

Abstract
The α4β2 nAChR is the most predominant subtype in the brain and is a well-known culprit for nicotine addiction. Previously we presented a series of α4β2 nAChR selective compounds that were discovered from a mixture-based positional-scanning combinatorial library. Here we report further optimization identified highly potent and selective α4β2 nAChR antagonists 5 (AP-202) and 13 (AP-211). Both compounds are devoid of in vitro agonist activity and are potent inhibitors of epibatidine-induced changes in membrane potential in cells containing α4β2 nAChR, with IC50 values of approximately 10 nM, but are weak agonists in cells containing α4β2 nAChR. In vivo studies show that 5 can. significantly reduce operant nicotine self-administration and nicotine relapse-like behavior in rats at doses of 0.3 and 1 mg/kg. The pharmacokinetic data also indicate that 5, via sc administration, is rapidly absorbed into the blood, reaching maximal concentration within 10 min with a half-life of less than 1 h.
Graphical Abstract
INTRODUCTION
Worldwide, 6 million people, including 480 000 in the United State, die premature deaths from smoking-induced disease each year.1 Despite decades of public health efforts, 17.8% of U.S. adults continue to smoke, with direct medical costs averaging $170 billion/year and lost productivity costs averaging $156 billion/year.1,2 Moreover, although 70% of smokers say they want to quit, smoking’s powerful dependence results in at least a 90% failure.1,3
Current FDA-approved pharmacotherapies to facilitate smoking cessation include nicotine replacement therapy (patches, gums, inhalers, and nasal sprays), the antidepressant bupropion, and varenicline. Unfortunately, cessation rates in most clinical trials of these therapies still average only from 10% to 20% for nicotine replacement therapy, from 15% to 25% for bupropion, and from 23% to 40% for varenicline.4–6 Although varenicline is the most successful treatment currently in clinical use, some users have become plagued by significant psychological problems including depression and thoughts of suicide.7 Additional nicotine pharmacotherapies are in clinical trials, including the nonselective nicotinic acetylcholine receptor (nAChR) antagonist mecamylamine, the opioid antagonist naltrexone, and nicotine monoclonal antibody therapy. Clearly new medications are a high priority.
Nicotine addiction, like addiction to other psychomotor stimulants, is thought to be due to the activation of the dopaminergic mesocorticolimbic pathway.8,9 The nicotinic receptor subtype most prevalent in this region is the α4β2 nAChR, although there is a significant level of the a6 subunit.10,11 A great deal of evidence suggests that the α4β2 receptor is involved in nicotine dependence. In particular, β2-knockout mice do not self-administer nicotine,8,12 the selective antagonists dihydro-β-erythroidine (DHβE) and 2-fluoro-3-(4-nitrophenyl)deschloroepibatidine (4-nitro-PFEB) block nicotine self-administration,13,14 and the α4β2 nAChR partial agonist varenicline is clinically used as a smoking cessation medication.4 More recently, the importance of other nAChR subunits in nicotine self-administration has been demonstrated, particularly α6 and β5 in the ventral tegmental area (VTA) and α5 and β4 in the habenular-interpeduncular pathway.15–18 Nevertheless, as demonstrated with varenicline, modulation of α4β2 nAChR clearly has promise as a mechanism to modulate smoking.
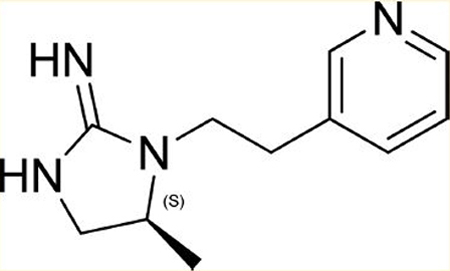
The Torrey Pines Institute for Molecular Studies (TPIMS) has a collection of small molecule libraries arranged in systematically formatted mixtures.19 Computational analysis of the TPIMS small molecule libraries demonstrates that the collection covers novel areas of chemical space as well as structural features not available in other compound collections.20 The TPIMS collection contains over 5 million individual small molecules; however, due to the formatting of these as mixtures, one can identify active individual compounds with moderate throughput capabilities (typically, fewer than 300–500 samples need to be tested). Utilizing the TPIMS small molecule library collection, compounds were screened for binding affinity at α4β2 nAChR and α4β2 nAChR. Initially, a “scaffold-ranking library” was tested. This library was made up of 37 mixtures, each mixture containing on average 135 000 structurally analogous compounds (identical scaffolds). One mixture, the one containing the bis-cyclic guanidines (Figure 1), showed significant inhibition of [3H]epibatidine binding. After screening the positional scanning library associated with this mixture, we identified a series of individual analogs of these compounds, some of which demonstrated low micromolar (μM) affinity at α4β2 nAChR. Several of these analogs show >10-fold selectivity at α4β2 over α4β2 nAChR. All of these steps are discussed in detail in our recent publication.21 From the compounds identified by library screening, traditional medicinal chemistry was initiated. Herein we discuss high affinity compounds that were identified as selective α4β2 nAChR pure antagonists and provide details of one compound in particular [5 (AP-202)] that showed significant activity in decreasing nicotine taking and seeking behaviors.
Figure 1.
Bis-cyclic guanidine scaffold contains compounds with moderate affinity for α4β2 nAChR.
RESULTS
SAR Studies.
By utilization of 16 R1 and 21 R2 functionalities, over 140 different analogs with core A were made (Figure 2; note that not all combinations of R groups were made). The compounds were initially tested for ability to inhibit [3H]epibatidine binding at a single concentration (10 μM) at both α4β2 and α4β2 nAChR, and representative data are shown in Figure 2 [8 R1 groups (x-axis) and 21 R2 groups (y-axis)]. Each dot in Figure 2 represents a compound tested. The dots are color coded by α4β2 affinity [using a color gradient: red (0%, no binding) to green (100%, binding affinity)] and sized by α4β2 affinity (larger is higher affinity). A larger green dot binds to both targets at 10 μM, a large red dot indicates binding only to α4β2 at 10 μM, and a small green dot indicates binding only to α3β4 at 10 μM. Many of the compounds that do not exhibit α4β2 selectivity at 10 μM (large green dots) are selective for α4β2 upon dose response. R group functionalities play a critical role in both affinity and selectivity. Increasing the size at the R1 position significantly reduces the affinity of the compound (this trend was seen for an additional 8 R1 groups tested, data not shown). For the R2 position, specific substituted aromatic functionalities provide the highest affinity compounds. Also, attachment position to the pyridyl ring (i.e., R2–2 to R2–3) or the length of the carbon chain (i.e. R2–2 to R2–9) had a significant effect on affinity. Of note, some of the compounds containing an aliphatic group at the R2 position maintained α4β2 affinity, albeit at reduced levels, while exhibiting no α3β4 affinity at 10 μM (R2–20).
Figure 2.
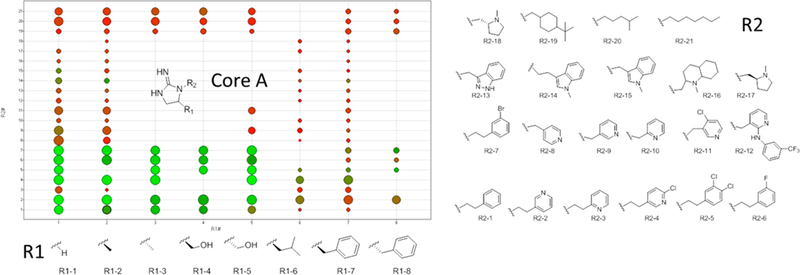
SAR of different analogs with core A utilizing the different R group functionalities shown. The compounds were tested for ability to inhibit [3H]epibatidine binding at a single concentration (10 μM) at both α4β2 and α3β4 nAChR. Each dot represents a compound tested. The dots are color coded by α3β4 affinity [using a color gradient: red (0%, no binding) to green (100%, binding affinity)] and sized by α4β2 affinity (larger is higher affinity). A larger green dot binds to both targets at 10 μM, a large red dot indicates binding only to α4β2 at 10 μM, and a small green dot indicates binding only to α3β4 at 10 μM.
On the basis of these data, compounds were selected for Ki determination. Table 1 contains the data reported in nanomolar (nM) from a series of compounds in which 2-(pyridine-3-yl)ethyl (R2–2 from Figure 2) is fixed in the R2 position. The compounds in this series all show selectivity toward α4β2 with a shift in affinity favoring the small R1 substitutions (hydrogen, methyl, and hydroxymethyl) as previously noted in Figure 2. We next determined the Ki values for a series of analogs where the R1 position was fixed with one of the small R1 functionalities and R2 was adjusted (Table 2). Using either 2-(pyridine-3-yl)ethyl or 2-(6-chloropyridin-3-yl)ethyl at the R2 position produced potent and α4β2 nAChR-selective compounds (24, 11 nM with an 11-fold selectivity, and 13 (AP-211), 13 nM with a 74-fold selectivity). Utilization of a non-pyridyl aromatic group at R2 either eliminates or slightly inverts the desired selectivity (i.e., compounds 1, 3, 17, and 21), and shortening the R2 linker carbon chain significantly reduces the affinity against both targets (15 vs 8 and 14 vs 5). Compounds with highest affinity and selectivity for α4β2 over α3β4 nAChR were 5 and 13.
Table 1.
Binding Affinity and Selectivity over α4β2 and α3β4 nAChR of a Series of Compounds Where 2-(Pyridine-3-yl)ethyl (R2–2 from Figure 2) Is Fixed in the R2 Positiona
 | |||||
|---|---|---|---|---|---|
| compd | R | α3β4 | α4β2 | selectivity | |
| 4 | S-benzyl | 12348 ± 1861 | 771 ± 334 | 16 | |
| 5 | S-methyl | 1026 ± 254 | 18 ± 3 | 57 | |
| 6 | S-isobutyl | 18683 ± 2058 | 4964 ± 103 | 4 | |
| 7 | S-2-butyl | 18107 ± 1529 | 863 ± 13 | 21 | |
| 8 | hydrogen | 644 ± 17 | 58 ± 3 | 11 | |
| 9 | S-isopropyl | 24487 ± 622 | 577 ± 25 | 42 | |
| 10 | S-3-propylguanidine | 24043 ± 1073 | 1531 ± 30 | 16 | |
| 11 | S-4-hydroxylbenzyl | 27420 ± 4396 | 1606 ± 41 | 17 | |
| 12 | S-2-(methylthio)ethyl | 35590 ± 173 | 448 ± 14 | 79 | |
| 13 | R-hydroxymethyl | 964 ± 193 | 13 ± 1 | 74 | |
All experiments are performed in duplicate or triplicate and repeated 2–3 times. Data are presented as the mean ± SEM.
Table 2.
Binding Affinity and Selectivity over α4β2 and α3β4 nAChR of a Series of Compounds Having Three Small R1 Functionalities with an Adjusted R2 Groupa
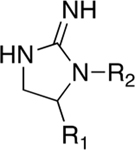 | |||||
|---|---|---|---|---|---|
| compd | R1 | R2 | α3β4 | α4β2 | selectivity |
| 1 | hydrogen | 2-(3,4-dichlorophenyl)ethyl | 711 ± 18 | 1296 ± 44 | 0.5 |
| 2 | hydrogen | pyridine-4-ylmethyl | 50107 ± 1806 | 652 ± 166 | 77 |
| 3 | hydrogen | phenylethyl | 325 ± 40 | 361 ± 1 | 0.9 |
| 5 | S-methyl | 2-(pyridine-3-yl)ethyl | 1026 ± 254 | 18 ± 3 | 57 |
| 8 | hydrogen | 2-(pyridine-3-yl)ethyl | 644 ± 17 | 58 ± 3 | 11 |
| 13 | R-hydroxymethyl | 2-(pyridine-3-yl)ethyl | 964 ± 193 | 13 ± 1 | 74 |
| 14 | S-methyl | pyridine-3-ylmethyl | 36547 ± 729 | 1358 ± 9 | 27 |
| 15 | hydrogen | pyridine-3-ylmethyl | 98927 ± 1106 | 3750 ± 81 | 26 |
| 16 | S-methyl | phenylethyl | N/A | 2250 ± 20 | N/A |
| 17 | R-hydroxymethyl | 2-(3,4-dichlorophenyl)ethyl | 321 ± 33 | 1045 ± 91 | 0.3 |
| 18 | hydrogen | 3-fluorophenylethyl | 633 ± 6 | 221 ± 17 | 3 |
| 19 | S-methyl | 3-fluorophenylethyl | N/A | 474 ± 12 | N/A |
| 20 | R-hydroxymethyl | 3-fluorophenylethyl | N/A | 704 ± 36 | N/A |
| 21 | hydrogen | 3-bromophenylethyl | 314 ± 26 | 601 ± 42 | 0.5 |
| 22 | S-methyl | 3-bromophenylethyl | 628 ± 61 | 361 ± 30 | 1.7 |
| 23 | R-hydroxymethyl | 3-bromophenylethyl | 1090 ± 123 | 640 ± 72 | 1.7 |
| 24 | S-methyl | 2-(6-chloropyridin-3-yl)ethyl | 125 ± 6 | 11 ± 1 | 11 |
| 25 | hydrogen | 2-(6-chloropyridin-3-yl)ethyl | 471 ± 16 | 80 ± 5 | 6 |
| 26 | R-hydroxymethyl | 2-(6-chloropyridin-3-yl)ethyl | 281 ± 19 | 20 ± 2 | 14 |
All experiments are performed in duplicate or triplicate and repeated 2–3 times. Data are presented as the mean ± SEM.
5 and 13 were tested for binding affinity to α4β2, α3β4, α4β4, α3β2, and α3β4α5 nAChR in membranes from cells transfected with these receptors. As seen in Table 3, 5 and 13 have higher affinity for β2-containing nAChR over β4-containing receptor, with 14-fold or greater selectivity for α4β2 over α3β4, α4β4, and α3β4α5 nAChR.
Table 3.
Ki (nM) Values for 5 and 13 at Various nAChR Subtypesa
| compd | α4β2 | α3β4 | α4β4 | α3β2 | α3β4α5 |
|---|---|---|---|---|---|
| 5 | 18.0 ± 2.68 | 1026 ± 254 | 716 ± 200 | 167 ± 20.4 | 1619 ± 93.7 |
| 13 | 13.06 ± 0.55 | 964 ± 193 | 184 ± 28.7 | 85.4 ± 9.64 | 957 ± 70.6 |
All experiments are performed in triplicate and repeated 3−4 times. Data are presented as the mean ± SEM.
Figure 3 shows the in vitro activity for 5 and 13. Compounds were tested for their ability to induce a change in membrane potential or Ca2+ flux in HEK cells transfected with rat α4β2 nAChR and α3β4 nAChR, respectively. Both 5 and 13 are devoid of agonist activity in this in vitro model (Figure 3A), and both are very potent inhibitors of epibatidine-induced changes in membrane potential in cells containing α4β2 nAChR, with IC50 values of approximately 10 nM (Figure 3B). In cells containing α3β4 nAChR, these compounds have weak agonist activity with EC50 values of 3509 nM and 2538 nM (Figure 3C) and also weakly desensitize the receptor with IC50 values of 6730 nM and 2717 nM, respectively (Figure 3D). Neither of these compounds have ability to activate α7 nAChR or block acetylcholine induced changes in membrane potential in cells transfected with α7 nAChR (data not shown). Therefore, these compounds are high affinity and selective α4β2 nAChR antagonists.
Figure 3.
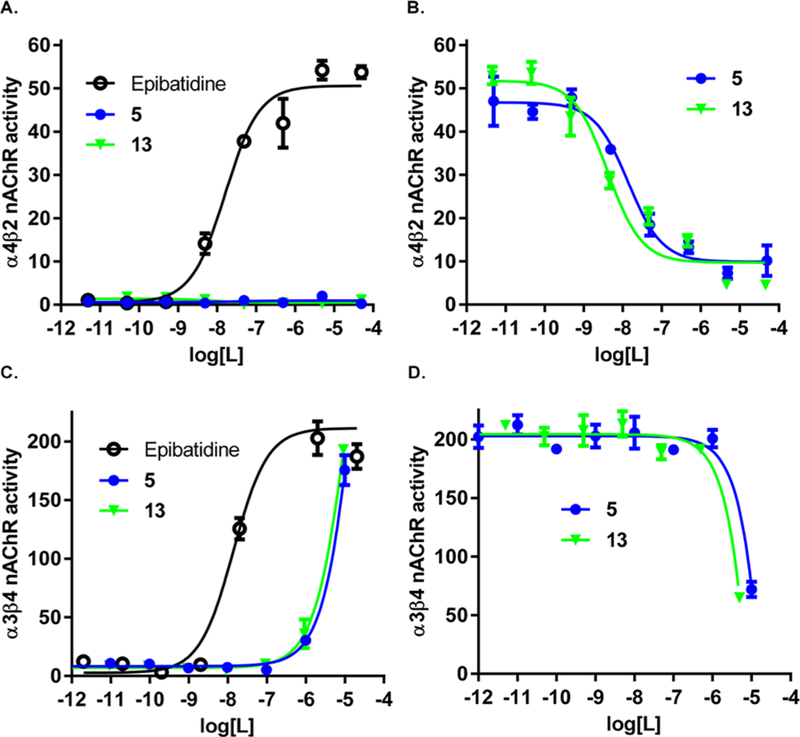
Activity of 5 and 13 on α4β2 nAChR and α3β4 nAChR in HEK cells. (A) Both 5 and 13 alone have no agonistic activity on α4β2 nAChR. (B) 5 and 13 potently inhibit epibatidine-induced α4β2 nAChR activation in a dose-dependent manner. (C) Both 5 and 13 alone only stimulate α3β4 nAChR at high dose. (D) 5 and 13 partially block epibatidine-induced α3β4 nAChR activation only at high dose. All experiments are performed in triplicate and repeated at least twice. Data are presented as mean ± SEM.
Our lead compounds 5 and 13 possess many properties that are in the desirable range for orally available central nervous system (CNS)-active drugs and are compared to the mean values for marketed CNS drugs22 shown in Table 4.
Table 4.
Properties of Compounds 5 and 13
| compd property | 5 | 13 | mean value of marketed CNS drugs |
|---|---|---|---|
| molecular weight | 204 | 220 | 310 |
| total no. of O and N | 4 | 5 | 4.3 |
| ClogP | 0.2 | −0.8 | 2.5 |
| tPSA | 51 | 72 | 60–70 (<90) |
In Vivo Activity.
5 was tested for its ability to block nicotine self-administration in 2 h operant sessions. Response rate at the end of the experiment was 25.4 ± 4.4 infusions for the vehicle group; 21.4 ± 4.4 for the 5, 0.3 mg/kg group; and 22.1 ± 4.8 for the 5, 1.0 mg/kg group. Initial one-way analysis of variance (ANOVA) conducted on the cumulative infusions obtained in 2 h sessions revealed no effect of 5 treatment (F(2,12) = 2.7, NS) (Figure 4A). However, by analysis of the infusions obtained in 30 min intervals, the experiment led to different results with 5 showing effectiveness in significantly attenuating nicotine self-administration. In fact, two-way ANOVA revealed significant interaction “time point X treatment” (F(636) = 4.1, p < 0.01). Post hoc analysis indicated that 5 treatment decreased nicotine self-administration during the first 30 min but not during the remaining 90 min period of the session, thus suggesting short-term activity of the antagonist (Figure 4B). Additionally, both doses examined of 5, 0.3 and 1.0 mg/kg, were effective (p < 0.01, p < 0.001, respectively). These data are consistent with other α4β2 nAChR antagonists, which also block nicotine self-administration.13,14
Figure 4.
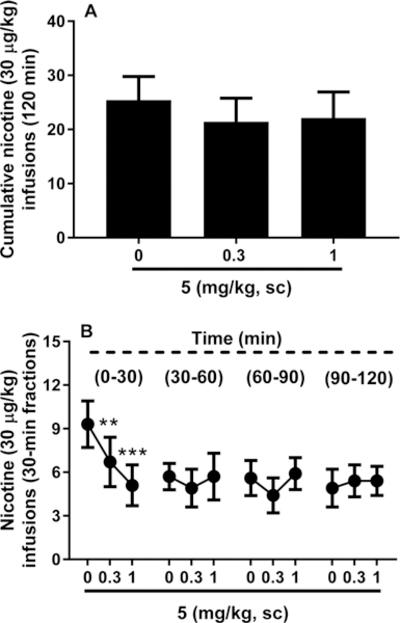
Effect of 5 on operant nicotine self-administration under a FR-3 reinforcement schedule. (A) Subcutaneous administration of 5 failed to alter the cumulative number of nicotine lever presses across a 120 min session. (B) 5 successfully decreased nicotine self-administration across the first 30 min (0.3 and 1 mg/kg) while leaving lever pressing unaltered across the remaining portion of the session. Results are described as mean (±SEM) cumulative number and 30 min fractions of nicotine infusions, respectively: (**) p < 0.01, (***) p < 0.001 difference from vehicle (5, 0 mg/kg). For detailed statistics, see “Results”.
Subsequently, 5 was tested for its ability to reduce nicotine-seeking behavior. As shown in Figure 5A, ANOVA analysis performed on the nicotine-associated lever indicated a main effect of 0.15 mg/kg nicotine priming in inducing nicotine-seeking behavior, as compared with extinction conditions (F(1,6) = 19.5, p < 0.01). In examining the effect of pretreatment of 5 before the priming injection of nicotine, the overall ANOVA indicated a significant effect that 5 decreased responding to a nicotine priming (F(2,12) = 3.9, p < 0.05). Post hoc comparisons revealed that both doses of 0.3 and 1.0 mg/kg significantly decreased responding (p < 0.05 for both doses).
Figure 5.
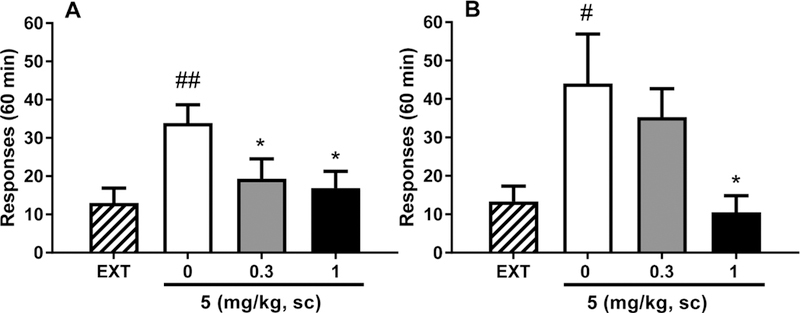
Effect of 5 on relapse-like behavior. 5 blocked nicotine priming-induced as well as cue-induced reinstatement of nicotine seeking. (A) There was significant reinstatement induction by subcutaneous administration of a nicotine priming dose of 0.15 mg/kg. Pretreatment with 5 (0.3 and 1 mg/kg) returned lever pressing to the extinction (EXT) levels. (B) Reinstatement of lever pressing was also obtained upon presentation of stimuli (cue light, orange odor) previously associated with nicotine. Pretreatment with 5 (1 mg/kg) abolished reinstatement. Values represent the mean (±SEM) number of total responses on the nicotine-associated lever in 60 min for both reinstatement paradigms: (#) p < 0.05, (##) p < 0.01 difference from EXT; (∗) p < 0.05 difference from vehicle (5, 0 mg/kg). For detailed statistics, see “Results”.
Conditioned stimuli previously associated with nicotine infusions were also able to elicit reinstatement of nicotine seeking (F(1,6) = 7.9, p < 0.05). 5 could attenuate this effect [F(2,6) = 5.1, p < 0.05]. Post hoc comparison tests revealed that 5 at the dose of 1 mg/kg resulted in a diminished number of responses as compared with those of the vehicle group (p < 0.05, Figure 5B).
5 was tested to determine whether it could block nicotine-induced hypothermia, an effect that can be mimicked by other selective and nonselective nAChR agonists.23 Overall treatments significantly modified body temperature. ANOVA showed “treatment” × “time point” interaction (F(12,84) = 7.3, p < 0.001) with pairwise comparisons showing reduced rat core body temperature due to subcutaneous (sc) administration of 0.5 mg/kg of nicotine (p < 0.001 at 15 and 30 min time point). However, 5 (1.0 mg/kg, sc) was neither able to induce hypothermia in rats nor able to block nicotine-induced hypothermia (Figure 6). These data suggest that nAChR subunits other than α4β2 could be implicated in mediating thermoregulatory effects of nicotine.
Figure 6.
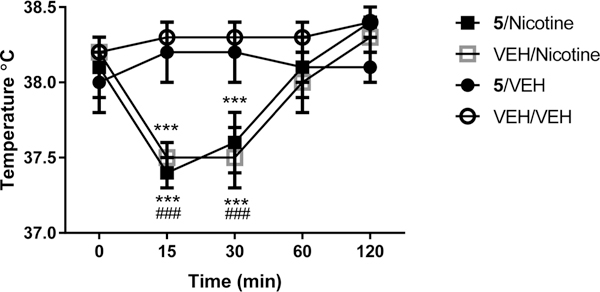
Assessment of body temperature. 5 failed to block nicotine- induced hypothermia and did not induce per se hypothermia. Baseline rectal temperature was measured just prior to injection of 5 (1.0 mg/ kg, sc) or vehicle (0.9% saline). Ten minutes later, nicotine (0.5 mg/ kg, sc) or vehicle (0.9% saline) was injected, and temperature was measured again after 15, 30, 60, and 120 min. Values represent the mean (±SEM) body temperature (°C): (###) p < 0.001 difference from T =0 for the nicotine group; (***) p < 0.001 difference from 5/ VEH and VEH/VEH at T =15 and T = 30. For detailed statistics, see “Results”.
To determine the pharmacokinetic parameters of 5, relative blood content and blood−brain barrier penetration were examined in rats following sc administration of 5 (2.0 mg/ kg). Results indicate that 5 is taken up rapidly into the blood (F(3,9) = 17.7, p < 0.001) and brain (F(3,9) = 8.7, p < 0.01), reaching maximal concentration within 10 min, with a half-life of less than 1 h (blood, T =10, p < 0.001; T =30, p < 0.05; brain, T = 10, p <0.01; T =30, p < 0.01; Figure 7). During this time, the brain concentration of 5 approached that of the blood concentration. These data are consistent with the short acting effect that we have demonstrated on nicotine self-administration.
Figure 7.
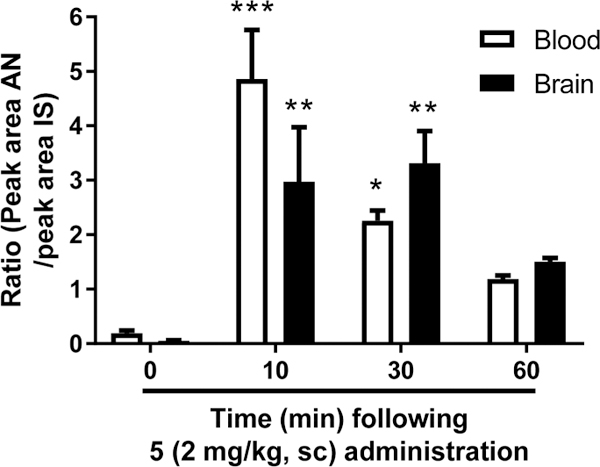
Detection of 5 in rat blood and brain using LC–MS–MS. 5 contents were determined in rat blood and brain 0, 10, 30, and 60 min following sc injection at the dose of 2 mg/kg. Values represent the ratio between the peak area of the analyte (AN) and the peak area of the internal standard (IS). Average (±SEM) for each time point is plotted. For detailed statistics, see “Results”.
DISCUSSION
Agents active at the nAChR have been actively studied for their ability to attenuate both nicotine and alcohol use in both preclinical and clinical settings. The most successful compound has been varenicline. This compound, which has had several billion dollars in sales as Chantix, is a potent α4β2 nAChR partial agonist. Although very potent and selective in binding studies (1000-fold selective for α4β2 over α3β4 or α7 nAChR), in vitro efficacy studies have determined that it is in fact a relatively low efficacy partial agonist at α4β2 nAChR, with EC50 in the 1 μM range. Surprisingly, varenicline has somewhat similar potency and full agonist activity at α3β4 and α7 nAChR.24 However, varenicline potently desensitizes the receptor and thereby acts as a functional antagonist at nanomolar concentrations in vitro.25,26 It may be desensitization combined with partial agonist activity27,28 that leads to the full complement of varenicline’s activities in vivo. Other investigators have suggested conversely that the α3β4 or α7 activity might be the component that reduces nicotine consumption.24,25 In fact varenicline blocks self-administration of both nicotine and alcohol, and it has been suggested that the α3β4 nAChR activity mediates the reduction in consumption of both drugs.25,29 Our previous work suggests otherwise, as we have demonstrated that the selective α3β4 nAChR partial agonist N-(2-bromophenyl)-9-methyl-9-azabicyclo[3.3.1]-nonan-3-amine (AT-1001) can block nicotine self-administration at doses that do not affect alcohol self-administration.26,30 Furthermore, activation of α4-containing nAChRs has been shown to be necessary and sufficient for varenicline to reduce alcohol consumption.31 Varenicline as a smoking cessation medication has also been plagued by reports of neuropsychiatric side effects.7,32 Although the black box warning has been removed by the FDA, labeling still states postmarketing studies have reported serious or clinically significant neuropsychiatric adverse events in patients treated with the drug. Finally, although better than placebo and other medications in double blind studies, a recent open label study for smoking cessation has found varenicline to be equivalent to nicotine replacement therapy at 6 month and year time points.33 Ultimately it is not clear whether the side effects and moderate efficacy are due to the fact that varenicline has partial agonist activity at α4β2 nAChR or that it has agonist activity at other nAChR such as α3β4 and α7 nAChR.
Many additional nAChR active agents have been tested in animal models and clinical trials. Cytisine, a nAChR partial agonist and a close analog of varenicline, is self-administered in mice34 and has shown some efficacy in a limited number of clinical trials for smoking cessation.35 The partial agonist sazetidine-A can also attenuate nicotine self-administration as well as alcohol self-administration in alcohol preferring rats.36,37 Positive allosteric modulators of α4β2 nAChRs attenuate nicotine taking and seeking.38,39 The classical nonselective nAChR antagonist mecamylamine has also shown efficacy in blocking nicotine self-administration14 and some efficacy in clinical trials when combined with a nicotine patch.40,41 The selective α4β2 nAChR antagonist DHβE has also been demonstrated to attenuate nicotine self-administration in rats.14 However, this compound, although very selective, has only moderate affinity and therefore rather poor potency for inhibition of the receptor in vitro.42 Over the past several years, Carroll and colleagues have identified a number of epibatidine analogs with very high affinity and selective α4β2 nAChR agonist and antagonist activity.43–45 One antagonist in particular, 4-nitro-PFEB, has near picomolar binding affinity to the α4β2 nAChR and inhibits receptor activity in vitro in the nanomolar range,46 although more recent studies have suggested partial agonist activity for this compound as well.47 4-Nitro-PFEB has also been demonstrated to block nicotine self-administration in rats and nicotine conditioned place preference in mice13
5 appears to have properties different from the compounds discussed above. 5 was not developed based upon known nicotinic structures but rather by traditional medicinal chemistry starting, a priori, from very large, small molecule, mixture libraries.21 By utilization of the hits that were identified in the original screening campaign as starting points, a set of truncated analogs were synthesized and screened in order to identify the critical structural features driving potency and selectivity. This led to the monocyclic guanidine hits. A further medicinal chemistry effort was then undertaken to assess the potency and selectivity changes observed from a range of different R1 and R2 functionalities around the core monocyclic guanidine. 5 and its analog 13 have high affinity and selectivity in binding to the α4β2 nAChR as compared to α4β4, α3β2, and α3β4α5 nAChR. Affinity and selectivity are considerably higher than the “prototypical” α4β2 nAChR antagonist DHyβE, though not as high as the epibatidine analogs. These compounds have no apparent agonist activity in cells transfected with α4β2 nAChR and potently inhibit receptor activity in vitro at concentration equivalent to their binding affinities. They have moderate activity at α3β4 nAChR at very high concentrations and no apparent effect on α7 nAChR. Lack of agonist activity in vitro is consistent with the inability of 5 to induce hypothermia.
When used in a preclinical model to determine potential efficacy as a smoking cessation medication, 5 potently (1 mg/ kg) attenuated nicotine self-administration. However, the duration of action was short, reducing nicotine self-administration only in the first 30 min of the self-administration session. This is consistent with the time course of blood and brain concentration after systemic administration. However, 5 was more effective in reducing nicotine seeking in a model of relapse, completely inhibiting both nicotine prime-induced and cue-induced reinstatement of nicotine seeking.
CONCLUSION
We have identified a novel high affinity and selective α4β2 nAChR antagonist (5) that was originally derived from a small molecule mixture combinatorial library. 5 and analog 13 have low nanomolar affinity for α4β2 nAChR. In vitro these compounds are devoid of agonist activity in cells transfected with rat α4β2 nAChR, act as antagonists of epibatidine at nanomolar concentrations, and have very weak partial agonist activity at α3β4 nAChR. In vivo 5 is also devoid of agonist activity and potently inhibits nicotine self-administration, as well as reinstatement of nicotine seeking. These results demonstrate that compounds like this could both reduce smoking and block relapse, suggesting that 5, with appropriate formulation to increase duration of action, has potential as a smoking cessation medication.
EXPERIMENTAL SECTION
Chemistry.
The compounds were synthesized as described in the Scheme 1. The synthetic approach utilized was modified from a previously reported approcah for obtaining cyclic guanidines from resin bound polyamines.21‘48 The solid phase synthesis was performed using the “tea-bag” methodology.49 Initially, 100 mg of p-methylbenz-hydrylamine (MBHA) resin (1.1 mmol/g, 100–200 mesh) was sealed in a mesh “tea bag”, neutralized with 5% diisopropylethylamine (DIEA) in dichloromethane (DCM) and subsequently swelled with additional DCM washes. A Boc-amino acid (6 equiv) was coupled in dimethylformamide (0.1 M DMF) for 120 min in the presence of diisopropylcarbodiimide (DIC, 6 equiv) and 1-hydroxybenzotriazole hydrate (HOBt, 6 equiv) (1, Scheme 1). The Boc protecting group was removed with 55% trifluoroacetic acid (TFA)/DCM for 30 min and subsequently neutralized with 5% DIEA/DCM (3×). Carboxylic acids were coupled using 6 equiv in the presence of DIC (10 equiv) and HOBt (10 equiv) in DMF (0.1M) for 120 min (2, Scheme 1). All coupling reactions were monitored for completion by the ninhydrin test. The reduction was performed in a 4000 mL Wilmad LabGlass vessel under nitrogen. Borane in 1.0 M tetrahydrofuran complex solution was used in 40-fold excess for each amide bond. The vessel was heated to 65 °C and maintained at temperature for 72 h. The solution was then discarded, and the bags were washed with THF and methanol. Once completely dry, the bags were treated overnight with piperidine at 65 °C and washed several times with methanol, DMF, and DCM (3, Scheme 1). As previously reported, the reduction of polyamides with borane is free of racemization.50,51 Before proceeding, completion of reduction was monitored by a control cleavage and analyzed by LCMS. Guanidine cyclization (d, Scheme 1) was performed with a 5-fold excess of cyanogen bromide (CNBr) in a 0.1 M anhydrous DCM solution. Following the cyclization, the bags were rinsed with DMF and DCM. The resin was cleaved with HF in the presence of anisole in an ice bath at 0 °C for 90 min (5, Scheme 1). The products were extracted using 95% acetic acid. Samples were then repeatedly frozen and lyophilized in 50% acetonitrile and water. Confirmation of the desired product was obtained by reverse phase LC−MS analysis using a Shimadzu 2010 LCMS system, consisting of a LC-20AD binary solvent pump, a DGU-20A degasser unit, a CTO- 20A column oven, SIL-20A HT autosampler, and SPD-M20A diode array set to scan 190–600 nm. Separation was achieved using a Phenomenex Luna C18 column (5 μm, 50 mm × 4.6 mm i.d.) protecting with a Phenomenex C18 column guard (5 μm, 4 mm × 3.0 mm i.d.).
Scheme 1. General Synthetic Scheme Used for the Synthesis of Core A Type Compoundsa.
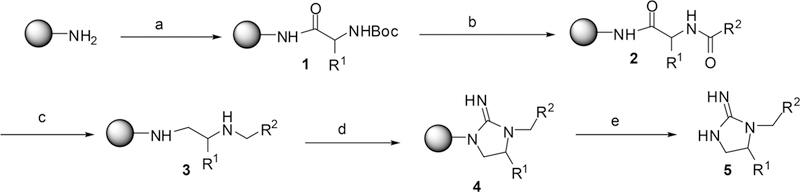
aStandard Boc coupling protocol utilizing repetitive (a1) 5% DIEA/95% DCM; (a2) Boc-AA, DIC, HOBt, DMF; 1. (b1) 55% TFA/45% DCM; (b2) 5% DIEA/95% DCM; COOH, DIC, HOBt, DMF; 2. (c) 40× BH3/THF (65 °C, 72 h); piperidine (65 °C, 18 h); 3. (d) 5× BrCN/0.1 M DCM (shaking12 h); 4. (e) HF/anisole, (0 °C, 7 hr); 5.
The crude product was purified using reverse phase mode on a Shimadzu Prominence preparative HPLC system consisting of LC-8A binary solvent pumps, a SCL-10A system controller, a SIL-10AP autosampler, a FRC-10A fraction collector, and a Shimadzu SPD-20A UV detector. The wavelength was set at 214 nm during analysis. Chromatographic separations were obtained using a Phenomenex Luna C18 preparative column (5 μm, 150 mm × 21.5 mm i.d.). The column was protected by a Phenomenex C18 column guard (5 μm, 15 mm × 21.2 mm i.d.). Prominence prep software was used to set all detection and collection parameters. The mobile phases for HPLC purification were HPLC grade obtained from Sigma-Aldrich and Fisher Scientific. The mobile phase A consisted of water with 0.1% TFA, and mobile phase B consisted of acetonitrile with 0.1% trifluoroacetic acid. Initial setting was set to 2% mobile phase B and was gradually increased over time to achieve ideal separation for each compound. The peak corresponding to calculated m/z of the desired product was collected and concentrated. Most of compounds exhibited a gooey (soft and sticky) status except compound 8 (white, solid powder). The concentrated pure product was analyzed on LC-MS and determined to be ≥95% purity based on peak area.
The 1H and 13C NMR spectra were obtained utilizing the Bruker 400 Ascend (400 and 100 MHz, respectively). 1H NMR chemical shifts were reported in δ (ppm) using the δ 7.26 signal of CHCl3 the δ 4.79 signal of D2O, and the δ 2.50 signal of DMSO-d6 as internal standards. 13C NMR chemical shifts were reported in δ (ppm) using the δ 77.16 signal of CDCl3 the δ 39.52 signal of DMSO-d6 as internal standards. Melting points were measured using Stuart’s SMP40 melting point apparatus (Staffordshire, U.K.), and specfic optical rotations were measured using Autopol IV automatic polarimeter (Rudolph Research Analytical, NJ, USA).
1-(3,4-Dichlorophenethyl)imidazolidin-2-imme (1) was synthesized with Boc-glycine for the R1 reagent used in step a and 3,4-dichlorophenylacetic acid used for the R2 reagent in step b. 1H NMR (400 MHz, DMSO-d6): δ 10.1 (s, 1H), 8.58 (s, 1H), 7.37 (d, J = 4.0 Hz, 1H), 7.36 (d, J = 2.0 Hz, 1H), 7.11 (dd, J = 8.4, 2.0 Hz, 1H), 3.61–3.56 (m, 4H), 3.49–3.45 (m, 2H), 2.89–2.95(m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 159.33, 139.49, 130.97, 130.87, 130.41, 129.40, 129.07, 47.04, 44.46, 40.34, 31.43. LC-MS (ESI+) (m/z): [M + H]+ calcd for C11H14Cl2N3 258.1; found, 257.9 and 259.9.
1-(Pyridin-4-ylmethyl)imidazolidin-2-imine (2) was synthesized with Boc-L-glycine for the R1 reagent used in step a and isonicotinic acid used for the R2 reagent in step b. 1H NMR (400 MHz, CDCl3): δ 8.60 (dd, J = 4.4, 1.6 Hz, 2H), 8.45 (s, 1H), 7.19 (d, J = 6.0 Hz, 2H), 4.59 (s, 2H), 3.67–3.60 (m, 2H), 3.52–3.48 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 160.34, 150.63, 143.75, 122.56, 47.82, 47.74, 41.20. LC-MS (ESI+) (m/z): [M + H]+ calcd for C9H13N4 177.1; found, 177.0.
1-Phenethylimidazolidin-2-imine (3) was synthesized with Boc- glycine for the R1 reagent used in step a and phenylacetic acid used for the R2 reagent in step b. 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.32 (t, J = 7.2 Hz, 2H), 7.25–7.22 (m, 2H), 3.57–3.54 (m, 4h), 3.42 (t, J = 8.0 Hz, 2H), 2.91 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 159.93, 137.76, 129.08, 128.81, 127.22, 48.38, 46.46, 41.06, 31.05. LC-MS (ESI+) (m/z): [M + H] + calcd for C11H16N3190.1; found,190.0.
(S)-5-Benzyl-1-(2-(pyridin-3-yl)ethyl)imidazolidin-2-imine (4) was synthesized with Boc-L-phenylalanine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 26.6 (0.5, DMSO). 1H NMR (400 MHz, CDCl3): δ 8.49 (s, 1H), 8.41 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.35–7.31 (m, 2H), 7.30–7.28 (m, 1H), 7.26–7.23 (m, 1H), 7.11–7.09 (m, 2H), 3.91–3.81 (m, 2H), 3.49 (t, J = 9.2 Hz, 1H), 3.34–3.28 (m, 2H), 3.01 (dd, J = 14.0, 5.6 Hz, 1H), 2.93–2.85 (m, 2H), 2.69 (q, J = 8.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 159.45, 150.00, 148.50, 136.72, 135.58, 133.27, 129.27, 129.07, 127.59, 123.89, 60.36, 46.55, 43.71, 38.83, 30.92. LC-MS (ESI+) (m/z): [M + H]+ calcd for C17H21N4 281.1; found, 281.0.
(S)-5-Methyl-1-(2-(pyridin-3-yl)ethyl)imidazolidin-2-imine (5) was synthesized with Boc-L-alanine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20 D 26.8 (0.25, 50% DMSO). 1H NMR (400 MHz, CDCl3): 8 8.49 (d, J = 6.0 Hz, 2H), 8.40 (s, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.28–7.25 (m, 1H), 3.89–3.81 (m, 2H), 3.71 (t, J = 9.2 Hz, 1H), 3.46–3.39 (m, 1H), 3.21 (q, J = 7.2 Hz, 1H), 2.98–2.85 (m, 2H), 1.25 (d, J = 6.0 Hz, 3h). 13C NMR (100 MHz, D2O) δ 158.12, 146.54, 144.99, 141.12, 135.73, 125.41, 55.30, 47.86, 42.25, 29.84, 16.93. LC-MS (ESI+) (m/z): [M + H]+ calcd for C11H17N4 205.1; found, 205.0.
(S)-5-Isobutyl-1-(2-(pyridin-3-yl)ethyl)imidazolidin-2-imine (6) was synthesized with Boc-L-leucine isoleucine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 24.6 (0.5, 50% DMSO). 1H NMR (400 MHz, CDCl3): δ 8.57 (s, 1H), 8.49 (s, 2H), 7.64 (d, J = 8.0 Hz, 1H), 7.27–7.23 (m, 1H), 3.83–3.76 (m, 1H), 3.66–3.60 (m, 2H), 3.42–3.34 (m, 1H), 3.24–3.18 (m, 1H), 2.96–2.83 (m, 2H), 1.54–1.50 (m, 1H), 1.45 (td, J =12.4, 3.5 Hz, 1H), 1.37–1.30 (m, 1H), 0.92 (d, J = 6.4 Hz, 3H), 0.83 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.46, 150.11, 148.52, 136.65, 133.43, 123.83, 57.96, 47.12, 43.15, 41.11, 30.99, 24.74, 23.78. LC-MS (ESI+) (m/z): [M + H]+ calcd for C14H23N4 247.1; found, 247.0.
(s)-5-((R)-sec-Butyl)-1-(2-(pyridin-3-yl)ethyl)imidazolidin-2- imine (7) was synthesized with Boc-L-isoleucine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 26.8 (0.5, 50% DMSO). 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 1H), 8.49 (d, J = 9.6 Hz, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.27–7.24 (m, 1H), 4.01–3.97 (m, 1H), 3.76–3.73 (m, 1H), 3.47 (t, J = 9.6 Hz, 1H), 3.66–3.25 (m, 2H), 2.92 (q, J = 6.4 Hz, 2H), 1.80–1.74 (m, 1H), 1.22–1.13 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 159.36, 150.06, 148.48, 136.83, 133.27, 123.87, 62.13, 42.93, 41.45, 34.39, 31.03, 30.57, 25.63, 12.00. LC-MS (ESI+) (m/z): [M + H] + calcd for C14H23N4 247.1; found, 247.0.
1-(2-(Pyridin-3-yl)ethyl)imidazolidin-2-imine (8) was synthesized with Boc-glycine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. Melting point: 197.1–197.6 °C. 1H NMR (400 MHz, CDCl3): δ 8.52 (d, J = 2.0 Hz, 1H), 8.45 (dd, J = 4.8, 1.6 Hz, 1H), 8.40 (s, 1H), 7.74 (dt, J = 8.0, 1.6 Hz, 1H), 7.34 (q, J = 4.8 Hz, 1H), 3.63–3.58 (m, 2H), 3.55–3.51 (m, 2H), 3.49–3.46 (m, 2H), 2.86 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 159.08, 150.04, 147.77, 136.40, 133.68, 123.42, 46.98, 44.54, 30.67, 29.56. LC-MS (eSI+) (m/z): [M + H]+ calcd for C10H15N4 191.1; found,191.0.
(S)-5-Isopropyl-1-(2-(pyridin-3-yl)ethyl)imidazolidin-2-imine (9) was synthesized with Boc-L-valine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 18.0 (1.0, DMSO). 1H NMR (400 MHz, DMSO-d6): δ 8.54 (d, J =1.6 Hz, 1H), 8.45 (dd, J = 4.8, 2.0 Hz, 1H), 8.41 (s, 1H), 7.77 (dt, J = 7.6, 2.0 Hz, 1H), 7.34 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 3.94–3.89 (m, 1H), 3.75–3.68 (m, 1H), 3.44 (t, J = 6.0 Hz, 1H), 3.31–3.30 (m, 1h), 3.30–3.27 (m, 1h), 2.95–2.88 (m, 1H), 2.81–2.74 (m, 1H), 2.18–2.13 (m, 1H), 0.85 (d, J = 7.2 Hz, 3H), 0.75 (d, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 158.84, 150.11, 147.75, 136.46, 133.60, 123.35, 61.65, 41.87, 30.66, 29.39, 26.76, 17.56. LC-MS (ESI+) (m/z): [M + H] + calcd for C13H21N4 233.2; found, 233.0.
(S)-1-(3-(2-Imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)- propyl)guanidine (10) was synthesized with Boc-L-arginine isoleucine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 68.8 (0.25, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.51 (s, 2H), 7.92 (d, J = 7.6 Hz, 1h), 7.56–7.53 (m, 1H), 4.01–3.95 (m, 1H), 3.75–3.68 (m, 2H), 3.66–3.59 (m, 1H), 3.38–3.34 (m, 1H), 3.22 (t, J = 6.4 Hz, 2H), 3.05 (t, J = 6.4 Hz, 2h), 1.73–1.68 (m, 2H), 1.56–1.50 (m, 2H). 13C NMR (100 MHz, D2o) S 158.22, 156.78, 148.14, 146.49, 139.08, 134.95, 124.73, 58.49, 45.54, 42.70, 40.77, 29.84, 27.92, 22.59. LC-MS (ESI+) (m/z): [M + H]+ calcd for C14H24N7 290.2; found, 290.
(S)-4-((2-Imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)- methyl)phenol (11) was synthesized with Boc-L-tyrosine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 14.4 (0.5, DMSO). 1H NMR (400 MHz, CDCl3): S 8.51 (d, J = 2.4 Hz, 1H), 8.44 (dd, J = 4.8, 1.6 Hz, 1H), 8.41 (s, 1H), 7.72 (dt, J = 8.0, 2.0 Hz, 1H), 7.34 (ddd, J = 7.6, 4.8, 0.8 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 4.12–4.06 (m, 1H), 3.74–3.67 (m, 1h), 3.45–3.42 (m, 1H), 3.41–3.38 (m, 1H), 3.21 (q, J = 6.0 Hz,1H), 2.96 (d, J = 13.6, 4.4 Hz, 1H), 2.89 (q, J = 5.2 Hz, 1H), 2.82–2.75 (m, 1H), 2.61 (q, J = 8.4 Hz, 1h). 13C NMR (100 MHz, DMSO-d6) δ 158.52, 156.26, 150.13, 147.75, 136.50, 133.60, 130.23, 126.09, 123.35, 115.30, 58.77, 45.17, 42.30, 36.26, 30.66. LC-MS (ESI+) (m/z): [M + H]+ calcd for C17H21N4O 297.1; found, 297.0.
(S)-5-(2-(Methylthio)ethyl)-1-(2-(pyridin-3-yl)ethyl)- imidazolidin-2-imine (12) was synthesized with Boc-L-methionine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 13.6 (0.25, 50% DMSO). 1H NMR (400 MHz, CDCl3): δ 8.53 (d, J = 2.0 Hz, 1H), 8.45 (dd, J = 4.8, 1.6 Hz, 1H), 8.41 (s, 1H), 7.75 (dt, J = 8.0, 2.0 Hz, 1H), 7.34 (ddd, J = 7.6, 4.8, 0.8 Hz, 1H), 3.99–3.93 (m, 1H), 3.70–3.64 (m, 1H), 3.59 (t, J = 9.6 Hz, 1H), 3.28 (q, J = 6.8 Hz, 1H), 3.20 (q, J = 6.8 Hz, 1H), 2.94–2.87 (m, 1H), 2.81–2.73 (m, 1H), 2.44 (t, J = 8.0 Hz, 2H), 2.17 (s, 3H), 2.00–1.94 (m, 1H), 1.76–1.71 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 158.76, 150.09, 147.77, 136.46, 133.58, 123.38, 57.03, 45.44, 42.01, 30.65, 30.61, 29.63, 27.94. LC-MS (ESI+) (m/z): [M + H]+ calcd for CUH21N4S 265.1; found, 265.0.
(R)-(2-Imino-3-(2-(pyridin-3-yl)ethyl)imidazolidin-4-yl)-methanol (13) was synthesized with Boc-L-serine for the R1 reagent used in step a and 3-pyridinylacetic acid for the R2 reagent used in step b. [α]20D 28.4 (0.25, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.59 (s, 1H), 8.49 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.69–7.66 (m, 1H), 4.12–4.08 (m, 1H), 3.90 (dd, J = 12.8, 3.2 Hz, 1H), 3.80–3.73 (m, 2H), 3.71–3.66 (m, 2H), 3.54 (dd, J = 10.0, 6.0 Hz, 1H), 3.13–3.10 (m, 2H). 13C NMR (100 MHz, D2O) δ 158.44, 146.43, 144.93, 141.04, 135.65, 125.34, 59.81, 59.52,43.04,42.72, 29.66. LC-MS (ESI +) (m/z): [M + H]+ calcd for C11H17N4O 221.1; found, 221.0.
(S)-5-Methyl-1-(pyridin-3-ylmethyl)imidazolidin-2-imine (14) was synthesized with Boc-L-alanine for the R1 reagent used in step a and nicotinic acid used for the R2 reagent in step b. [α]20D −2.8 (0.25, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.55–8.41 (m, 2H), 7.86 (d, J = 8.0 Hz, 1H), 7.50 (dd, J = 7.6, 5.2 Hz, 1h), 4.65–4.53 (m, 2H), 4.03–3.98 (m, 1H), 3.77 (t, J = 9.6 Hz, 1H), 3.29–3.25 (m, 1H), 1.22 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, D2O) δ 158.60, 147.67, 146.72, 137.31, 131.97, 124.78, 55.82, 48.00, 43.30, 17.13. MS (ESi) m/z [M + H]+: 191. LC-MS (ESI+) (m/z): [M + H]+ calcd for C10H15N4 191.1; found, 191.0.
1-(Pyridin-3-ylmethyl)imidazolidin-2-imine (15) was synthesized with Boc-glycine for the R1 reagent used in step a and nicotinic acid used for the R2 reagent in step b. 1H NMR (400 MHz, CDCl3): δ 8.60 (dd, J = 4.4, 1.6 Hz, 2H), 8.45 (s, 1H), 7.19 (d, J = 6.0 Hz, 2H), 4.59 (s, 2H), 3.67–3.60 (m, 2H), 3.52–3.48 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 160.34, 150.63, 143.75, 122.56, 47.82, 47.74, 41.20. LC-MS (ESI+) (m/z): [M + H]+ calcd for C9H13N4 177.1; found,177.0
(S)-5-Methyl-1-phenethylimidazolidin-2-imine (16) was synthesized with Boc-L-alanine for the R1 reagent used in step a and phenylacetic acid used for the R2 reagent in step b. LC-MS (ESI+) (m/z): [M + H]+ calcd for C12H18N3 204.1; found, 204.0. 16 has a very weak binding affinity and was not further purified for additional chemical characteraization.
(R)-(3-(3,4-Dichlorophenethyl)-2-iminoimidazolidin-4-yl)- methanol (17) was synthesized with Boc-L-serine serine for the R1 reagent used in step a and 3,4-dichlorophenylacetic acid used for the R2 reagent in step b. [α]20D 26.4 (0.25, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.52 (s, 1H), 7.54 (d, J = 8.0 Hz,1H), 7.52 (d, J = 2.4 Hz, 1H), 4.01–3.96 (m, 1H), 3.86 (dd, J = 12.8, 3.6 Hz, 1h), 3.74–3.69 (m, 2H), 3.67–3.64 (m, 1H), 3.63–3.57 (m, 1H), 3.51 (q, J = 6.0 Hz, 1H), 2.96 (td, J = 6.0, 2.0 Hz, 2H). 13C NMR (100 MHz, D2O) δ 158.40, 138.72, 131.76, 130.80, 130.64, 130.18, 128.84, 59.74, 59.49,43.01, 43.00, 31.58. LC-MS (ESI+) (m/z): [M + H]+ calcd for C12H16Cl2N3O 288.1; found, 287.8 and 289.9.
1-(3-Fluorophenethyl)imidazolidin-2-imine (18) was synthesized with Boc-glycine for the R1 reagent used in step a and 3-fluorophenylacetic acid for the R2 reagent used in step b. 1H NMR (400 MHz, DMSO-d6): δ 9.41 (s, 1H), 8.06 (s, 1H), 7.32–7.28 (m, 1H), 7.01 (d, J = 7.6 Hz, 1H), 6.95 (d, J = 9.2 Hz, 2h), 3.61 (t, J = 6.8 Hz, 2H), 3.57–3.55 (m, 2h), 3.45–3.41 (m, 2h), 2.91 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 159.96, 140.15, 131.87, 130.50, 130.19, 127.58, 122.85, 48.30, 45.91, 41.01, 33.44. LC-MS (ESI+) (m/z): [M + H]+ calcd for C11H15FN3 208.1; found, 208.0.
(S)-1-(3-Fluorophenethyl)-5-methylimidazolidin-2-imine (19) with Boc-L-alanine for the R1 reagent used in step a and 3-fluorophenylacetic acid used for the R2 reagent in step b. [α]20D 13.6 (0.5, 50% DMSO). 1H NMR (400 MHz, DMSO-d6): δ 8.50 (s, 1H), 7.45–7.39 (m, 1H), 7.01 (d, J = 7.6 Hz, 1H), 7.17 (d, J = 8.0 Hz, 1h), 7.14–7.07 (m, 2H), 4.02–3.95 (m, 1H), 3.71 (t, J = 9.6 Hz, 1h), 3.62–3.55 (m, 2H), 3.22 (q, J = 6.8 Hz, 1H), 2.99–2.95 (m, 2H), 1.27 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, D2O) δ 162.79 (d, J = 242 Hz), 157.96, 140.95 (d, J = 7 Hz), 130.49 (d, J = 9 Hz), 124.91 (d, J = 3 Hz), 115.67 (d, J =21 Hz), 113.62 (d, J = 21 Hz), 55.13, 47.77, 42.63, 32.25, 16.80. MS (ESi) m/z [M + H]+: 222. LC-MS (ESI+) (m/z): [M + H]+ calcd for C12H17FN3 222.1; found, 222.0.
(R)-(3-(3-Fluorophenethyl)-2-iminoimidazolidin-4-yl)methanol (20) was synthesized with Boc-L-serine for the R1 reagent used in step a and 3-fluorophenylacetic acid used for the R2 reagent in step b. [α]20D 3.4 (0.5, DMSO). 1H NMR (400 MHz, CDCl3): δ 8.50 (s, 1H), 7.43 (q, J = 7.6 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 7.16–7.09 (m, 2h), 4.01–3.95 (m, 1H), 3.88 (dd, J = 12.8, 3.6 Hz,1H), 3.78–3.73 (m, 1H), 3.70 (d, J = 10.0 Hz, 1H), 3.67–3.65 (m, 1h), 3.64–3.60 (m, 1H), 3.51 (q, J = 6.0 Hz, 1H), 3.03–2.99 (m, 2H). 13C NMR (100 MHz, D2O) δ 162.5 (J = 242.1 Hz), 158.44, 140.77 (J = 7.4 Hz), 130.55 (J =8.3 Hz), 124.87 (J = 2.7 Hz), 115.64 (J = 21.2 Hz), 113.69 (J = 20.8 Hz), 59.70, 59.51, 43.16, 43.00, 32.13 (J = 1.3 Hz). MS (ESI) m/z [M + H]+: 238. LC-MS (ESI+) (m/z): [M + H] + calcd for C12H17FN3O 238.1; found, 238.0.
1-(3-Bromophenethyl)imidazolidin-2-imine (21) was synthesized with Boc-glycine for the R1 reagent used in step a and 3-bromophenylacetic acid used for the R2 reagent in step b. 1H NMR (400 MHz, CDCl3): δ 8.59 (s, 1H), 7.39 (s, 1H), 7.36 (dt, J = 6.8, 2.0 Hz, 1H), 7.18–7.14 (m, 2H), 3.58 (t, J = 7.2 Hz, 2H), 3.53 (d, J = 8.8 Hz, 2H), 3.41 (t, J = 8.4 Hz, 2H), 2.86 (t, J = 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 159.95, 140.15, 131.87, 130.50, 130.18, 127.58, 122.85, 48.30, 45.90, 41.01, 33.44. LC-MS (ESI+) (m/z): [M + H]+ calcd for C11H15BrN3 268.1; found, 267.9 and 269.9.
(S)-1-(3-Bromophenethyl)-5-methylimidazolidin-2-imine (22) was synthesized with Boc-L-alanine for the R1 reagent used in step a and 3-bromophenylacetic acid used for the R2 reagent in step b. [α]20D 19.3 (1.0, DMSO). 1H NMR (400 MHz, CDCl3):δ 8.50 (s, 1H), 7.55 (s, 1H), 7.53–7.52 (m, 1H), 7.33 (d, J = 4.8 Hz, 2H), 4.01–3.95 (m, 1H), 3.72 (t, J = 9.6 Hz, 1h), 3.66–3.52 (m, 2H), 3.21 (q, J = 7.6 Hz, 1H), 2.94 (t, J = 6.4 Hz, 2H), 1.26 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, D2O) δ 157.94, 140.80, 131.87, 130.58, 129.83, 127.92, 122.03, 55.12, 47.76, 42.64, 32.16, 16.82. LC-MS (ESI+) (m/z): [M + H]+ calcd for C12H17BrN3 282.1; found, 28.8 and 283.9.
(R)-(3-(3-Bromophenethyl)-2-iminoimidazolidin-4-yl)methanol (23) was synthesized with Boc-L-serine for the R1 reagent used in step a and 3-bromophenylacetic acid used for the R2 reagent in step b.[α]20D 7.4 (0.5, DMSO). 1H NMR (400 MHz, CDCl3): δ 8.53 (s, 1H), 7.56 (s, 1H), 7.54–7.3 (m, 1H), 7.34 (s, 1H), 7.31 (d, J = 0.8 Hz, 1H), 3.99–3.94 (m, 1H), 3.86 (ddd, J = 12.4, 3.2, 0.8 Hz, 1H), 3.74–3.72 (m, 1H), 3.70–3.69 (m, 1H), 3.65–3.58 (m, 2H), 3.51 (ddd, J = 9.6, 5.6, 0.8 Hz, 1H), 3.00–2.96 (m, 2H). 13C NMR (100 MHz, D2O) δ 158.42, 140.62, 131.85, 130.63, 129.90, 127.88, 122.08, 59.74, 59.48, 43.18, 43.00, 32.03. LC-MS (ESI+) (m/z): [M + H]+ calcd for C12H17BrN3O 298.1; found, 297.8 and 299.7.
(S)-1-(2-(6-Chloropyridin-3-yl)ethyl)-5-methylimidazolidin-2-imine (24) was synthesized with Boc-L-alanine for the R1 reagent used in step a and 2-(6-chloropyridin-3-yl)acetic acid used for the R2 reagent in step b. [α]20D 20.4 (0.25, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.28 (d, J = 2.4 Hz, 1H), 7.82 (dd, J = 8.0, 2.4 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 4.07–4.02 (m, 1H), 3.74 (t, J = 9.6 Hz, 1h), 3.64–3.60 (m, 2h), 3.24 (q, J = 7.2 Hz, 1h), 3.00 (q, J = 6.8 Hz, 2h), 1.27 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, D2O) 8 158.10, 149.36, 149.04, 141.04, 133.65, 124.80, 55.26, 47.84, 42.30, 29.17, 16.92. LC-MS (ESI+) (m/z): [M + H]+ calcd for C11H16ClN4 239.1; found,239.0.
1-(2-(6-Chloropyridin-3-yl)ethyl)imidazolidin-2-imine (25) was synthesized with Boc-L-glycine tyrosine for the R1 reagent used in step a and 2-(6-chloropyridin-3-yl)acetic acid for the R2 reagent used in step b. 1H NMR (400 MHz, D2O): δ 8.29 (d, J = 2.4 Hz, 1H), 7.83 (dd, J = 8.4, 2.4 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 3.74–3.69 (m, 2H), 3.64–3.63 (m, 2H), 3.62–3.60 (m, 2H), 3.02 (q, J = 6.8 Hz, 2H). 13C NMR (100 MHz, D2O) δ 158.70, 149.30, 149.04, 140.93, 133.65, 124.80, 47.69, 45.00, 40.63, 29.03. LC-MS (ESI+) (m/z): [M + H]+ calcd for C10H14ClN4 225.1; found,225.0.
(R)-(3-(2-(6-Chloropyridin-3-yl)ethyl)-2-iminoimidazolidin-4-yl)methanol (26) was synthesized with Boc-L-serine for the R1 reagent used in step a and 2-(6-chloropyridin-3-yl)acetic acid used for the R2 reagent in step b. [α]20D 39.52 (0.17, 50% DMSO). 1H NMR (400 MHz, D2O): δ 8.31 (s, 1H), 7.85 (dd, J = 8.4, 2.4 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 4.10–4.06 (m, 1H), 3.90 (dd, J = 12.4, 3.2 Hz, 1H), 3.78–3.73 (m, 2H), 3.71–3.67 (m, 2H), 3.54 (dd, J = 10.0, 5.6 Hz, 1H), 3.05 (t, J = 6.4 Hz, 2H). 13C NMR (100 MHz, D2O) δ 158.43, 149.24, 149.04, 140.93, 133.40, 124.76, 59.76, 59.42, 42.99, 42.71, 28.96. LC-MS (ESI+) (m/z): [M + H]+ calcd for CuH16C11N4O 255.1; found, 255.0.
Cell Culture.
HEK cells, stably expressing rat α3β4, α4β4, α4β2, and α3β2 nAChR (obtained from Drs. Kenneth Kellar and Yingxian Xiao, Georgetown University) and α3β4α5 nAChR (obtained from Dr. Jon Martin Lindstrom) were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 0.5% penicillin/streptomycin, and 0.4 mg/mL of Geneticin and were maintained in an atmosphere of 7.5% CO2 in a humidified incubator at 37 °C. For binding assays, cells were passaged on 150 mm dishes and harvested when confluent.
Binding Assays.
Cells were harvested by scraping the plates with a rubber policeman, suspended in 50 mM Tris buffer, pH 7.4, homogenized using a Polytron homogenizer, and the centrifugation was repeated twice at 20 000g (13 500 rpm) for 20 min. For binding, the cell membranes were incubated with the test compounds or mixtures in the presence of 0.3 nM [3H]epibatidine. After 2 h of incubation, at room temperature, samples were filtered, using a Tomtec cell harvester, through glass fiber filters that had been presoaked in 0.05% polyethylenimine. Filters were counted on a betaplate reader (Wallac). Nonspecific binding was determined by using 0.1 μM unlabeled epibatidine for α3β4 and α4β2 nAChR, respectively. IC50 values were determined by using the program GraphPad Prism. Ki values were calculated using the Cheng-Prusoff transformation: Ki= IC50/(1 + L/Kd), where L is radioligand concentration and Kd is the binding affinity of the radioligand, as determined previously by saturation analysis.
nAChR Functional Assays.
nAChR functional activity was determined by measuring nAChR-induced membrane potential change, which can be directly read by Molecular Devices membrane potential assay kit (Blue Dye) (Molecular Devices, Sunnyvale, CA) using the FlexStation 3 microplate reader (Molecular Devices, Sunnyvale, CA). The HEK cells with stably expressing α3β4 or α4β2 nAChR were seeded in a 96-well plate (4000 cells per well) 1 day prior to the experiments. For agonist assays, after brief washing, the cells were loaded with 225 μL of HBSS assay buffer (Hank’s balanced salt solution with 20 mM of HEPES, pH 7.4), containing the blue dye, and incubated at 37 °C. After 30 min, 25 μL of the appropriate compounds were dispensed into the wells by the FlexStation and nAChR stimulation-mediated membrane potential change was recorded every 3 s for 120 s by reading 565 nm fluorescence excited at 530 nm wavelength. For the antagonist assay, the cells were loaded with 200 μL HBSS buffer containing the blue dye and incubated at 37 °C. After 20 min, 25 μL of test compounds was added, and after another 10 min, 25 μL of epibatidine (or nicotine) was added by the FlexStation, to a final concentration of 100 nM, with fluorescence measured as described above. The change in fluorescence represents the maximum response minus the minimum response for each well. GraphPad Prism was used to determine the EC50 and IC50 values.
Animals.
Male Sprague Dawley rats obtained from Charles River (Portage, MI) and weighing 200–225 g at their arrival were used in this study. Rats were housed in groups of two in a room with a reverse 12 h light/12 h dark cycle (lights off at 07:30 a.m.). All behavioral experiments were conducted during the dark phase of the cycle. Animals were acclimatized for 7 days with water and chow (Teklad Diets, Madison, WI) and handled 3 times before the experiments were started. All animal experiments performed in this manuscript were conducted in compliance with U.S. Institutional Animal Care and Use Committee (IACUC)
Drugs and Chemicals.
5 was dissolved in a vehicle of 0.9% saline and injected subcutaneously at doses of (0.0, 0.3, 1.0 mg/kg). The injection volume was 1 mL/kg. Nicotine hydrogen tartrate salt was purchased from Sigma (St. Louis, MO). Nicotine solution for iv injection (30 (μg/kg)/0.1 mL infusion) was obtained by dissolving the salt in 0.9% saline and the pH adjusted to 7.0–7.4 with 3 M sodium hydroxide. Nicotine self-administration dose is reported as free base concentration.
Apparatus.
The self-administration boxes consisted of operant conditioning chambers (Med Associates, Inc., St. Albans, VT) enclosed in lit, sound attenuating, ventilated environmental cubicles. Each chamber was equipped with two retractable levers located in the front panel, laterally to a food pellet magazine. A pellet dispenser was positioned behind the front panel of the boxes. Chambers were also equipped with auditory stimuli presented via a speaker and visual stimuli located above the levers (cue light). Infusions occurred by means of syringe pumps (Med Associates, Inc., St. Albans, VT) and liquid swivels (Instech Solomon, Plymouth Meeting, PA) connected to plastic tubing protected by a flexible metal sheath for attachment to the external catheter terminus. To allow delivery of nicotine, an infusion pump was activated by responses on the right (active) lever, while responses on the left (inactive) lever were recorded but did not result in any programmed consequences. Activation of the pump resulted in a delivery of 0.1 mL of the reinforcer. A microcomputer controlled the delivery of nicotine, presentation of auditory and visual stimuli, and recording of the behavioral data.
Food Training and Intravenous (iv) Catheterization.
One week after arrival, all rats were trained to lever-press for 45 mg of food pellets (test diet, 5-TUM, Richmond, IN) under a fixed ratio 1 (FR-1) schedule of reinforcement in 30 min sessions for 3 days. Then, animals underwent iv surgery that occurred under isoflurane anesthesia. Incisions were made to expose the right jugular vein. A catheter made from microrenathane tubing (inner diameter = 0.020 in., outside diameter = 0.037 in.) was subcutaneously positioned between the vein and the back as described in refs 26 and 52. For the duration of the experiment, the catheters were flushed daily with 0.2 mL of heparinized saline solution containing enrofloxacin (0.7 mg/mL). The self-administration experiments began 1 week after recovery from surgery.
Effect of 5 on Nicotine Self-Administration.
Rats (n = 7) were trained to self-administer nicotine under a FR-1 schedule of reinforcement for 6 days of 2 h daily sessions and under a fixed ratio 3 (FR-3) for additional 9 sessions. Every three active lever presses resulted in the delivery of one nicotine dose (0.03 (mg/kg)/0.1 mL infusion). Following each nicotine infusion, a 20 s time out (TO) period occurred, during which responses at the active lever did not lead to programmed consequences. This TO period was concurrent with illumination of a cue light located above the active lever to signal delivery of the positive reinforcement. Additionally, an intermittent tone (7 kHz, 70 dB) was sounded throughout the 2 h nicotine sessions. The rats were trained to self-administer nicotine until a stable baseline of reinforcement was established. A Latin square within-subject design was used for the drug treatment. The rats were injected subcutaneously with the drug (0.0, 0.3, and 1.0 mg/kg) 10 min before the beginning of the session. The animals were subjected to all treatments in counterbalanced order at least at 48 h intervals between the drug test days.
Effect of 5 on Nicotine Priming-Induced Reinstatement of Nicotine Seeking.
Rats (n = 7) were trained to self-administer nicotine at the dose of 0.03 (mg/kg)/infusion in daily 2 h sessions under a FR-1 followed by FR-3 schedule of reinforcement as described above. Following each nicotine infusion (0. 1 mL), a 20 s TO period occurred, during which pressing the active lever did not lead to programmed consequences. The TO was accompanied by illumination of a cue light located above the active lever to signal delivery of the positive reinforcement, while an intermittent tone was sounded throughout the sessions. Then, an extinction phase was conducted for 14 consecutive sessions. During 1 h extinction sessions, the lever presses were no longer associated with nicotine delivery while all cues were presented to allow for their concomitant extinction. The day after the last extinction session, the rats were subjected to the reinstatement test by the subcutaneous injection of nicotine at the dose of 0.15 mg/ kg. To evaluate the effect of 5 on nicotine priming-induced reinstatement, rats were administered the drug (0.3 and 1.0 mg/kg) or its vehicle (0.0 mg/kg) in a counterbalanced order (Latin square) 10 min before nicotine injection that was in turn administered 10 min before the 1 h reinstatement session. A 3-day interval occurred between drug tests, during which the animals were subjected to extinction sessions. The nicotine dose, time of injection, and experimental design have been previously described.26
Effect of 5 on Cue-Induced Reinstatement of Nicotine Seeking.
Rats (n = 7) were trained to lever-press for nicotine at the dose of 0.03 mg/kg in daily 2 h sessions under a FR-1 schedule of reinforcement for 6 days and under FR-3 for additional 9 days. Concurrently with the lever pressing, a 20 s TO period that was concurrent to presentation of a cue light was in effect. A stimulus predictive of nicotine (orange odor) was also presented immediately after the animals were placed in the operant chambers and immediately before the onset of every conditioning session.30 Furthermore, an intermittent tone (7 kHz, 70 dB) was sounded throughout the 2 h nicotine sessions. Nicotine-reinforced responding was then extinguished in daily 1 h sessions that continued for 14 days. In this phase, neither nicotine nor the tone, the cue light, or the orange odor was available. On the day following the last extinction session, a 1 h reinstatement session was carried out without any drug treatment. Tone, odor, and cue light, but not nicotine, were presented, and reinstatement response rates (i.e., responses on the previously nicotine-associated lever) were recorded. These response rates were used to assign animals to treatment groups balanced for response rates for the drug treatment experiment that followed. To assess the effects of 5, reinstatement experiments were then conducted every third day. In a Latin-square counterbalanced order that paralleled that used for the self-administration studies, animals were pretreated with 5 (0.0, 0.3, 1.0 mg/kg, sc) 10 min prior the onset of the reinstatement sessions.
Assessment of Body Temperature.
Baseline rectal temperature was measured in rats (n = 8) just prior to injection of 5 (1.0 mg/kg, sc) or vehicle (0.9% saline) using a Latin square within-subject design. Ten minutes later, nicotine (0.5 mg/kg, sc) or vehicle (0.9% saline) was injected, and temperature was measured again after 15, 30, 60, and 120 min.
Detection of Compound in Rat Blood and Brain Using LC- MS/MS.
Rats (n = 3–4 per group) were sc injected with 5 (2 mg/kg) and blood as well as brain samples taken at various time points (10, 30, and 60 min) following drug administration. Samples of rats treated with vehicle (time point 0) were also taken. Blood (100 μL) was obtained by cardiac puncture and collected into a corresponding microcentrifuge tube that contained 1.0 mL of cold acetonitrile with 10 ng/mL of cotinine which was used as internal standard. Samples were then centrifuged at 3000 rpm for 5 min. The supernatant was dried in a speed vacuum. Samples were reconstituted in 100 μL of 5% acetonitrile in water. Next, samples were centrifuged at 13 000 rpm for 5 min. A volume of 10 μL of supernatant was used for analysis. Brain samples were collected following perfusion with PBS and homogenized on ice in 500 μL of PBS buffer. Protein precipitation was achieved by adding 1.0 mL of cold acetonitrile with 10 ng/mL internal standard. Samples were centrifuged at 10 000 rpm for 10 min. The supernatant was dried in a speed vacuum. Samples were reconstituted in 100 μL of 5% acetonitrile in water. Analysis was conducted by using high performance liquid chromatography (HPLC, 20AD Shimadzu Prominence)/tandem mass spectrometry (LC-MS/MS) AbSciex 3200 QTrap triple-quadrupole linear ion trap mass spectrometer fitted with a TurboIonSpray interface (Applied Biosystems/MDS Sciex, Darmstadt, Germany). In brief, to achieve separation on the HPLC of the analyte and the internal standard, reverse phase mode with a gradient of 1–10% acetonitrile over 15 min was used on a C-18 reverse phase column (Phenomenex Gemini NX 110A 50 mm × 4.6 mm). Mobile phase A was LCMS grade water with 0.1% formic acid (Fisher Optima catalog no. LS118–4). Mobile phase B was acetonitrile with 0.1% formic acid (Fluka catalog no. 34668). The MS/MS analysis was performed in multiple reaction monitoring (MRM) mode using the three largest fragments of the parent ions. MS instrument parameters were spray voltage 5.5 kV, curtain gas 20 psi, source temperature 650 °C, ion source gas 150 psi and gas 240 psi. The ion transitions monitored were 205.225/106.1, 205.225/100.3, 205.225/ 78.2 and 177.163/80.2, 177.163/98.1, 177.163/53.2. Blank solvent injections were run between each sample to minimize analyte carry over. The counts for the ion transitions were summed to give the peak area. The ratio of the analyte peak area to the internal standard peak area was calculated for each rat. The average was calculated and plotted for each time point.
Data Analysis.
Data were analyzed by using Statistica 7 software.53 The effects of 5 on nicotine self-administration and nicotine-induced changes in body temperature were analyzed by means of a two-way ANOVA that used two within-subject factors (i.e., “time interval”, which is the number of infusions in fractions of 30 min, or “time point” and “treatment dose” or “treatment”, respectively). To establish that reinstatement was successfully induced, responding during the last EXT session was compared to the respective reinstatement session of the vehicle-treated group by one-way within-subject ANOVA. The effect of systemic 5 on reinstatement experiments was analyzed using one-way repeated measures ANOVA with treatment (drug dose) as a within-subject factor. Blood and brain contents were analyzed by oneway ANOVA that used time point as between factor. The level of significance was set at p < 0.05. ANOVAs were followed, where appropriate, by Student-Newman-Keuls post hoc tests.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by both NIH Grants R43 DA036968 and R44 DA036968 to J.W. and the State of Florida, Executive Office of the Governor’s Department of Economic Opportunity. We thank Drs. Yingxian Xiao and Kenneth Kellar (Georgetown University, Washington, DC) for kindly providing the ratα3β4, α4β4, α4β2, and α3β2 nAChR HEK293 cell lines, and we thank Dr. Jon Lindstrom (University of Pennsylvania, PA) for providing α3β4α5 nAChR HEK293 cell line used in the functional assays.
ABBREVIATIONS USED
- nAChR
nicotinic acetylcholine receptor
- DhβE
dihydro-β-erythroidine
- 4-nitro-PFEB
2-fluoro-3-(4-nitrophenyl)-deschloroepibatidine
- VTA
ventral tegmental area
- TPIMS
The Torrey Pines Institute for Molecular Studies
- ANOVA
analysis of variance
- MBHA
p-methylbenzhydrylamine
- DIEA
diisopropylethylamine
- DCM
dichloromethane
- Boc
tert-butyloxycarbonyl
- DIC
diisopropylcarbodiimide
- HOBt
hy-droxybenzotriazole hydrate
- TFA
trifluoroacetic acid
- CNBr
cyanogen bromide
- LC-MS
liquid chromatography-mass spectrometry
- sc
subcutaneous
- FR-1
fixed ratio 1
- FR-3
fixed ratio 3
- AN
analyte
- IS
internal control
- TO
time out
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.7b01250.
Molecular formula strings and some data (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).The health consequences of smoking—50 years of progress: A report of the surgeon general U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion: Office on Smoking and Health, U.S. Government Printing Office: Washington, DC, 2014,. [Google Scholar]
- (2).Xu X; Bishop E; Kennedy S; Simpson S; Pechacek T Annual healthcare spending attributable to cigarette smoking: An update. Am. J. Prev. Med. 2015, 48, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).WHO report on the global tobacco epidemic. World Health Organization: Geneva, 2011. [Google Scholar]
- (4).Jorenby DE; Hays JT; Rigotti NA; Azoulay S; Watsky EJ; Williams KE; Billing CB; Gong J; Reeves KR Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: A randomized controlled trial. JAMA 2006, 296, 56–63. [DOI] [PubMed] [Google Scholar]
- (5).Reus VI; Smith BJ Multimodal techniques for smoking cessation: A review of their efficacy and utilisation and clinical practice guidelines. Int. J. Clin. Pract. 2008, 62, 1753–1768. [DOI] [PubMed] [Google Scholar]
- (6).Rigotti NA Clinical practice. Treatment of tobacco use and dependence. N. Engl. J. Med. 2002, 346, 506–512. [DOI] [PubMed] [Google Scholar]
- (7).Moore TJ; Furberg CD; Glenmullen J; Maltsberger JT; Singh S Suicidal behavior and depression in smoking cessation treatments. PLoS One 2011, 6, e27016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Picciotto MR; Zoli M; Rimondini R; Lena C; Marubio LM; Pich EM; Fuxe K; Changeux JP Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998, 391, 173–177. [DOI] [PubMed] [Google Scholar]
- (9).Stolerman IP; Shoaib M The neurobiology of tobacco addiction. Trends Pharmacol. Sci. 1991, 12, 467–473. [DOI] [PubMed] [Google Scholar]
- (10).Perry DC; Mao D; Gold AB; McIntosh JM; Pezzullo JC; Kellar KJ Chronic nicotine differentially regulates alpha6-and beta3-containing nicotinic cholinergic receptors in rat brain. J. Pharmacol. Exp. Ther. 2007, 322, 306–315. [DOI] [PubMed] [Google Scholar]
- (11).Perry DC; Xiao Y; Nguyen HN; Musachio JL; Davila- Garcia MI; Kellar KJ Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J. Neurochem. 2002, 82, 468–481. [DOI] [PubMed] [Google Scholar]
- (12).Epping-Jordan MP; Picciotto MR; Changeux JP; Pich EM Assessment of nicotinic acetylcholine receptor subunit contributions to nicotine self-administration in mutant mice. Psychopharmacology (Berl) 1999, 147, 25–26. [DOI] [PubMed] [Google Scholar]
- (13).Tobey KM; Walentiny DM; Wiley JL; Carroll FI; Damaj MI; Azar MR; Koob GF; George O; Harris LS; Vann RE Effects of the specific alpha4beta2 nachr antagonist, 2-fluoro-3-(4-nitrophenyl) deschloroepibatidine, on nicotine reward-related behaviors in rats and mice. Psychopharmacology (Berl) 2012, 223, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Watkins SS; Epping-Jordan MP; Koob GF; Markou A Blockade of nicotine self-administration with nicotinic antagonists in rats. Pharmacol Biochem. Behav. 1999, 62, 743–751. [DOI] [PubMed] [Google Scholar]
- (15).Fowler CD; Lu Q; Johnson PM; Marks MJ; Kenny PJ Habenular a5 nicotinic receptor subunit signalling controls nicotine intake. Nature 2011, 471, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Frahm S; Slimak MA; Ferrarese L; Santos-Torres J; Antolin-Fontes B; Auer S; Filkin S; Pons S; Fontaine JF; Tsetlin V; Maskos U; Ibanez-Tallon I Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron 2011, 70, 522–535. [DOI] [PubMed] [Google Scholar]
- (17).Harrington L; Vinals X; Herrera-Solis A; Flores A; Morel C; Tolu S; Faure P; Maldonado R; Maskos U; Robledo P Role of beta4* nicotinic acetylcholine receptors in the habenulo-interpeduncular pathway in nicotine reinforcement in mice. Neuropsychopharmacology 2016, 41, 1790–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Morel C; Fattore L; Pons S; Hay YA; Marti F; Lambolez B; De Biasi M; Lathrop M; Fratta W; Maskos U; Faure P Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol. Psychiatry 2014, 19, 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Houghten RA; Pinilla C; Giulianotti MA; Appel JR; Dooley CT; Nefzi A; Ostresh JM; Yu Y; Maggiora GM; Medina-Franco JL; Brunner D; Schneider J Strategies for the use of mixture-based synthetic combinatorial libraries: Scaffold ranking, direct testing in vivo, and enhanced deconvolution by computational methods. J. Comb. Chem. 2008, 10, 3–19. [DOI] [PubMed] [Google Scholar]
- (20).Lopez-Vallejo F; Giulianotti MA; Houghten RA; Medina-Franco JL Expanding the medicinally relevant chemical space with compound libraries. Drug Discovery Today 2012, 17, 718–726. [DOI] [PubMed] [Google Scholar]
- (21).Wu J; Zhang Y; Maida LE; Santos RG; Welmaker GS; LaVoi TM; Nefzi A; Yu Y; Houghten RA; Toll L; Giulianotti MA Scaffold ranking and positional scanning utilized in the discovery of nachr-selective compounds suitable for optimization studies. J. Med. Chem. 2013, 56, 10103–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pajouhesh H; Lenz GR Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Carroll FI; Ware R; Brieaddy LE; Navarro HA; Damaj MI; Martin BR Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2/-fluoro-3/-(substituted phenyl)-deschloroepibatidine analogues. Novel nicotinic antagonist. J. Med. Chem. 2004, 47, 4588–4594. [DOI] [PubMed] [Google Scholar]
- (24).Mihalak KB; Carroll FI; Luetje CW Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 801–805. [DOI] [PubMed] [Google Scholar]
- (25).Chatterjee S; Steensland P; Simms JA; Holgate J; Coe JW; Hurst RS; Shaffer CL; Lowe J; Rollema H; Bartlett SE Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuro- psjchopharmacology 2011, 36, 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cippitelli A; Wu J; Gaiolini KA; Mercatelli D; Schoch J; Gorman M; Ramirez A; Ciccocioppo R; Khroyan TV; Yasuda D, Zaveri NT; Pascual C; Xie XS; Toll L At-1001: A high-affinity alpha3beta4 nachr ligand with novel nicotine-suppressive pharmacology. Br. J. Pharmacol. 2015, 172, 1834–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rollema H; Chambers LK; Coe JW; Glowa J; Hurst RS; Lebel LA; Lu Y; Mansbach RS; Mather RJ; Rovetti CC; Sands SB; Schaeffer E; Schulz DW; Tingley FD 3rd; Williams KE Pharmacological profile of the alpha4beta2 nicotinic acetylcho-line receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 2007, 52, 985–994. [DOI] [PubMed] [Google Scholar]
- (28).Rollema H; Coe JW; Chambers LK; Hurst RS; Stahl SM; Williams KE Rationale, pharmacology and clinical efficacy of partial agonists of alpha4beta2 nach receptors for smoking cessation. Trends Pharmacol. Sci. 2007, 28, 316–325. [DOI] [PubMed] [Google Scholar]
- (29).Chatterjee S; Bartlett SE Neuronal nicotinic acetylcholine receptors as pharmacotherapeutic targets for the treatment of alcohol use disorders. CNS Neurol. Disord.: Drug Targets 2010, 9, 60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cippitelli A; Brunori G; Gaiolini KA; Zaveri NT; Toll L Pharmacological stress is required for the anti-alcohol effect of the alpha3beta4* nachr partial agonist at-1001. Neuropharmacology 2015, 93, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hendrickson LM; Zhao-Shea R; Pang X; Gardner PD; Tapper AR Activation of alpha4* nachrs is necessary and sufficient for varenicline-induced reduction of alcohol consumption. J. Neurosci. 2010, 30, 10169–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ahmed AI; Ali AN; Kramers C; Harmark LV; Burger DM; Verhoeven WM Neuropsychiatric adverse events of varenicline: A systematic review of published reports. J. Clin. Psychopharmacol. 2013, 33, 55–62. [DOI] [PubMed] [Google Scholar]
- (33).Baker TB; Piper ME; Stein JH; Smith SS; Bolt DM; Fraser DL; Fiore MC Effects of nicotine patch vs varenicline vs combination nicotine replacement therapy on smoking cessation at 26 weeks: A randomized clinical trial. JAMA 2016, 315, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rasmussen T; Swedberg MD Reinforcing effects of nicotinic compounds: Intravenous self-administration in drug-naive mice. Pharmacol., Biochem. Behav. 1998, 60, 567–573. [DOI] [PubMed] [Google Scholar]
- (35).Cahill K; Lindson-Hawley N; Thomas KH; Fanshawe TR; Lancaster T Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst. Rev. 2016, CD006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Levin ED; Rezvani AH; Xiao Y; Slade S; Cauley M; Wells C; Hampton D; Petro A; Rose JE; Brown ML; Paige MA; McDowell BE; Kellar KJ Sazetidine-a, a selective alpha4beta2 nicotinic receptor desensitizing agent and partial agonist, reduces nicotine self-administration in rats. J. Pharmacol. Exp. Ther. 2010, 332, 933–939. [DOI] [PubMed] [Google Scholar]
- (37).Rezvani AH; Slade S; Wells C; Petro A; Lumeng L; Li TK; Xiao Y; Brown ML; Paige MA; McDowell BE; Rose JE; Kellar KJ; Levin ED Effects of sazetidine-a, a selective alpha4beta2 nicotinic acetylcholine receptor desensitizing agent on alcohol and nicotine self-administration in selectively bred alcohol-preferring (p) rats. Psychopharmacology (Berl) 2010, 211, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hopkins TJ; Rupprecht LE; Hayes MR; Blendy JA; Schmidt HD Galantamine, an acetylcholinesterase inhibitor and positive allosteric modulator of nicotinic acetylcholine receptors, attenuates nicotine taking and seeking in rats. Neuropsychopharmacology 2012, 37, 2310–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Maurer JJ; Sandager-Nielsen K; Schmidt HD Attenuation of nicotine taking and seeking in rats by the stoichiometry-selective alpha4beta2 nicotinic acetylcholine receptor positive allosteric modulator ns9283. Psychopharmacology (Berl) 2017, 234, 475–484. [DOI] [PubMed] [Google Scholar]
- (40).Rose JE; Behm FM; Westman EC Nicotine- mecamylamine treatment for smoking cessation: The role of precessation therapy. Exp. Clin. Psjchopharmacol. 1998, 6, 331–343. [DOI] [PubMed] [Google Scholar]
- (41).Rose JE; Behm FM; Westman EC; Levin ED; Stein RM; Ripka GV Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin. Pharmacol. Ther. 1994, 56, 86–99. [DOI] [PubMed] [Google Scholar]
- (42).Fitch RW; Xiao Y; Kellar KJ; Daly JW Membrane potential fluorescence: A rapid and highly sensitive assay for nicotinic receptor channel function. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 4909–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Carroll FI; Lee JR; Navarro HA; Brieaddy LE; Abraham P; Damaj MI; Martin BR Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2- exo-2-(2ʹ-substituted-3ʹ-phenyl-5ʹ-pyridinyl)-7-azabicyclo[2.2.1]- heptanes. Novel nicotinic antagonist. J. Med. Chem. 2001, 44, 4039–4041. [DOI] [PubMed] [Google Scholar]
- (44).Carroll FI; Liang F; Navarro HA; Brieaddy LE; Abraham P; Damaj MI; Martin BR Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 2- exo-2-(2ʹ-substituted 5ʹ-pyridinyl)-7-azabicyclo[2.2.1]heptanes. Epiba- tidine analogues. J. Med. Chem. 2001, 44, 2229–2237. [DOI] [PubMed] [Google Scholar]
- (45).Ondachi P; Castro A; Luetje CW; Damaj MI; Mascarella SW; Navarro HA; Carroll FI Synthesis and nicotinic acetylcholine receptor in vitro and in vivo pharmacological properties of 2ʹ-fluoro-3ʹ-(substituted phenyl)deschloroepibatidine analogues of 2ʹ-fluoro-3ʹ-(4-nitrophenyl)deschloroepibatidine. J. Med. Chem. 2012, 55, 6512–6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Abdrakhmanova GR; Damaj MI; Carroll FI; Martin BR 2- fluoro-3-(4-nitro-phenyl)deschloroepibatidine is a novel potent competitive antagonist of human neuronal alpha4beta2 nachrs. Mol. Pharmacol. 2006, 69, 1945–1952. [DOI] [PubMed] [Google Scholar]
- (47).Carroll FI; Ma W; Deng L; Navarro HA; Damaj MI; Martin BR Synthesis, nicotinic acetylcholine receptor binding, and antinociceptive properties of 3ʹ-(substituted phenyl)epibatidine analogues. Nicotinic partial agonists. J. Nat. Prod. 2010, 73, 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Giulianotti MA; Vesely BA; Azhari A; Souza A; LaVoi T; Houghten RA; Kyle DE; Leahy JW Identification of a hit series of antileishmanial compounds through the use of mixture-based libraries. ACS Med. Chem. Lett. 2017, 8, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Houghten RA General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 5131–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Manku S; Laplante C; Kopac D; Chan T; Hall DG A mild and general solid-phase method for the synthesis of chiral polyamines. Solution studies on the cleavage of borane-amine intermediates from the reduction of secondary amides. J. Org. Chem. 2001, 66, 874–885. [DOI] [PubMed] [Google Scholar]
- (51).Ostresh JM; Schoner CC; Hamashin VT; Nefzi A; Meyer J-P; Houghten RA Solid-phase synthesis of trisubstituted bicyclic guanidines via cyclization of reduced n-acylated dipeptides. J. Org. Chem. 1998, 63, 8622–8623. [Google Scholar]
- (52).Cippitelli A; Schoch J; Debevec G; Brunori G; Zaveri NT; Toll L A key role for the N/OFQ-NOP receptor system in modulating nicotine taking in a model of nicotine and alcohol co-administration. Sci. Rep. 2016, 6, 26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hilbe JM Statistica 7. Am. Stat. 2007, 61, 91–94. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.