expression was found in nonsurviving animals (indicated as up and down in Fig. 7A, 7B). The proteins whose expression is restricted to nonsurviving animals induce activation of TWISTNB, MECOM, FOS, STAT1, and other proteins/transcription factors resulting in selective production of TNF-α and key cytokines that participate in the storm that precedes death in sepsis (Fig. 6B, Supplemental Fig. 2A).
Altogether, the accumulated data suggest that the IRSh-induced fine-tuning of the immune response suppresses the pathological proinflammatory reaction in sepsis most likely through the engagement of the memory compartment TCR repertoire specific to the IRSh antigenic components that generate a new secondary activation. This secondary activation induces a new and effective immune response with low inflammatory profile and immune regeneration activity as suggested by our in silico analysis. In this new context, the IRSh plus the antibiotic drug may shift the host–pathogen balance toward cure from an otherwise lethal outcome. Thus, there is a molecular signature of uncontrolled sepsis with a bias toward proinflammatory reaction, proapoptotic effect, suppressed immune regeneration, and reduced metabolism recovery leading to death, as well as a molecular signature dedicated to disease resolution that is characterized by controlled low inflammation, antiapoptotic effect, ongoing immune regeneration, and metabolism recovery conducive to survival. The choice between these two possible opposite outcomes is likely to be determined by the pathogen recognition receptors, danger recognition receptors, stress signals, and TCR repertoire naturally engaged by the original pathogens or by the same type of receptor engagements in the context of the IRSh-driven resetting. In our model, IRSh favors survival by overlaying multiple stimuli that reactivate memory responses. The latter are likely to be the ultimate guide for immune resetting and survival in sepsis.
Label-free quantitative MS: quality of proteome data
Collectively, 29,715 peptides were identified and quantified with an average of six peptides/protein. The used proteomic approach revealed a broad dynamic range of detected proteins with more than seven orders of magnitude as expected for this biological matrix and considering the instrument technology. More detailed information can be found in Supplemental Table II and references (75, 81, 88).
Lipidomics results
The global lipid profile data display the metabolites that symmetrically increase or decrease in surviving and in nonsurviving animals. In total, 314 lipids were detected by considering three out of three replicates that have an absolute coefficient of variance <0.3. Forty-three lipids were greater than log2 ratio = ±1.6 with the following additional criteria: isotopologue pattern match (considering both intensity and m/z), elemental composition, and mass error <2 ppm for precursor ions and 5 ppm for the fragments. Considering the survival over nonsurvival group ratio, there were 22 metabolites significantly increased (ANOVA, p < 0.0026, n = 56 for survival and n = 7 for nonsurvival) and 21 metabolites significantly decreased (ANOVA, p < 0.01, n = 56 for survival and n = 7 for nonsurvival) (Fig. 8A, Supplemental Table II). The lysophosphatidylcholine (LPC)/phosphatidylcholine (PC) ratio was ∼3-fold higher in surviving mice than in the nonsurvivors (Fig. 8B). It is noteworthy that in clinical settings, the LPC/PC ratio tends to be narrower in hyperinflammatory conditions such as sepsis and correlates with worse prognosis because LPC drops and PC ascends in these conditions. Alternatively, hypoinflammatory states are associated to wider ratio and a better prognosis and the LPC increases and PC decreases (89–91). In the cecum ligation/perforation murine experimental sepsis model without treatment, LPC is significantly decreased in plasma as measured by MS (92). Our results are in agreement with the latter findings and support the possibility of replacing the hyper for hypoinflammatory state in ongoing disease.
FIGURE 8.

Differential lipid expression in survival and death outcomes. (A) The volcano plot shows the distribution of differentially expressed lipids in survival over death outcomes. Differences with ANOVA p value < 0.05 and log2 ≥ ±1.6 were considered significant. The most up- and downregulated lipids are shown in green and red, respectively. (B) LPC/PC ratios in survivors compared with nonsurvivors in sepsis. The ratios and the respective p values calculated by the Mann–Whitney U test are presented (n = 56 for survival and n = 7 for nonsurvival).
Discussion
This is a proof of concept article in which we demonstrate that it is possible to reset an improper proinflammatory pathological immune response in the course of high-grade sepsis into an effective hypoinflammatory secondary immune response. The resetting was achieved in real time by repeated stimulation with a selected memory antigenic pool (IRSh) in a highly stringent murine sepsis model. Most importantly, this immune intervention increased the survival rate obtained with imipenem alone over five times, with a clear synergic effect of the therapeutic combination on the mean expected lifetime of the animals, which has doubled. It should be stressed that the beneficial effect of the proposed immune intervention was not dependent on the generation of a focused, pathogen-driven immune response itself but rather on the reactivation of memory to a set of diverse and mostly unrelated Ags to compose a new image of the aggressor pathogen as hypothesized. The resulting secondary activation can drive an effective immune response to the primary pathogen in presence of the antimicrobial equilibrium shifter. Thus, IRSh alone changes the ongoing pathological immune response but does not impact on survival because it needs the antimicrobial action to shift the host–pathogen balance in favor of the host.
There were three players in the petri dish in the classical experimental model used by Fleming to discover penicillin: the pathogen (bacteria), the antimicrobial drug (represented by the mold-infused broth), and the inert culture medium, which did not interfere with the interaction between the first two other components. If the drug was effective in this model, there was pathogen clearance in vitro (1). Comparatively, there are also three performers during the treatment of an infection in vivo: the pathogen, the antimicrobial drug, and the localized or systemic environment provided by the host, which is not inert because of the evolutionary acquisition of an immune system that shares the same task of blocking and/or destroying the pathogen (55, 93, 94). Only two out of three components of the in vitro model are variable, as opposed to all three components of the in vivo counterpart. Thus, the efficacy of a given drug to induce pathogen clearance in vitro does not necessarily translate into cure in vivo, because it depends on the joint action of such a drug with the third variable, namely the immune system.
In strong support of this view, infections and sepsis in both ends of the age spectrum are usually more severe, more frequent, and exhibit higher morbidity and mortality, even when appropriate antimicrobials are used in the correct indication, dosing and timing, possibly reflecting the fact that the immune system is dysfunctional in elders and immature in the young (8, 95–97). A complex pattern of susceptibility to infections associated to pathogen multiplicity and diversity also occurs in secondary and primary severe immune deficiencies, leading to higher morbidity and mortality. Thus, broad systemic antimicrobial prophylaxis is required in immune compromised patients because it greatly increases their survival. Moreover, the treatment of overt infections or fever in neutropenic and other severe immune-compromised patients require the use of broad spectrum antimicrobials early on, before the identification of the etiological agent, and often in combination, higher doses, and longer time as compared with the immune competent patient for the very same clinical disease presentation (98–113). Cure of severe infections and sepsis with antimicrobial drugs in terminal immune compromised patients is virtually unachievable (114–116). Finally, in poor income populations, malnutrition may act synergistically with circumstantial factors, such as lack of sewerage, drinkable water, and protection to environmental aggression, to compromise fitness and functionality of the immune system, decreasing the efficiency of antimicrobial drugs (117, 118). In line with these observations, there is no solid evidence in the literature of pathogen clearance in vivo by the sole action of antimicrobial drugs because there is always an immune reaction to the invader agent (6, 25, 26, 32, 51, 52, 55, 66). In contrast, the cure of some infections is possible, relying solely on the immune system, as can be inferred from the survival of humans and other animal species in natural selection (93, 94, 119–121). Therefore, joint action is an absolute requirement for cure because there is no antimicrobial drug effect in vivo without an accompanying immune response.
The nature of the immune response against pathogens can be ineffective, partially effective, or fully curative. The first two instances require an antimicrobial drug, when available, to attempt a curative outcome. The missing concept in the current understanding of the dynamics of the therapy of infectious diseases is that the success of antimicrobial drugs is ultimately dependent on the underlying immune response: antimicrobials are often necessary but never sufficient without a proper immune response. This conceptual mistake is probably responsible for the virtual absence of effective treatments for sepsis, septic shock, and severe infections and for the countless clinical trial failures of medicines that were promising in preclinical studies (7, 8, 12, 14–23).
Antimicrobial drugs as an equilibrium shifter do not change the nature of the immune response, only facilitate pathogen elimination or containment by the immune system. Thus, we have shown in our sepsis experiments that saline- and antibiotic-treated animals responded similarly as regards to the induction of hyperactivated conventional and nonconventional splenic T cell subpopulations. Addition of IRSh stimulation to the antibiotic therapy reverted course, with a substantial drop in all activated T cells, most evident in the CD8+ compartment. This is in line with the observation that activation of noncognate memory CD8+ T cells by the inflammasome plays an important role in the amplification of their effector function (54, 122).
It should be noted that an effective immune response may occur spontaneously in the antibiotic-treated animals without IRSh stimulation, but with very low frequency, which explains their dismal survival. The few animals that survive with antibiotic treatment alone share the same splenic expansion and also hypoactivation of all T cell subsets (Supplemental Fig. 1E). Thus, there was no survival without an effective immune response, spontaneous or induced, in antibiotic-treated animals. It is worth noting that late immune suppression in conventionally treated sepsis has been in part attributed to a contraction by apoptosis of the CD4+ naive T cell population that is also less functional (8, 15, 21, 23). IRSh induced a clear homeostatic recovery of these cells, which was lacking in animals that have only been treated with antibiotic. The use of repeated IRSh injections separated by short periods (3–4 d) for memory reactivation resembles a drawn-out disease or a protracted antigenic stimulus that is likely to inhibit the reactive immune suppressive wave elicited by the combined antigenic profile of IRSh and of the pathogen.
Our interpretation that IRSh-induced survival is a consequence of the replacement of a hyperinflammatory reaction by a contained, memory-driven, and effective hypoinflammatory immune response is supported primarily by the limitation of total CD3+ T cell expansion and the drop in expression of surface activation markers on the recovered splenic T cells. It is also corroborated by the quantitative proteomics and metabolomics (lipidomics) analyses, which revealed the reversion of the signatures of cytokine storm, deregulated inflammatory reaction, proapoptotic environment, and narrow LPC/PC ratio, with a central role for TNF-α neutralization in animals that did respond to combined therapy as opposed to those that died. In addition, the upregulated proteins in IRSh-induced survival may also mediate regulation of inflammation, lymphoid recovery and maintenance, intracellular detoxification, muscle metabolism improvement, and cell regeneration. Interestingly, the opposite proteomic profile found in natural sepsis may help to explain the severity of evolution of antimicrobial-treated sepsis after apparent primary pathogen clearance. Furthermore, lipid metabolites segregated very distinctively into survival and nonsurviving groups. It is of particular interest that LPC is a lipid second messenger that may be generated from PC in conditions of low inflammatory and effective immune responses, and that the LPC/PC ratio is higher in the surviving animals in our experiments, with absolute increase of LPC and decrease of PC. These findings are reminiscent of data reported by others on surviving sepsis patients treated with antimicrobial drugs (89–91). Also, there is some experimental evidence elsewhere that LPC has immune modulatory and host protection properties that have been tested therapeutically in preclinical models with modest results (123–125). The latter references support the interpretation that the plasma LPC upregulation that we have observed in surviving mice may have had an active mechanistic role in rescuing animals from sepsis, but this assumption remains to be tested in future studies.
The proteins and metabolites identified in this study reveal a glimpse of the default molecular program set in place by the immune response in sepsis that may often lead to death with or without antimicrobial treatment. The trigger of this response is the pathogen’s antigenic image. However, the default program can be reverted or replaced by another that is indeed compatible with survival, and the switch to the curative immune response can be achieved by overlaying the pathogen’s original antigenic image with that of the IRSh memory Ag pool. Thus, the current medical strategies to treat infection mimic the reactive defenses established along evolution of the immune system, which are primarily induced or guided by the pathogen. The two most conserved of these natural defenses are 1) the host’s resistance to infection or direct antipathogen action, which may be achieved clinically by antimicrobial drugs helping the host to reduce pathogen burden; and 2) the control of the host’s tissue damage definable as tolerance to the aggression, which limits the severity of the disease regardless of the pathogen load and may be accomplished clinically by life support measures (119–121). The price paid by the host for the immune response reactive pattern is immunopathology, when an improper response is induced by the pathogen. The central fault of this exclusive reactive pattern, and of the current medical approaches that resemble it, is to let the pathogen in command of the immune response. It leaves the host organism to bear the pathological consequences, without disclosing the entire healing potential of the immune system that, as we show in this study, may be proactively reset to adopt a curative path guided by cellular memory.
It is reasonable that our strategy mimics and possibly synergizes with the natural microbiota, which might have been evolutionarily selected among other things to provide ongoing homeostatic antigenic stimuli to commensal-specific memory immune cells, allowing the simultaneous elimination of the occasional invading pathogen with limited inflammation. The commensal and pathogen image perceived by the immune system would partially overlap, leading to a lower probability of a life-threatening response. This hypothesis is supported by recent findings on the interplay between microbiota and the innate and adaptive arms of the immune system, in particular the memory cells (126).
The IRSh immune modulation is no more than a therapeutic vaccination or memory recall in real time, with mostly unrelated Ags that overlap and modify the perceived antigenic image of the pathogen by the immune system. Thus, the IRSh removes the aggressor agent from the driver’s seat by resetting a new secondary activation state capable of guiding and redirecting an effective hypoinflammatory secondary pathogen-specific response, when used in combination with antimicrobial drugs. The applicability of the IRSh approach beyond sepsis in other disease models in which the immune system may be implicated remains to be tested.
Supplementary Material
Acknowledgments
We thank Christiano Chaccur Chadad, Celso Colombo Neto, and Marcelo Gonçalves de Campos Pinto for critical comments on the manuscript. They also thank Flávio Baggio Pires (IT Department at Integrated Center for Pediatric OncoHaematological Research, UNICAMP, Campinas, SP, Brazil) for informatics support as well as Simone Simonek Nowill, Mônica Keiko Maeda, Selma Maria Fontana, Beatriz Garcia Martins, and Yria Lungow Magro Andreoli for logistics and office support. The authors are grateful to Michael Murgu (Waters Corporation, Sao Paulo, SP, Brazil) for technical support and MS analysis for lipidomics studies. The authors also thank Prof. Daniel Martins-de-Souza and Juliana Silva Cassoli (Neuroproteomics Laboratory, Department of Biochemistry and Tissue Biology, Institute of Biology, UNICAMP, Campinas, SP, Brazil) for Ingenuity Pathway Analysis support. The following colleagues are also recognized: Angelo Brunelli Albertoni Larangeira for animal experimentation technical support and Ronei Luciano Mamoni (Clinical Pathology, Faculty of Medical Sciences, UNICAMP, Campinas, SP, Brazil) for flow cytometry support and MS sample preparation. The authors are grateful to Waters Corporation for the use of its São Paulo MS facilities and for the technical support. We thank Prof. Roy Edward Bruns for statistics support.
This work was supported by a Salvador Arena Foundation grant and Integrated Center for Pediatric OncoHaematological Research intramural funds at the State University of Campinas. Daniel Martins-de-Souza is a recipient of São Paulo Research Foundation Grant 2014/10068-4.
The online version of this article contains supplemental material.
- CV
- coefficient of variation
- FCA
- Freund’s complete adjuvant
- IRSh
- Immune Response Shifter
- LPC
- lysophosphatidylcholine
- MS
- mass spectrometry
- MS/MS
- tandem MS
- PC
- phosphatidylcholine
- ppm
- part per million
- UDMSE
- MSE multiplexed mode with ultradefinition ion mobility
- UNICAMP
- State University of Campinas
- UPLC
- ultraperformance liquid chromatography.
Disclosures
A.E.N. holds a patent on IRSh (US patent application no. US 15/431,329 and international patent no. PCT/BR2018/000004). The other authors have no financial conflicts of interest.
References
- 1.Alexander F. 1929. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of b. influenzae. Br. J. Exp. Pathol. 10: 226–236. [Google Scholar]
- 2.Waksman S. A., Woodruff H. B. 1942. Selective antibiotic action of various substances of microbial origin. J. Bacteriol. 44: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson R. L., Wright A. J. 1998. General principles of antimicrobial therapy. Mayo Clin. Proc. 73: 995–1006. [DOI] [PubMed] [Google Scholar]
- 4.Barlam T. F., Cosgrove S. E., Abbo L. M., MacDougall C., Schuetz A. N., Septimus E. J., Srinivasan A., Dellit T. H., Falck-Ytter Y. T., Fishman N. O., et al. 2016. Implementing an antibiotic stewardship program: guidelines by the infectious diseases society of America and the society for healthcare epidemiology of America. Clin. Infect. Dis. 62: e51–e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Society for Healthcare Epidemiology of America; Infectious Diseases Society of America; Pediatric Infectious Diseases Society . 2012. Policy statement on antimicrobial stewardship by the society for healthcare epidemiology of America (SHEA), the infectious diseases society of America (IDSA), and the pediatric infectious diseases society (PIDS). Infect. Control Hosp. Epidemiol. 33: 322–327. [DOI] [PubMed] [Google Scholar]
- 6.Angus D. C., van der Poll T. 2013. Severe sepsis and septic shock. N. Engl. J. Med. 369: 840–851. [DOI] [PubMed] [Google Scholar]
- 7.Gotts J. E., Matthay M. A. 2016. Sepsis: pathophysiology and clinical management. BMJ 353: i1585. [DOI] [PubMed] [Google Scholar]
- 8.Hotchkiss R. S., Moldawer L. L., Opal S. M., Reinhart K., Turnbull I. R., Vincent J. L. 2016. Sepsis and septic shock. Nat. Rev. Dis. Primers 2: 16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A., Zarychanski R., Light B., Parrillo J., Maki D., Simon D., Laporta D., Lapinsky S., Ellis P., Mirzanejad Y., et al. Cooperative Antimicrobial Therapy of Septic Shock (CATSS) Database Research Group 2010. Early combination antibiotic therapy yields improved survival compared with monotherapy in septic shock: a propensity-matched analysis. Crit. Care Med. 38: 1773–1785. [DOI] [PubMed] [Google Scholar]
- 10.Micek S. T., Welch E. C., Khan J., Pervez M., Doherty J. A., Reichley R. M., Kollef M. H. 2010. Empiric combination antibiotic therapy is associated with improved outcome against sepsis due to gram-negative bacteria: a retrospective analysis. Antimicrob. Agents Chemother. 54: 1742–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okamoto K., Tamura T., Sawatsubashi Y. 2016. Sepsis and disseminated intravascular coagulation. J. Intensive Care 4: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent J. L., De Backer D. 2013. Circulatory shock. N. Engl. J. Med. 369: 1726–1734. [DOI] [PubMed] [Google Scholar]
- 13.Singer M., Deutschman C. S., Seymour C. W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G. R., Chiche J. D., Coopersmith C. M., et al. 2016. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yo C. H., Hsu T. C., Gabriel Lee M. T., Porta L., Tsou P. Y., Wang Y. H., Lee W. C., Chen S. T., Lee C. C., National Taiwan University Hospital Health Economics and Outcome Research Group 2018. Trend and outcome of sepsis in children: a nationwide cohort study. J. Paediatr. Child Health 54: 776–783. [DOI] [PubMed] [Google Scholar]
- 15.Hotchkiss R. S., Monneret G., Payen D. 2013. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 13: 862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hotchkiss R. S., Monneret G., Payen D. 2013. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 13: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Poll T., van de Veerdonk F. L., Scicluna B. P., Netea M. G. 2017. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17: 407–420. [DOI] [PubMed] [Google Scholar]
- 18.Horiguchi H., Loftus T. J., Hawkins R. B., Raymond S. L., Stortz J. A., Hollen M. K., Weiss B. P., Miller E. S., Bihorac A., Larson S. D., et al. Sepsis and Critical Illness Research Center Investigators 2018. Innate immunity in the persistent inflammation, immunosuppression, and catabolism syndrome and its implications for therapy. Front. Immunol. 9: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasten K. R., Tschöp J., Adediran S. G., Hildeman D. A., Caldwell C. C. 2010. T cells are potent early mediators of the host response to sepsis. Shock 34: 327–336. [DOI] [PubMed] [Google Scholar]
- 20.Langley R. J., Tsalik E. L., van Velkinburgh J. C., Glickman S. W., Rice B. J., Wang C., Chen B., Carin L., Suarez A., Mohney R. P., et al. 2013. An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 5: 195ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmoeckel K., Traffehn S., Eger C., Pötschke C., Bröker B. M. 2015. Full activation of CD4+ T cells early during sepsis requires specific antigen. Shock 43: 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deutschman C. S., Tracey K. J. 2014. Sepsis: current dogma and new perspectives. Immunity 40: 463–475. [DOI] [PubMed] [Google Scholar]
- 23.Cabrera-Perez J., Condotta S. A., Badovinac V. P., Griffith T. S. 2014. Impact of sepsis on CD4 T cell immunity. J. Leukoc. Biol. 96: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modi S. R., Collins J. J., Relman D. A. 2014. Antibiotics and the gut microbiota. J. Clin. Invest. 124: 4212–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janeway C. A., Jr., Medzhitov R. 2002. Innate immune recognition. Annu. Rev. Immunol. 20: 197–216. [DOI] [PubMed] [Google Scholar]
- 26.Medzhitov R. 2008. Origin and physiological roles of inflammation. Nature 454: 428–435. [DOI] [PubMed] [Google Scholar]
- 27.Ahmed R., Gray D. 1996. Immunological memory and protective immunity: understanding their relation. Science 272: 54–60. [DOI] [PubMed] [Google Scholar]
- 28.Willis N. J. 1997. Edward Jenner and the eradication of smallpox. Scott. Med. J. 42: 118–121. [DOI] [PubMed] [Google Scholar]
- 29.Crookshank E. M. 1889. The Historical Medical Library of The College of Physicians of Philadelphia. H.K. Lewis, London. [Google Scholar]
- 30.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401: 708–712. [DOI] [PubMed] [Google Scholar]
- 31.Zinkernagel R. M. 2002. On differences between immunity and immunological memory. Curr. Opin. Immunol. 14: 523–536. [DOI] [PubMed] [Google Scholar]
- 32.Steinman R. M., Banchereau J. 2007. Taking dendritic cells into medicine. Nature 449: 419–426. [DOI] [PubMed] [Google Scholar]
- 33.Miller J. D., van der Most R. G., Akondy R. S., Glidewell J. T., Albott S., Masopust D., Murali-Krishna K., Mahar P. L., Edupuganti S., Lalor S., et al. 2008. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 28: 710–722. [DOI] [PubMed] [Google Scholar]
- 34.Sallusto F., Lanzavecchia A., Araki K., Ahmed R. 2010. From vaccines to memory and back. Immunity 33: 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C. M., Quigley M. F., Almeida J. R., Gostick E., Yu Z., Carpenito C., et al. 2011. A human memory T cell subset with stem cell-like properties. Nat. Med. 17: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zielinski C. E., Mele F., Aschenbrenner D., Jarrossay D., Ronchi F., Gattorno M., Monticelli S., Lanzavecchia A., Sallusto F. 2012. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484: 514–518. [DOI] [PubMed] [Google Scholar]
- 37.Rosalia R. A., Quakkelaar E. D., Redeker A., Khan S., Camps M., Drijfhout J. W., Silva A. L., Jiskoot W., van Hall T., van Veelen P. A., et al. 2013. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur. J. Immunol. 43: 2554–2565. [DOI] [PubMed] [Google Scholar]
- 38.Oliveira G., Ruggiero E., Stanghellini M. T., Cieri N., D’Agostino M., Fronza R., Lulay C., Dionisio F., Mastaglio S., Greco R., et al. 2015. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. [Published erratum appears in 2015 Sci. Transl. Med. 7: 319er9.] Sci. Transl. Med. 7: 317ra198. [DOI] [PubMed] [Google Scholar]
- 39.Cerwenka A., Lanier L. L. 2016. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 16: 112–123. [DOI] [PubMed] [Google Scholar]
- 40.Laidlaw B. J., Craft J. E., Kaech S. M. 2016. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 16: 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller S. N., Mackay L. K. 2016. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 16: 79–89. [DOI] [PubMed] [Google Scholar]
- 42.Haks M. C., Bottazzi B., Cecchinato V., De Gregorio C., Del Giudice G., Kaufmann S. H. E., Lanzavecchia A., Lewis D. J. M., Maertzdorf J., Mantovani A., et al. 2017. Molecular signatures of immunity and immunogenicity in infection and vaccination. Front. Immunol. 8: 1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K. D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y. X. 2007. Adaptive immune cells temper initial innate responses. Nat. Med. 13: 1248–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martín-Fontecha A., Baumjohann D., Guarda G., Reboldi A., Hons M., Lanzavecchia A., Sallusto F. 2008. CD40L+ CD4+ memory T cells migrate in a CD62P-dependent fashion into reactive lymph nodes and license dendritic cells for T cell priming. J. Exp. Med. 205: 2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palm N. W., Medzhitov R. 2007. Not so fast: adaptive suppression of innate immunity. Nat. Med. 13: 1142–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guarda G., Dostert C., Staehli F., Cabalzar K., Castillo R., Tardivel A., Schneider P., Tschopp J. 2009. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 460: 269–273. [DOI] [PubMed] [Google Scholar]
- 47.Rosenblum M. D., Way S. S., Abbas A. K. 2016. Regulatory T cell memory. Nat. Rev. Immunol. 16: 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farber D. L., Netea M. G., Radbruch A., Rajewsky K., Zinkernagel R. M. 2016. Immunological memory: lessons from the past and a look to the future. Nat. Rev. Immunol. 16: 124–128. [DOI] [PubMed] [Google Scholar]
- 49.Narni-Mancinelli E., Campisi L., Bassand D., Cazareth J., Gounon P., Glaichenhaus N., Lauvau G. 2007. Memory CD8+ T cells mediate antibacterial immunity via CCL3 activation of TNF/ROI+ phagocytes. J. Exp. Med. 204: 2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guarda G., Hons M., Soriano S. F., Huang A. Y., Polley R., Martín-Fontecha A., Stein J. V., Germain R. N., Lanzavecchia A., Sallusto F. 2007. L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat. Immunol. 8: 743–752. [DOI] [PubMed] [Google Scholar]
- 51.Matzinger P. 2002. The danger model: a renewed sense of self. Science 296: 301–305. [DOI] [PubMed] [Google Scholar]
- 52.Brodsky I. E., Medzhitov R. 2009. Targeting of immune signalling networks by bacterial pathogens. Nat. Cell Biol. 11: 521–526. [DOI] [PubMed] [Google Scholar]
- 53.Martinon F., Mayor A., Tschopp J. 2009. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27: 229–265. [DOI] [PubMed] [Google Scholar]
- 54.Kupz A., Guarda G., Gebhardt T., Sander L. E., Short K. R., Diavatopoulos D. A., Wijburg O. L., Cao H., Waithman J. C., Chen W., et al. 2012. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8+ T cells. Nat. Immunol. 13: 162–169. [DOI] [PubMed] [Google Scholar]
- 55.Pulendran B. 2015. The varieties of immunological experience: of pathogens, stress, and dendritic cells. Annu. Rev. Immunol. 33: 563–606. [DOI] [PubMed] [Google Scholar]
- 56.den Haan J. M., Arens R., van Zelm M. C. 2014. The activation of the adaptive immune system: cross-talk between antigen-presenting cells, T cells and B cells. Immunol. Lett. 162(2 Pt B): 103–112. [DOI] [PubMed] [Google Scholar]
- 57.Blasius A. L., Beutler B. 2010. Intracellular toll-like receptors. Immunity 32: 305–315. [DOI] [PubMed] [Google Scholar]
- 58.Canton J., Neculai D., Grinstein S. 2013. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13: 621–634. [DOI] [PubMed] [Google Scholar]
- 59.Chen G., Shaw M. H., Kim Y. G., Nuñez G. 2009. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4: 365–398. [DOI] [PubMed] [Google Scholar]
- 60.Dixit E., Kagan J. C. 2013. Intracellular pathogen detection by RIG-I-like receptors. Adv. Immunol. 117: 99–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elinav E., Strowig T., Henao-Mejia J., Flavell R. A. 2011. Regulation of the antimicrobial response by NLR proteins. Immunity 34: 665–679. [DOI] [PubMed] [Google Scholar]
- 62.Martinez-Pomares L. 2012. The mannose receptor. J. Leukoc. Biol. 92: 1177–1186. [DOI] [PubMed] [Google Scholar]
- 63.Osorio F., Reis e Sousa C. 2011. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 34: 651–664. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi O., Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140: 805–820. [DOI] [PubMed] [Google Scholar]
- 65.Wu J., Chen Z. J. 2014. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32: 461–488. [DOI] [PubMed] [Google Scholar]
- 66.Bottazzi B., Doni A., Garlanda C., Mantovani A. 2010. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu. Rev. Immunol. 28: 157–183. [DOI] [PubMed] [Google Scholar]
- 67.Subramanian N., Natarajan K., Clatworthy M. R., Wang Z., Germain R. N. 2013. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A. 2009. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Benjamim C. F., Ferreira S. H., Cunha F. Q. 2000. Role of nitric oxide in the failure of neutrophil migration in sepsis. J. Infect. Dis. 182: 214–223. [DOI] [PubMed] [Google Scholar]
- 70.Alves-Filho J. C., Sônego F., Souto F. O., Freitas A., Verri W. A., Jr., Auxiliadora-Martins M., Basile-Filho A., McKenzie A. N., Xu D., Cunha F. Q., Liew F. Y. 2010. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16: 708–712. [DOI] [PubMed] [Google Scholar]
- 71.Manley M. O., O’Riordan M. A., Levine A. D., Latifi S. Q. 2005. Interleukin 10 extends the effectiveness of standard therapy during late sepsis with serum interleukin 6 levels predicting outcome. Shock 23: 521–526. [PubMed] [Google Scholar]
- 72.Vyas D., Javadi P., Dipasco P. J., Buchman T. G., Hotchkiss R. S., Coopersmith C. M. 2005. Early antibiotic administration but not antibody therapy directed against IL-6 improves survival in septic mice predicted to die on basis of high IL-6 levels. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289: R1048–R1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu Y. Q., Gilar M., Lee P. J., Bouvier E. S., Gebler J. C. 2003. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal. Chem. 75: 6023–6028. [DOI] [PubMed] [Google Scholar]
- 74.Gilar M., Olivova P., Daly A. E., Gebler J. C. 2005. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 28: 1694–1703. [DOI] [PubMed] [Google Scholar]
- 75.Souza G. H. M. F., Guest P. C., Martins-de-Souza D. 2017. LC-MSE, multiplex MS/MS, ion mobility, and label-free quantitation in clinical proteomics. Methods Mol. Biol. 1546: 57–73. [DOI] [PubMed] [Google Scholar]
- 76.Gilar M., Olivova P., Chakraborty A. B., Jaworski A., Geromanos S. J., Gebler J. C. 2009. Comparison of 1-D and 2-D LC MS/MS methods for proteomic analysis of human serum. Electrophoresis 30: 1157–1167. [DOI] [PubMed] [Google Scholar]
- 77.Cassoli J. S., Brandão-Teles C., Santana A. G., Souza G. H. M. F., Martins-de-Souza D. 2017. Ion mobility-enhanced data-independent acquisitions enable a deep proteomic landscape of oligodendrocytes. [Published erratum appears in 2018 Proteomics 18.] Proteomics 17: 1700209. [Google Scholar]
- 78.Distler U., Kuharev J., Navarro P., Levin Y., Schild H., Tenzer S. 2014. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 11: 167–170. [DOI] [PubMed] [Google Scholar]
- 79.Lalli P. M., Corilo Y. E., Fasciotti M., Riccio M. F., de Sa G. F., Daroda R. J., Souza G. H., McCullagh M., Bartberger M. D., Eberlin M. N., Campuzano I. D. 2013. Baseline resolution of isomers by traveling wave ion mobility mass spectrometry: investigating the effects of polarizable drift gases and ionic charge distribution. J. Mass Spectrom. 48: 989–997. [DOI] [PubMed] [Google Scholar]
- 80.Curty N., Kubitschek-Barreira P. H., Neves G. W., Gomes D., Pizzatti L., Abdelhay E., Souza G. H., Lopes-Bezerra L. M. 2014. Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J. Proteomics 97: 126–140. [DOI] [PubMed] [Google Scholar]
- 81.Li G. Z., Vissers J. P., Silva J. C., Golick D., Gorenstein M. V., Geromanos S. J. 2009. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9: 1696–1719. [DOI] [PubMed] [Google Scholar]
- 82.Geromanos S. J., Hughes C., Golick D., Ciavarini S., Gorenstein M. V., Richardson K., Hoyes J. B., Vissers J. P., Langridge J. I. 2011. Simulating and validating proteomics data and search results. Proteomics 11: 1189–1211. [DOI] [PubMed] [Google Scholar]
- 83.Silva J. C., Gorenstein M. V., Li G.-Z., Vissers J. P. C., Geromanos S. J. 2006. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5: 144–156. [DOI] [PubMed] [Google Scholar]
- 84.Nascimento J. M., Garcia S., Saia-Cereda V. M., Santana A. G., Brandao-Teles C., Zuccoli G. S., Junqueira D. G., Reis-de-Oliveira G., Baldasso P. A., Cassoli J. S., Martins-de-Souza D. 2016. Proteomics and molecular tools for unveiling missing links in the biochemical understanding of schizophrenia. Proteomics Clin. Appl. 10: 1148–1158. [DOI] [PubMed] [Google Scholar]
- 85.Garcia S., Silva-Costa L. C., Reis-de-Oliveira G., Guest P. C., Baldasso P. A., Cassoli J. S., Martins-de-Souza D. 2017. Identifying biomarker candidates in the blood plasma or serum proteome. Adv. Exp. Med. Biol. 974: 193–203. [DOI] [PubMed] [Google Scholar]
- 86.Ahmed A. R., Blose D. A. 1983. Delayed-type hypersensitivity skin testing. A review. Arch. Dermatol. 119: 934–945. [PubMed] [Google Scholar]
- 87.Valdés-Ferrer S. I., Rosas-Ballina M., Olofsson P. S., Lu B., Dancho M. E., Ochani M., Li J. H., Scheinerman J. A., Katz D. A., Levine Y. A., et al. 2013. HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C(high) inflammatory monocytes in murine sepsis survivors. J. Intern. Med. 274: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murad A. M., Rech E. L. 2012. NanoUPLC-MSE proteomic data assessment of soybean seeds using the Uniprot database. BMC Biotechnol. 12: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Das U. N. 2004. Role of lipids in sepsis. Crit. Care Shock 7: 87–92. [Google Scholar]
- 90.Park D. W., Kwak D. S., Park Y. Y., Chang Y., Huh J. W., Lim C. M., Koh Y., Song D. K., Hong S. B. 2014. Impact of serial measurements of lysophosphatidylcholine on 28-day mortality prediction in patients admitted to the intensive care unit with severe sepsis or septic shock . J. Crit. Care 29: 882.e5–882.e11. [DOI] [PubMed] [Google Scholar]
- 91.Drobnik W., Liebisch G., Audebert F.-X., Fröhlich D., Glück T., Vogel P., Rothe G., Schmitz G. 2003. Plasma ceramide and lysophosphatidylcholine inversely correlate with mortality in sepsis patients. J. Lipid Res. 44: 754–761. [DOI] [PubMed] [Google Scholar]
- 92.Ahn W. G., Jung J. S., Kwon H. Y., Song D. K. 2017. Alteration of lysophosphatidylcholine-related metabolic parameters in the plasma of mice with experimental sepsis. Inflammation 40: 537–545. [DOI] [PubMed] [Google Scholar]
- 93.Travis J. 2009. Origins. On the origin of the immune system. Science 324: 580–582. [DOI] [PubMed] [Google Scholar]
- 94.Litman G. W., Rast J. P., Fugmann S. D. 2010. The origins of vertebrate adaptive immunity. Nat. Rev. Immunol. 10: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martín S., Pérez A., Aldecoa C. 2017. Sepsis and immunosenescence in the elderly patient: a review. Front. Med. (Lausanne) 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Collins A., Weitkamp J. H., Wynn J. L. 2018. Why are preterm newborns at increased risk of infection? Arch. Dis. Child. Fetal Neonatal. Ed. 103: F391–F394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shane A. L., Stoll B. J. 2014. Neonatal sepsis: progress towards improved outcomes. J. Infect. 68(Suppl. 1): S24–S32. [DOI] [PubMed] [Google Scholar]
- 98.Conley M. E., Dobbs A. K., Farmer D. M., Kilic S., Paris K., Grigoriadou S., Coustan-Smith E., Howard V., Campana D. 2009. Primary B cell immunodeficiencies: comparisons and contrasts. Annu. Rev. Immunol. 27: 199–227. [DOI] [PubMed] [Google Scholar]
- 99.Durandy A., Kracker S., Fischer A. 2013. Primary antibody deficiencies. Nat. Rev. Immunol. 13: 519–533. [DOI] [PubMed] [Google Scholar]
- 100.Milner J. D., Holland S. M. 2013. The cup runneth over: lessons from the ever-expanding pool of primary immunodeficiency diseases. Nat. Rev. Immunol. 13: 635–648. [DOI] [PubMed] [Google Scholar]
- 101.Notarangelo L. D. 2013. Functional T cell immunodeficiencies (with T cells present). Annu. Rev. Immunol. 31: 195–225. [DOI] [PubMed] [Google Scholar]
- 102.Puel A., Yang K., Ku C. L., von Bernuth H., Bustamante J., Santos O. F., Lawrence T., Chang H. H., Al-Mousa H., Picard C., Casanova J. L. 2005. Heritable defects of the human TLR signalling pathways. J. Endotoxin Res. 11: 220–224. [DOI] [PubMed] [Google Scholar]
- 103.Dobbs K., Domínguez Conde C., Zhang S. Y., Parolini S., Audry M., Chou J., Haapaniemi E., Keles S., Bilic I., Okada S., et al. 2015. Inherited DOCK2 deficiency in patients with early-onset invasive infections. N. Engl. J. Med. 372: 2409–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.von Bernuth H., Picard C., Jin Z., Pankla R., Xiao H., Ku C. L., Chrabieh M., Mustapha I. B., Ghandil P., Camcioglu Y., et al. 2008. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321: 691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holland S. M. 2010. Chronic granulomatous disease. Clin. Rev. Allergy Immunol. 38: 3–10. [DOI] [PubMed] [Google Scholar]
- 106.Casanova J. L. 2015. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc. Natl. Acad. Sci. USA 112: E7128–E7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grumach A. S., Kirschfink M. 2014. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol. Immunol. 61: 110–117. [DOI] [PubMed] [Google Scholar]
- 108.Rubin R. H., Wolfson J. S., Cosimi A. B., Tolkoff-Rubin N. E. 1981. Infection in the renal transplant recipient. Am. J. Med. 70: 405–411. [DOI] [PubMed] [Google Scholar]
- 109.Collin B. A., Ramphal R. 1998. Pneumonia in the compromised host including cancer patients and transplant patients. Infect. Dis. Clin. North Am. 12: 781–805, xi. [DOI] [PubMed] [Google Scholar]
- 110.Fishman J. A. 2007. Infection in solid-organ transplant recipients. N. Engl. J. Med. 357: 2601–2614. [DOI] [PubMed] [Google Scholar]
- 111.Kamboj M., Sepkowitz K. A. 2009. Nosocomial infections in patients with cancer. Lancet Oncol. 10: 589–597. [DOI] [PubMed] [Google Scholar]
- 112.Freifeld A. G., Bow E. J., Sepkowitz K. A., Boeckh M. J., Ito J. I., Mullen C. A., Raad I. I., Rolston K. V., Young J. A., Wingard J. R., Infectious Diseases Society of Americaa 2011. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the infectious diseases society of America. Clin. Infect. Dis. 52: 427–431. [DOI] [PubMed] [Google Scholar]
- 113.Dumford D. M., III, Skalweit M. 2016. Antibiotic-resistant infections and treatment challenges in the immunocompromised host. Infect. Dis. Clin. North Am. 30: 465–489. [DOI] [PubMed] [Google Scholar]
- 114.Thompson A. J., Silveira M. J., Vitale C. A., Malani P. N. 2012. Antimicrobial use at the end of life among hospitalized patients with advanced cancer. Am. J. Hosp. Palliat. Care 29: 599–603. [DOI] [PubMed] [Google Scholar]
- 115.Taverner J., Ross L., Bartlett C., Luthe M., Ong J., Irving L., Smallwood N. 2019. Antimicrobial prescription in patients dying from chronic obstructive pulmonary disease. Intern. Med. J. 49: 66–73. [DOI] [PubMed] [Google Scholar]
- 116.Macedo F., Nunes C., Ladeira K., Pinho F., Saraiva N., Bonito N., Pinto L., Gonçalves F. 2018. Antimicrobial therapy in palliative care: an overview. Support. Care Cancer 26: 1361–1367. [DOI] [PubMed] [Google Scholar]
- 117.Schaible U. E., Kaufmann S. H. E. 2007. Malnutrition and infection: complex mechanisms and global impacts. PLoS Med. 4: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rytter M. J., Kolte L., Briend A., Friis H., Christensen V. B. 2014. The immune system in children with malnutrition--a systematic review. PLoS One 9: e105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schneider D. S., Ayres J. S. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Graham A. L., Allen J. E., Read A. F. 2005. Evolutionary causes and consequences of immunopathology. Annu. Rev. Ecol. Evol. Syst. 36: 373–397. [Google Scholar]
- 121.Råberg L., Graham A. L., Read A. F. 2009. Decomposing health: tolerance and resistance to parasites in animals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lauvau G., Soudja S. M. 2015. Mechanisms of memory T cell activation and effective immunity. Adv. Exp. Med. Biol. 850: 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yan J. J., Jung J. S., Lee J. E., Lee J., Huh S. O., Kim H. S., Jung K. C., Cho J. Y., Nam J. S., Suh H. W., et al. 2004. Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat. Med. 10: 161–167. [DOI] [PubMed] [Google Scholar]
- 124.Parra Millán R., Jiménez Mejías M. E., Sánchez Encinales V., Ayerbe Algaba R., Gutiérrez Valencia A., Pachón Ibáñez M. E., Díaz C., Pérez Del Palacio J., López Cortés L. F., Pachón J., Smani Y. 2016. Efficacy of lysophosphatidylcholine in combination with antimicrobial agents against Acinetobacter baumannii in experimental murine peritoneal sepsis and pneumonia models. Antimicrob. Agents Chemother. 60: 4464–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murch O., Collin M., Sepodes B., Foster S. J., Mota-Filipe H., Thiemermann C. 2006. Lysophosphatidylcholine reduces the organ injury and dysfunction in rodent models of gram-negative and gram-positive shock. Br. J. Pharmacol. 148: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mao K., Baptista A. P., Tamoutounour S., Zhuang L., Bouladoux N., Martins A. J., Huang Y., Gerner M. Y., Belkaid Y., Germain R. N. 2018. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554: 255–259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.