

Abstract
Twenty eight new aryloxybenzene analogues were synthesized and their in vitro binding potencies toward S1PR2 were determined using a [32P]S1P competitive binding assay. Out of these new analogues, three compounds, 28c (IC50 = 29.9 ± 3.9 nM), 28e (IC50 = 14.6 ± 1.5 nM), and 28g (IC50 = 38.5 ± 6.3 nM) exhibited high binding potency toward S1PR2 and high selectivity over the other four receptor subtypes (S1PR1, 3, 4, and 5; IC50 > 1000 nM). Each of the three potent compounds 28c, 28e, and 28g contains a fluorine atom that will allow developing F-18 labeled PET radiotracers for imaging S1PR2.
Keywords: Sphingosine 1-phosphate receptor 2, Binding potency, Selectivity, Positron emission tomography
Graphical abstract
1. Introduction
Sphingosine 1-phosphate (S1P) is an essential membrane-derived bioactive sphingolipid mediator which plays critical regulatory functions in the cardiovascular, nervous, and immune systems.1 S1P is generated from the phosphorylation of sphingosine by sphingosine kinases (Sphk1 and 2) and presents at high levels in the blood and lymph.2 S1P exerts its functions by interacting with a family of five G protein-coupled receptor subtypes, S1PR1, 2, 3, 4, and 5, originally named as EDG-1, 5, 3, 6, and 8.3 Each subtype has its specific tissue expression and response to different biological process.4, 5 Out of these five subtypes, S1PR1, 2, and 3 are extensive ubiquitously expressed in a variety of cell types and tissues; S1PR4 expression is primarily in hematopoietic and immune system cells; S1PR5 is primarily expressed in the spleen and central nervous system (CNS).6, 7 Among these receptors, S1PR2 plays a pivotal role in mediating different cellular functions and pathologies, such as endothelial,8 metabolic,9, 10 muscle,11 neuronal,12, 13 and kidney functions.14 Conjugated bile acid induced activation of the S1PR2 signaling pathway plays a critical role in obstructive cholestasis and invasive growth of esophageal adenocarcinoma (EAC) cells, which represent a novel therapeutic target for cholestatic liver diseases and EAC.15, 16 During demyelination process in CNS diseases, S1PR2 regulates blood-brain barrier (BBB) function and an increased expression of S1PR2 is observed in disease-susceptible regions of both female SJL experimental allergic encephalomyelitis (EAE) mice and female multiple sclerosis (MS) patients with compared to their male counterparts.17 In the kidney of diabetic rats and mesangial cells under high glucose condition, the highest S1PR2 mRNA expression was observed compared to other four S1P subtypes, S1PR1, 3, 4, and 5.18 Together, S1PR2 protein may serve a promising biomarker for assessing the progress of diseases such as cholangiopathies, MS, and diabetic nephropathy. Positron emission tomography (PET) is a unique imaging modality and can provide a non-invasive method to quantitatively assess the target protein distribution in vivo in tissues. It was widely used in preclinical and clinical investigations. PET imaging with a suitable S1PR2 specific radiotracer could help advance our understanding of S1PR2 modulating functions in related diseases. Therefore, identification of S1PR2 specific ligands is imperative. Despite recognition of this potential, only a few compounds have been reported specifically for S1PR2 (Figure 1). JTE-013, a well-known antagonist for S1PR2, is widely used in preclinical studies19; CYM-5520 is an allosteric S1PR2 selective agonist, but it was not competitive with S1P in a [33P]S1P competitive binding assay.20 Takuya Seko’s group recently, reported four potent S1PR2 compounds 1–4 (Figure 1) that have high antagonist activity. Compounds 2, 3, and 4 showed improved metabolic stability compared to compound 1.21–23 Our group reported the radiosynthesis of [11C]TZ34125, a JTE-013 analogue, and performed in vivo evaluation in SJL mice that confirmed the sexual dimorphism of S1PR2 expression in the cerebellum.24 Nevertheless, no F-18 S1PR2 radiotracer was reported. To identify an F-18 PET radiotracer for imaging S1PR2 expression in vitro and in vivo for related diseases, herein we reported our recent efforts on the exploration of the new structural S1PR2 ligands.
Figure 1.

Structures of S1PR2 compounds
After analyzing the structure of compound 1, it was dissected into three fragments A, B, and C as shown in Figure 2. Our exploration of new S1PR2 analogues used three strategies. First, we focused on the optimization of fragment A: the proton at the 5-position of benzyl ring was replaced with different moieties including alkoxy heterocycle, trifluoromethyl, aryloxy, and arylthio groups. Second, we focused on the optimization of fragment B: different electronegativity functional groups were used to replace the fluorine at 4’-position of the aromatic ring. Third, we focused on the optimization of fragment C: the piperdin-4-ol was replaced with pyrrolidin-3-ol and azetidin-3-ol and different alkyl chains were used to replace the 2-(ethyl)butyl tail. The goal of our efforts was to identify S1PR2 ligands with high binding potency and high selectivity. The newly synthesized analogues were screened for their in vitro binding potency and selectivity using [32P]S1P as the competitive radioligand in an assay with cell membranes that were enriched for the different receptor subtypes. Out of these new analogues, three new compounds were discovered having high S1PR2 binding potency (IC50 < 50 nM) and high selectivity for S1PR2. Consequently, the structure-activity relationship analysis of these new analogues was explored.
Figure 2.

Design strategy of new S1PR2 analogues
2. Results and Discussion
2.1. Chemistry
Our exploration of new S1PR2 analogues was accomplished by following our abovementioned three strategies. Firstly, fluorine substituted alkoxy, 1-methyl-4-piperazine, 1H-pyrazole, and trifluoromethyl groups was used to replace the hydrogen atom at the 5-position of the aromatic ring in the fragment A to generate compounds 11a-b and 17a-c. Briefly, the synthesis of compounds 11a-b were accomplished by following Scheme 1. Briefly, benzyl 4-oxopiperidine-1-carboxylate (5) was treated with 2-ethylbutylmagnesium bromide in the presence of lanthanum trichloride-lithium chloride complex at 0 °C to afford alcohol intermediate, following by removal of carboxybenzyl (Cbz) group through Pd-catalyzed hydrogenation in methanol to yield 4-(2- ethylbutyl)piperidin-4-ol (6). Meanwhile, commercially available 1,3-difluoro-5-nitrobenzene (7) was treated with 2-fluorethanol or 3-fluoro-1-propanol and sodium hydride (NaH) in N,N-dimethylformamide (DMF) to yield 8a-b, which were subjected to a substitution reaction using 4-fluorophenol to afford aryloxy intermediates 9a-b in the presence of K3PO4 at 100 °C in N,N-dimethylacetamide (DMA). The Pdcatalyzed hydrogenation of 9a-b gave anilines, followed by treating with 2,2,2-trichloroethyl chloroformate and NaHCO3 gave intermediates 10a-b. The subsequent coupling reaction of 10a-b with 4-(2-ethylbutyl)piperidin-4-ol (6) in the presence of N,N-diisopropylethylamine (DIPEA) at 100 °C in DMA afforded target compound 11a-b.
Scheme 1.

Synthesis of 11a-b. Reagents, conditions, and yields: (a) LaCl3-2LiCl solution, 2-ethylbutylmagnesium bromide, 0 °C - RT; (b) Pd/C, H2, methanol, RT; 81%; (c) 2-fluorethanol or 3-fluoropyopan-1-ol, NaH, DMF, RT; 33–35%; (d) 4-fluorophenol, K3PO4, DMA, 100 °C; 91–99%; (e) (i) Pd/C, H2, ethyl acetate, RT; (ii) 2,2,2-trichloroethyl chloroformate, ethyl acetate, NaHCO3, 0 °C – RT; (f) 6, DIPEA, DMA, 100 °C; 97–98%.
The synthesis of 17a-c were achieved by following Scheme 2. Commercially available 4-fluorophenol (12) reacted with 1-fluoro-3-nitro-5-(trifluoromethyl)benzene or 1,3-difluoro-5-nitrobenzene afforded 13 or 14, respectively. Intermediate 14 was coupled with 1-methylpiperozine or 1H-pyrazole using K2CO3 as a base in dimethyl sulfoxide (DMSO) to afford 15a or 15b. After palladium-catalyzed reduction of 13 and 15a-b, the resulting anilines were reacted with 2,2,2-trichloroethyl chloroformate to afford 16a-c. The target compounds 17a-c was prepared by reacting the carbamates 16a-c with 4-(2-ethylbutyl)piperidin-4-ol (6) as described for 11a-b.
Scheme 2.

Synthesis of 17a-c. Reagents, conditions, and yields: (a) 1-fluoro-3-nitro-5-(trifluoromethyl)benzene, K3PO4, DMA, 100 °C; 93%; (b) 1,3-difluoro-5-nitrobenzene, K3PO4, DMA, 100 °C; 95%; (c) 1-methylpiperozine or 1H-pyrazole, K2CO3, DMSO, 65 °C; 42–53%; (d) (i) Pd/C, H2, ethyl acetate, RT; (ii) 2,2,2-trichloroethyl chloroformate, ethyl acetate, NaHCO3, 0 °C - RT; (e) 6, DIPEA, DMA, 100 °C ; 22–44%.
Secondly, to explore the impact of the substitution group at 4’-position of the aromatic ring in fragment B, we retained the trifluoromethyl group because trifluoromethyl-containing compound 17c showed moderate binding potency. Compounds 21a-f were synthesized with different substitution groups at 4’-position as shown in Scheme 3. Briefly, commercially available 1- fluoro-3-nitro-5-(trifluoromethyl)benzene (18) reacted with differently substituted phenols in the presence of K3PO4 in DMA afforded 19a-f. The palladium-catalyzed hydrogenation of 19a-f gave anilines, followed by condensation with 2,2,2-trichloroethyl chloroformate to afford key intermediates 20a-f. Coupling of intermediates 20a-f with 4-(2-ethylbutyl)piperidin-4-ol (6) yielded the target compounds 21a-f.
Scheme 3.

Synthesis of target compounds 21a-f. Reagents, conditions, and yields: (a) phenols, K3PO4, DMA, 100 °C; 60–99%; (b) (i) Pd/C, H2, ethyl acetate, RT; (ii) 2,2,2-trichloroethyl chloroformate, ethyl acetate, NaHCO3, 0 °C - RT; (c) 6, DIPEA, DMA, 100 °C; 39–81%.
Thirdly, to further evaluate the impact of the side chain and N-containing heterocycle in fragment C, we changed 2-(ethyl)butyl side chain with different alkyl chains and replaced the piperdin-4-ol with pyrrolidin-3-ol and azetidin-3-ol. Compounds 25a-j were synthesized by following Scheme 4. The amines (22a-d and 24ab) were prepared ahead using the same procedure as described for compound 6 with corresponding Grignard’s agents and ketones. The subsequent coupling two carbamates 20c and 20f with corresponding amines (22a-d and 24a-b) afforded compounds 25a-j.
Scheme 4.

Synthesis of different amines 22a-d, 24a-b, and target compounds 25a-j. Reagents, conditions, and yields: (a) LaCl3-2LiCl solution, Grignard’s agents, 0 °C - RT; (b) Pd/C, H2, methanol, RT; 92–98%; (c) amines (22a-d or 24a-b), DIPEA, DMA, 100 °C; 42–99%
Finally, to further investigate the substituted group at the 5-position of the middle aromatic ring in fragment A, a series of substituted aryloxy group and arylthio group were used to replace the trifluoromethyl group. New analogues 28a-g were synthesized by following Scheme 5. Briefly, compound 14 reacted with 4-methoxylphenol, 6-hydroxy-2-methyl-3,4-dihydroisoquinolin-1(2H)-one, 6-hydroxy-3,4-dihydroisoquinolin-1(2H)-one, N-(2-fluoroethyl)-4-hydroxybenzamide, or 4-(methylsulfonyl)benzene-thiol in the presence of K3PO4 gave compounds 26a-e, followed by palladium-catalyzed hydrogenation afforded corresponding anilines. The resulting anilines were treated with 2,2,2-trichloroethyl chloroformate and NaHCO3 to afford intermediates 27a-e. The subsequent condensation reaction of 27a-e with 6 or 22d in the presence of DIPEA at 100 °C in DMA yielded target compounds 28a-g.
Scheme 5.

Synthesis of 28a-g. Reagents, conditions, and yields: (a) phenols or thiophenol, K3PO4, DMA, 100 °C; 15–99%; (b) (i) Pd/C, H2, ethyl acetate, RT; (ii) 2,2,2-trichloroethyl chloroformate, ethyl acetate,NaHCO3, 0 °C - RT; 90–99%; (c) 6 or 22d, DIPEA, DMA, 100 °C; 24–88%.
2.2. Biological binding studies
In vitro binding of these newly synthesized compounds 11a-b, 17a-c, 21a-f, 25a–j, and 28a–g toward S1PR2 was determined by radioligand [32P]S1P competitive cell membrane binding assay following our published protocol.20 The results are shown in Table 1–2. The strategy of introducing alkoxy, heterocycle, or trifluoromethyl group in fragment A gave compounds 11a-b and 17a-c. The in vitro S1PR2 binding data showed that compound 17c, with a trifluoromethyl group at the 5-position exhibit moderate binding activity with an IC50 value of 362.3 nM, which is comparable to the lead compound 1 having IC50 value of 310 nM for S1PR2. The fluorine-containing alkoxy compounds, 11a and 11b showed low binding activities with IC50 > 1000 nM. The substitution with another two N-containing heterocycles, 1-methyl-4-piperazine and 1H-pyrazole didn’t improve the binding activity either; both 15a and 15b had IC50 > 1000 nM. Subsequently, our further exploration of new analogues focused on structural optimization of compound 17c. From one side, as shown in Scheme 3, we first retained trifluoromethyl group at 5-position in the fragment A and checked the impact of various substituted groups at 4’-position in fragment B (21a-f). Our binding data indicated that mono-methyl carbamide (CONHCH3) and methylsulfonyl (SO2CH3) group improved the binding activity, compound 21c and 21f had IC50 values of 278.5 and 270.3 nM, respectively. The other functional groups, like OCH3, CONH2, CON(CH3)2, and COOCH3 in 21a, 21b, 21d, and 21e decreased the binding activity with IC50 >1000 nM. To investigate the impact of the side chain in the fragment C, compounds 25a-h were synthesized and tested. We observed that the replacement of 2-(ethyl)butyl chain with methyl, ethyl, or isopropyl chains caused the loss of biological activity, compounds 25a-f had IC50 > 1000 nM. Interestingly, we observed compound 25g (IC50 = 359.3 nM) and 25h (IC50 = 296.5 nM) with a isobutyl side chain resulted in comparable binding activity compared to the 2-(ethyl)butyl compounds 21c and 21f. It suggested that the size and the steric hindrance of the side chain lead to increase the S1PR2 binding activity. Another strategy to modify the fragment C was to change the piperidine heterocycle moiety. The results revealed that the compounds 25i and 25j, with pyrrolidine and azetidine heterocycles, respectively, showed IC50 values that were over 1000 nM, which indicated the piperidine heterocycle was the favorable heterocycle moiety. From the other side, compound 2, having a 4-pyridineoxyl moiety showed potent binding activity with an IC50 value of 45 nM, we replaced the trifluoromethyl group at 5-position with different alkoxy moieties in fragment A and identified compounds 28a-g. The piperdine heterocycle moiety with isobutyl or 2-(ethyl)butyl side chain was retained because these two side chains were discovered as favorable pharmacophores in current analogues. As shown in Table 2, the aryloxy moieties containing compounds exhibited improved S1PR2 binding activity compared to compound 17c. Compound 28a showed an IC50 value of 188.5 nM; compounds 28b and 28c, bearing a cyclization of carbamide group exhibited good S1PR2 binding potency that compound 28b had an IC50 value of 73.3 nM and compound 28c had an IC50 value of 29.9 nM; Compound 28d possessing a fluoroethyl carbamide group had an IC50 value of 66.7 nM; compound 28e, containing a 4-(methylsulfonyl)benzene-thiol ether moiety was the most potent S1PR2 compound with an IC50 value of 14.6 nM, which is more potent than the 4-pyridineoxyl lead compound 2 having IC50 value of 45 nM. Additionally, both of compounds 28f (IC50 = 194.5 nM) and 28g (IC50 = 38.5 nM) possessing an isobutyl side chain exhibited less potency for S1PR2 than the corresponding 28d and 28e, suggesting 2-(ethyl)butyl side chain is the most favorable moiety for S1PR2 binding activity. The representative competitive binding curves of compounds JTE-013, 28b, 28c, 28d, 28e, and 28g toward S1PR2 were shown in Figure 3. Six different concentrations from 0.01 to 1000 nM were used to determine the binding potencies. As shown in the competitive binding curves, compound 28e has the best inhibition effect at 1.0 nM compared to the other compounds and it gave the best binding potency toward S1PR2 with an average IC50 value of 14.6 nM.
Table 1.
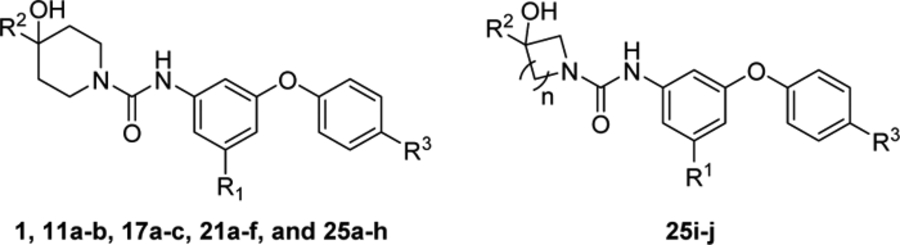
Structures and binding potencies (IC50 ± SD) of compounds 11a-b, 17a-c, 21a-f, and 25a-j toward S1PR2.a
 | |||||
|---|---|---|---|---|---|
| Compd. | n | R1 | R2 | R3 | SlPR2IC50(nM) |
| JTE-013 | - | - | - | - | 66.8 ±7.8 |
| 1b | - | II | 2-(ethyl)butyl | F | 310 |
| 11a | - | OCH2CH2F | 2-(ethyl)butyl | F | >1000 |
| 11b | - | O(CH2)3F | 2-(ethyl)butyl | F | > 1000 |
| 17a | - | 1 -methylpiperazine | 2-(ethyl)butyl | F | >1000 |
| 17b | - | 1H-pyrazole | 2-(ethyl)butyl | F | > 1000 |
| 17c | - | CF3 | 2-(ethyl)butyl | F | 362.3 ± 85.0 |
| 21a | - | CF3 | 2-(ethyl)butyl | OCH3 | > 1000 |
| 21b | - | CF3 | 2-(ethyl)butyl | CONH2 | >1000 |
| 21c | - | CF3 | 2-(ethyl)butyl | CONHCH3 | 278.5 ± 27.2 |
| 21d | - | CF3 | 2-(ethyl)butyl | CON(CH3)2 | >1000 |
| 21e | - | CF3 | 2-(ethyl)butyl | COOCH3 | > 1000 |
| 21f | - | CF3 | 2-(ethyl)butyl | SO2CH3 | 270.3 ± 72.0 |
| 25a | - | CF3 | methyl | CONHCH3 | > 1000 |
| 25b | - | CF3 | methyl | SO2CH3 | >1000 |
| 25c | - | CF3 | ethyl | CONHCH3 | > 1000 |
| 25d | - | CF3 | ethyl | SO2CH3 | >1000 |
| 25e | - | CF3 | isopropyl | CONHCH3 | > 1000 |
| 25f | - | CF3 | isopropyl | SO2CH3 | >1000 |
| 25g | - | CF3 | isobutyl | CONHCH3 | 359.3 ±50.1 |
| 25h | - | CF3 | isobutyl | SO2CH3 | 296.5 ± 32.3 |
| 25i | 1 | CF3 | isobutyl | CONHCH3 | > 1000 |
| 25j | 2 | CF3 | isobutyl | CONHCH3 | >1000 |
IC50 values were determined by at least two independent experiments, each run was performed in duplicate.
50 reference21
Table 2.
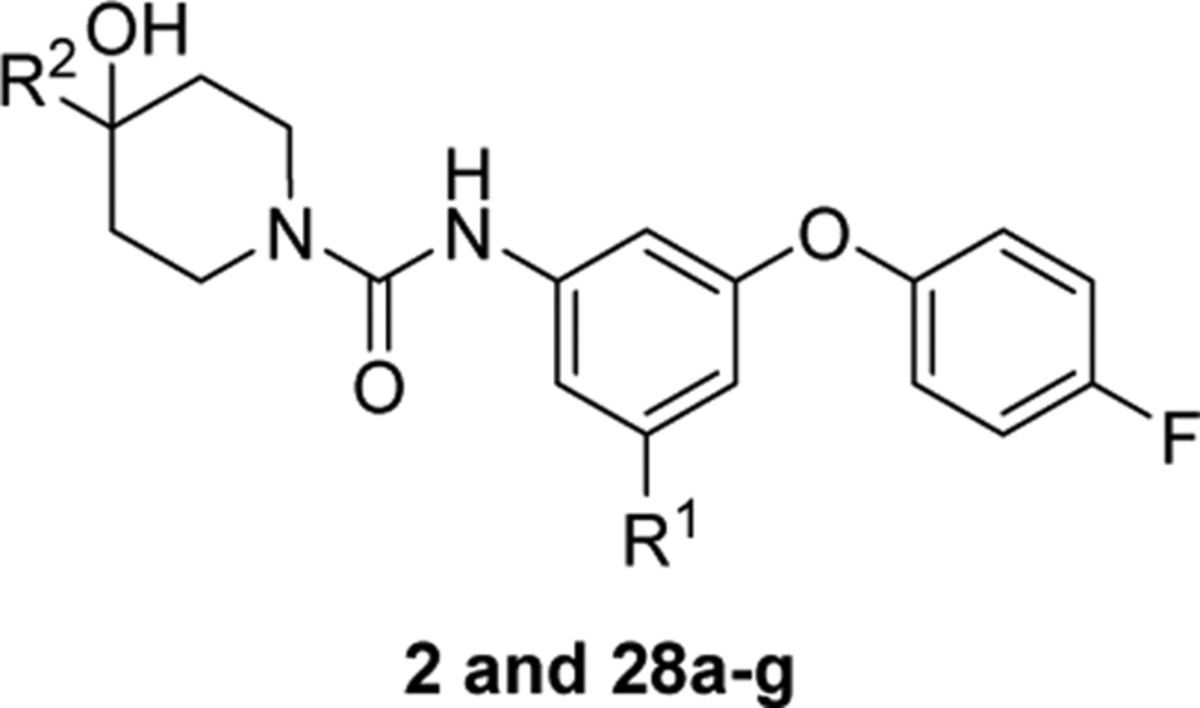
Structures and binding affinities (IC50 ± SD) of 28a-g toward S1PRs.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | IC50 (nM) | ||||
| S1PR1 | S1PR2 | S1PR3 | S1PR4 | S1PR5 | |||
| JTE-013 | - | - | >1000 | 66.8 ±7.8 | >1000 | >1000 | >1000 |
| SIPb | - | - | 1.4 ±0.3 | 3.6 ±0.5 | 0.4 ± 0.2 | 151 ±82 | 3.1 ± 1.1 |
| 2c | 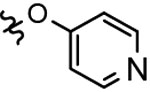 |
2-(ethyl)butyl | >10000 | 45 | > 10000 | > 10000 | > 10000 |
| 28a | 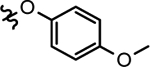 |
2-(ethyl)butyl | N.T. | 188.5 ±32.9 | N.T. | N.T. | N.T. |
| 28b | 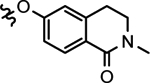 |
2-(ethyl)butyl | > 1000 | 73.3 ±10.6 | >1000 | >1000 | >1000 |
| 28c | 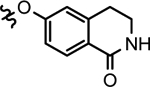 |
2-(ethyl)butyl | > 1000 | 29.9 ±3.9 | > 1000 | > 1000 | > 1000 |
| 28d | 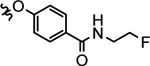 |
2-(ethyl)butyl | > 1000 | 66.7 ± 7.8 | > 1000 | > 1000 | > 1000 |
| 28e | 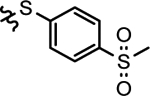 |
2-(ethyl)butyl | > 1000 | 14.6 ± 1.5 | > 1000 | > 1000 | > 1000 |
| 28f |  |
isobutyl | N.T. | 194.5 ±35.9 | N.T. | N.T. | N.T. |
| 28g | 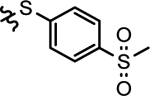 |
isobutyl | > 1000 | 38.5 ±6.3 | > 1000 | > 1000 | > 1000 |
Figure 3.

The representative competitive binding curves of compounds JTE-013, 28b, 28c, 28d, 28e, and 28g toward S1PR2. The averaged IC50 values were obtained from three independent experiments.
From the in vitro binding data shown in Table 1 and Table 2, the following structure-activity reationship information was generated: a) when the 5-position substitution groups were introduced ino the fragment A, the binding potency order of the substituents is aryloxy > CF3 > alkoxyl and N-containing heterocycles, the aryloxy group play an important role in regulating the S1PR2 binding ativity; b) when the 4’-position substitution groups were introduced into the fragment B, the stronger ectronegativity of substituents offered the higher S1PR2 binding activity with the order as SO2CH3 > ONHCH3 > F > CONH2, CON(CH3)2, COOCH3, OCH3; c) when optimizing the fragment C, comparing with different group, the piperidine ring was the most favorable heterocycle and the steric hindrance of side chains resulted in increased S1PR2 binding potency with the order as 2-(ethyl)butyl > isobutyl > isopropyl, ethyl, and methyl. The structure-activity relationship information of this scaffold is valuable for guiding the future design of new S1PR2 compounds.
Because compounds 28b, 28c, 28d, 28e, and 28g showed high S1PR2 binding potency with IC50 < 100 nM, their binding potencies toward the other S1P receptor subtypes, S1PR1, 3, 4, and 5 were also assessed to determine their in vitro binding selectivity for S1PR2. As shown in Table 2, all five compounds had no significant binding toward the other four subtypes S1PR1,3,4, and 5 (IC50 > 1000 nM), indicating that they are highly selective for S1PR2.
3. Conclusion
In summary, we successfully synthesized a series of ligands for S1PR2. The in vitro data suggested that compounds 28c, 28e, and 28g exhibit high S1PR2 binding potencies with IC50 values of 29.9, 14.6, and 38.5 nM, respectively. They are more potent for S1PR2 than compound JTE-013 and lead compounds 1 and 2. In addition, compounds 28c, 28e, and 28g also displayed high selectivity over S1PR 1, 3, 4, and 5. The initial structure-activity relationship analysis indicates that aryloxy substitution at 5-position of fragment A plays a key role in retaining the high potency for S1PR2; the strong electronegativity substituent at 4’-position of fragment B is favorable to the S1PR2 binding; the piperidine ring with steric hindrance side chain in fragment C also provides S1PR2 favorable binding. Three S1PR2 potent compounds 28c, 28e, and 28g contain fluorine atom that provides the position for introducing F-18 isotope to make the F-18 labeling counterparts. Further evaluation of the F-18 labeled radiotracer may lead to identify an F-18 PET radiotracer for imaging S1PR2 in vivo. Further characterizations of their in vitro and in vivo suitability to be PET radiotracers for assessing the S1PR2 expression in living animals are ongoing in our lab.
4. Experimental
4.1. Chemistry
Commercially available starting materials, reagents, and solvents were used as received. Unless otherwise indicated, all reactions were conducted in oven-dried glassware. In general, anhydrous reactions were performed under nitrogen. Reactions were monitored by thin-layer chromatography (TLC) carried out on pre-coated glass plates of silica gel (0.25 mm) 60 F254 from EMD Chemicals Inc. Visualization was accomplished with ultraviolet light (UV 254 nm), or by shaking the TLC plate in a sealed jar containing silica gel and iodine. Flash column chromatography was performed using 230–400 mesh silica gel purchased from Silicycle. All work-up and purification procedures were carried out with reagent grade solvents in the air. Yields refer to isolate yield unless otherwise stated. Melting points were determined on a MEL-TEMP 3.0 apparatus. 1H NMR and 13C NMR spectra were recorded on Varian 400 MHz instrument. Chemical shifts are reported in parts per million (ppm) and are calibrated using residual undeuterated solvent as an internal reference (CDCl3: δ 7.26 ppm; CD3OD: δ 3.31 ppm; DMSO: δ 2.50 ppm). Data are reported as follows: chemical shift, multiplicity, coupling constants (Hz), and integration. High resolution positive ion mass (HRMS) analyses were conducted on a Bruker MaXis 4G Q-TOF mass spectrometer with electrospray ionization source.
4.1.1. Synthesis of 4-(2-ethylbutyl)piperidin-4-ol (6)
To a dried round two neck bottomed flask equipped with a magnetic stir bar were added 0.6 M LaCl3-2LiCl in THF (12.5 mL, 7.5 mmol) under nitrogen, 0.25 M 2-ethylbutylmagnesium bromide in THF (30 mL, 7.5 mmol) was added slowly through syringe at 0 °C. After stirring at room temperature for 3 h, a solution of benzyl 4-oxopiperidine-1-carboxylate (5) (1.2 g, 5.0 mmol) in THF (5.0 mL) was added into the mixture. The reaction was stirred for another18 h until the reaction was completed as determined by TLC and then quenched with 25% acetic acid. The mixture was extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was used directly for the next step without purification.
To a round-bottomed flask equipped with a magnetic stir bar were added above crude product, 10% Pd/C (0.2 g) and methanol (10.0 mL). The reaction was bubbled with hydrogen gas for 6 h at room temperature until the reaction was completed as determined by TLC and then filtered through celite. The filtrate was concentrated under reduced pressure to afford yellow oil product 6. Yield: 81%. 1H NMR (400 MHz, CDCl3) δ 3.18 (s, 1H), 2.98 (d, J = 33.4 Hz, 4H), 1.96 (s, 1H), 1.63 (d, J = 24.9 Hz, 4H), 1.46 – 1.30 (m, 7H), 0.86 (t, J = 7.1 Hz, 6H).
4.1.2. General procedure to synthesize 8a-b.
A solution of 2-fluoroethanol or 3-fluoropyopan-1-ol (1.0 eq) in DMF (0.25 M) was stirred and cooled to 0 °C, then NaH (2.0 eq) was added. After stirring for 15 min, a solution of 1,3-difluoro-5-nitrobenzene (7) (1.0 eq) in DMF (0.5 M) was added to the mixture and stirred at room temperature for 12 h until the reaction was completed as determined by TLC. The reaction then was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 8a-b.
4.1.2.1. 1-Fluoro-3-(2-fluoroethoxy)-5-nitrobenzene (8a).
Compound 8a was eluted with hexane/ethyl acetate (10/1, V/V) as yellow oil. Yield: 33%. 1H NMR (400 MHz, CDCl3) δ 7.60 – 7.55 (m, 2H), 6.99 (dt, J = 9.6, 2.3 Hz, 1H), 4.87 – 4.83 (m, 1H), 4.75 – 4.71 (m, 1H), 4.35 – 4.31 (m, 1H), 4.29 – 4.25 (m, 1H).
4.1.2.2. 1-Fluoro-3-(3-fluoropropoxy)-5-nitrobenzene (8b)
Compound 8b was eluted with hexane/ethyl acetate (10/1, V/V) as yellow oil. Yield: 35%. 1H NMR (400 MHz, CDCl3) δ 7.58 – 7.56 (m, 1H), 7.55 – 7.50 (m, 1H), 6.98 – 6.92 (m, 1H), 4.71 (t, J = 5.7 Hz, 1H), 4.59 (t, J = 5.7 Hz, 1H), 4.17 (t, J = 6.1 Hz, 2H), 2.29 – 2.14 (m, 2H).
4.1.3. General procedure to synthesize 9a-b.
To a round-bottomed flask equipped with a magnetic stir bar was added 8a–b (1.0 eq), 4-fluorophenol (1.2 eq), potassium phosphate (2.0 eq), and DMA (1.0 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was pure enough for the next step.
4.1.3.1. 1-(2-Fluoroethoxy)-3-(4-fluorophenoxy)-5-nitrobenzene (9a).
Yellow oil, yield: 99%. 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 2.1 Hz, 1H), 7.37 (t, J = 2.1 Hz, 1H), 7.14 – 6.99 (m, 4H), 6.83 (t, J = 2.3 Hz, 1H), 4.83 – 4.79 (m, 1H), 4.71 – 4.68 (m, 1H), 4.31 – 4.26 (m, 1H), 4.24 – 4.20 (m, 1H).
4.1.3.2. 1-(4-Fluorophenoxy)-3-(3-fluoropropoxy)-5-nitrobenzene (9b).
Yellow oil, yield: 91%. 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.34 (s, 1H), 7.14 – 6.98 (m, 4H), 6.80 (s, 1H), 4.70 (t, J = 5.9 Hz, 1H), 4.58 (t, J = 5.9 Hz, 1H), 4.19 – 4.11 (m, 2H), 2.25 – 2.14 (m, 2H).
4.1.4 General procedure to synthesize 10a-b.
To a round-bottomed flask equipped with a magnetic stir bar were added 9a-b (1.0 eq), 10% Pd/C, and ethyl acetate (0.05 M). The reaction was bubbled with hydrogen gas for 12 h at room temperature until the reaction was completed as determined by TLC. The mixture then was filtered through celite. To the filtrate was added NaHCO3 (2.0 eq) followed by adding 2,2,2-trichloroethyl chloroformate (1.0 eq) slowly through syringe under nitrogen at 0 °C. The reaction was warmed to room temperature and stirred for 3 h until the reaction was completed as determined by TLC. The mixture then was washed with water, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was used directly for the next step without further purification.
4.1.5. General procedure to synthesize 11a-b.
To a round-bottomed flask equipped with a magnetic stir bar were added 10a–b (1.0 eq), 6 (1.2 eq), DIPEA (2.0 eq), and DMA. The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction mixture was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with 1 N HCl, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 11a-b.
4.1.5.1. 4-(2-Ethylbutyl)-N-(3-(2-fluoroethoxy)-5-(4-fluorophenoxy)phenyl)-4-hydroxypiperidine-1-carboxamide (11a).
Compound 11a was eluted with hexane/ethyl acetate (2/3, V/V) as yellow oil. Yield: 97%. 1H NMR (400 MHz, CDCl3) δ 7.08 – 6.74 (m, 5H), 6.57 (s, 1H), 6.51 (s, 1H), 6.21 (s, 1H), 4.78 – 4.72 (m, 1H), 4.67 – 4.60 (m, 1H), 4.22 – 4.16 (m, 1H), 4.15 – 4.09 (m, 1H), 3.83 – 3.72 (m, 2H), 3.35 – 3.22 (m, 2H), 1.64 – 1.57 (m, 4H), 1.42 – 1.31 (m, 8H), 0.85 (t, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.93 (d, J = 243.4 Hz), 159.90, 159.04, 154.32, 152.31 (d, J = 2.0 Hz), 141.31, 120.89 (d, J = 8.1 Hz), 116.25 (d, J = 24.2 Hz), 102.18, 100.41, 99.87, 81.74 (d, J = 171.7 Hz), 70.17, 67.23 (d, J = 21.2 Hz), 46.74, 40.59, 37.00, 35.37, 27.30, 10.81. HRMS (ESI) m/z [M + H]+ calcd. for C26H35F2N2O4 477.2559, found 477.2550.
4.1.5.2. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-(3-fluoropropoxy)phenyl)-4-hydroxypiperidine-1-carboxamide (11b).
Compound 11b was eluted with hexane/ethyl acetate (1/1, V/V) as yellow oil. Yield: 98%, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.10 – 6.95 (m, 4H), 6.92 – 6.89 (m, 1H), 6.47 (s, 1H), 6.36 (s, 1H), 6.19 (s, 1H), 4.66 (t, J = 5.8 Hz, 1H), 4.54 (t, J = 5.8 Hz, 1H), 4.04 (t, J = 6.1 Hz, 2H), 3.78 (d, J = 12.8 Hz, 2H), 3.33 – 3.24 (m, 2H), 2.18 – 2.06 (m, 2H), 1.63 – 1.58 (m, 4H), 1.41 – 1.34 (m, 7H), 1.08 (s, 1H), 0.85 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 160.28, 158.93, 158.85 (d, J = 242.4 Hz), 154.47, 152.45 (d, J = 2.0 Hz), 141.37, 120.77 (d, J = 8.1 Hz), 116.21 (d, J = 23.2 Hz), 101.95, 100.60, 99.58, 80.65 (d, J = 164.6 Hz), 70.20, 63.63 (d, J = 6.1 Hz), 46.75, 40.44, 37.05, 35.35, 30.26 (d, J = 20.2 Hz), 27.29, 10.81. HRMS (ESI) m/z [M + H]+ calcd. for C27H37F2N2O4 491.2716, found 491.2709.
4.1.6. 1-(4-Fluorophenoxy)-3-nitro-5-(trifluoromethyl)benzene (13).
To a round-bottomed flask equipped with a magnetic stir bar was added 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (2.5 g, 12 mmol), 4-fluorophenol (12) (1.1 g, 10 mmol), potassium phosphate (4.2 g, 20 mmol), and DMA (15 mL). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate (50/1, V/V) to afford 13. Yield: 93%. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.89 (t, J = 2.1 Hz, 1H), 7.52 (s, 1H), 7.21 – 7.12 (m, 2H), 7.12 – 7.05 (m, 2H).
4.1.7. Synthesis of 1-fluoro-3-(4-fluorophenoxy)-5-nitrobenzene (14).
To a round-bottomed flask equipped with a magnetic stir bar were added 1,3-difluoro-5-nitrobenzene (7) (8.0 g, 50 mmol), 4-fluorophenol (12) (6.2 g, 55 mmol), Cs2CO3 (17.9 g, 55 mmol), and DMA (80 mL). The reaction vessel was immersed in a 65 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was pure enough for the next step. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.1 Hz, 1H), 7.54 (s, 1H), 7.13 – 7.03 (m, 4H), 6.96 (d, J = 9.2 Hz, 1H).
4.1.8. General procedure to synthesize 15a-b.
To a round-bottomed flask equipped with a magnetic stir bar was added 14 (1.0 eq), 1-methylpiperazine or 1H-pyrazole (1.0 eq), K2CO3 (1.0 eq), and DMSO (2.0 M). The reaction vessel was immersed in a 65 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 15a–b.
4.1.8.1. 1-(3-(4-Fluorophenoxy)-5-nitrophenyl)-4-methylpiperazine (15a).
Compound 15a was eluted with hexane/ethyl acetate (1/5, V/V) as yellow oil. Yield: 42%. 1H NMR (400 MHz, CDCl3) δ 7.46 (s, 1H), 7.14 – 6.97 (m, 5H), 6.79 (s, 1H), 3.34 – 3.22 (m, 4H), 2.61 – 2.51 (m, 4H), 2.36 (s, 3H).
4.1.8.2. 1-(3-(4-Fluorophenoxy)-5-nitrophenyl)-1H-pyrazole (15b).
Compound 15b was eluted with hexane/ethyl acetate (5/1, V/V) as yellow semi-solid. Yield: 53%. 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.98 (d, J = 2.4 Hz, 1H), 7.73 (d, J = 12.4 Hz, 2H), 7.62 (s, 1H), 7.18 – 7.05 (m, 4H), 6.53 (s, 1H).
4.1.9. General procedure to synthesize 16a-c.
To a round-bottomed flask equipped with a magnetic stir bar were added 13 or 15a–b (1.0 eq), 10% Pd/C, and ethyl acetate (0.2 M). The reaction was bubbled with hydrogen gas for 12 h at room temperature until the reaction was completed as determined by TLC. The mixture then was filtered through celite. To the filtrate was added NaHCO3 (2.0 eq) followed by adding 2,2,2-trichloroethyl chloroformate (1.0 eq) slowly through syringe under nitrogen at 0 °C. The reaction was warmed to room temperature and stirred for 3 h until the reaction was completed as determined by TLC. Then, the mixture was washed with water, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was used directly for the next step without further purification.
4.1.10. General procedure to synthesize 17a-c.
To a round-bottomed flask equipped with a magnetic stir bar were added 16a–c (1.0 eq), 6 (1.2 eq), DIPEA (2.0 eq) and DMA (0.5 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column to afford 17a–c.
4.1.10.1. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-(4-methylpiperazin-1-yl)phenyl)-4-hydroxypiperidine-1-carboxamide (17a).
Compound 17a was eluted with ethyl acetate/methanol (5/1, V/V) as yellow oil. Yield: 20%, 1H NMR (400 MHz, CDCl3) δ 7.00 – 6.91 (m, 5H), 6.45 (s, 1H), 6.32 (s, 1H), 6.21 (s, 1H), 3.80 – 3.71 (m, 2H), 3.31 – 3.20 (m, 2H), 3.16 (s, 4H), 2.50 (s, 4H), 2.31 (s, 3H), 1.65 – 1.56 (m, 4H), 1.40 – 1.25 (m, 8H), 0.83 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 158.69, 158.68 (d, J = 242.4), 154.78, 153.02 (d, J = 2.02), 152.93, 141.33, 120.40 (d, J = 8.08), 116.19 (d, J = 23.2), 102.20, 100.93, 100.78, 70.22, 54.99, 48.58, 46.86, 46.11, 40.54, 37.14, 35.42, 27.37, 10.92. HRMS (ESI) m/z [M + H]+ calcd. for C29H42FN4O3 513.3235, found 513.3226.
4.1.10.2. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-(1H-pyrazol-1-yl)phenyl)-4-hydroxypiperidine-1-carboxamide (17b).
Compound 17b was eluted with hexane/ethyl acetate (2/1, V/V) as yellow semi-solid. Yield: 30%, 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 7.65 (s, 1H), 7.50 (s, 1H), 7.08 – 6.90 (m, 6H), 6.80 (s, 1H), 6.41 (s, 1H), 3.85 – 3.70 (m, 2H), 3.32 – 3.21 (m, 2H), 1.57 (s, 4H), 1.42 – 1.30 (m, 7H), 1.26 (s, 1H), 0.84 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.17 (d, J = 243.4 Hz), 159.12, 154.56, 152.32 (d, J = 3.0 Hz), 141.87, 141.52, 141.21, 127.21, 121.03 (d, J = 8.8 Hz), 116.54 (d, J = 23.2 Hz), 107.82, 107.19, 105.13, 103.44, 70.31, 46.89, 40.58, 37.18, 35.47, 27.43, 10.96. HRMS (ESI) m/z [M + H]+ calcd. for C27H34FN4O3 481.2609, found 481.2603.
4.1.10.3. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-(trifluoromethyl)phenyl)-4-hydroxypiperidine-1-carboxamide (17c).
Compound 17c was eluted with hexane/ethyl acetate (2/1, V/V) as yellow solid. Yield: 44%, M.P. 108 – 111 °C. 1H NMR (400 MHz, CDCl3) δ 7.23 (d, J = 21.6 Hz, 2H), 7.04 – 6.84 (m, 5H), 6.75 (s, 1H), 3.84 – 3.67 (m, 2H), 3.32 – 3.12 (m, 2H), 1.81 (s, 1H), 1.62 – 1.44 (m, 4H), 1.37 – 1.21 (m, 7H), 0.90 – 0.72 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 159.18 (d, J = 244.4 Hz), 158.54, 154.26, 151.73, 141.51, 132.12 (d, J = 32.3 Hz), 123.55 (d, J = 273.7 Hz), 121.04 (d, J = 8.1 Hz), 116.53 (d, J = 23.2 Hz), 112.04, 110.77, 108.63, 70.14, 46.73, 40.43, 37.01, 35.32, 27.27, 10.78. HRMS (ESI) m/z [M + Na]+ calcd. for C25H30F4N2NaO3 505.2085, found 505.2078.
4.1.11. General procedure to synthesize 19a-f.
To a round-bottomed flask equipped with a magnetic stir bar was added 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.0 eq), 4-substituted phenol (1.0 eq), potassium phosphate (2 eq), and DMA (2.0 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 19a–f.
4.1.11.1. 1-(4-Methoxyphenoxy)-3-nitro-5-(trifluoromethyl)benzene (19a).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.0 g, 4.8 mmol) with 4-methoxyphenol (0.5 g, 4.0 mmol) yielded 19a (1.4 g, 99%), eluted with hexane/ethyl acetate (20/1, V/V). 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.86 (s, 1H), 7.50 (s, 1H), 7.10 – 6.92 (m, 5H), 3.85 (s, 3H).
4.1.11.2. 4-(3-Nitro-5-(trifluoromethyl)phenoxy)benzamide (19b).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.1 g, 5.0 mmol) with 4-hydroxybenzamide (0.7 g, 5.0 mmol) yielded 19b (1.6 g, 99%), eluted with hexane/ethyl acetate (1/2, V/V). 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.93 (s, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.53 (s, 1H), 7.08 (d, J = 8.8 Hz, 2H).
4.1.11.3. N-methyl-4-(3-nitro-5-(trifluoromethyl)phenoxy)benzamide (19c).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.8 g, 8.7 mmol) with 4-hydroxy-N-methylbenzamide (1.1 g, 7.3 mmol) yielded 19c (2.5 g, 99%), eluted with hexane/ethyl acetate (1/2, V/V). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 7.97 (s, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.58 (s, 1H), 7.13 (d, J = 8.9 Hz, 2H), 6.13 (s, 1H), 3.04 (d, J = 4.9 Hz, 3H).
4.1.11.4. N, N-dimethyl-4-(3-nitro-5-(trifluoromethyl)phenoxy)benzamide (19d).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.1 g, 5.0 mmol) with 4-hydroxy-N,N-dimethylbenzamide (0.8 g, 5.0 mmol) yielded 19d (1.8 g, 99%), eluted with hexane/ethyl acetate (1/1, V/V). 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 7.98 (s, 1H), 7.60 – 7.51 (m, 3H), 7.15 – 7.08 (m, 2H), 3.09 (s, 6H).
4.1.11.5. Methyl 4-(3-nitro-5-(trifluoromethyl)phenoxy)benzoate (19e).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (1.1 g, 5.0 mmol) with methyl- 4-hydroxybenzoate (0.8 g, 5.0 mmol) yielded 19e (1.0 g, 60%), eluted with hexane/ethyl acetate (10/1, V/V). 1H NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 8.13 (d, J = 8.2 Hz, 2H), 8.00 (s, 1H), 7.60 (s, 1H), 7.12 (d, J = 8.9 Hz, 2H), 3.94 (s, 3H).
4.1.11.6. 1-(4-(Methylsulfonyl)phenoxy)-3-nitro-5-(trifluoromethyl)benzene (19f).
Coupling of 1-fluoro-3-nitro-5-(trifluoromethyl)benzene (18) (2.5 g, 12.0 mmol) with 4-(methylsulfonyl)phenol (1.7 g, 10.0 mmol) yielded 19f (2.8 g, 78%), eluted with hexane/ethyl acetate (3/2, V/V). 1H NMR (400 MHz, CDCl3) δ 8.33 – 8.30 (m, 1H), 8.07 – 8.01 (m, 3H), 7.67 – 7.63 (m, 1H), 7.26 – 7.20 (m, 2H), 3.11 (s, 3H).
4.1.12. General procedure to synthesize 20a-f.
To a round-bottomed flask equipped with a magnetic stir bar were added 19a-f (1.0 eq), 10% Pd/C and ethyl acetate (0.2 M). The reaction was bubbled with hydrogen gas for 12 h at room temperature until the reaction was completed as determined by TLC. The mixture then was filtered through celite. To the filtrate was added NaHCO3 (2.0 eq) followed by adding 2,2,2-trichloroethyl chloroformate (1.0 eq) slowly through syringe under nitrogen at 0 °C. The reaction was warmed to room temperature and stirred for 3 h until the reaction was completed as determined by TLC. Then, the mixture was washed with water, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was used directly for the next step without further purification.
4.1.13. General procedure to synthesize 21a-f.
To a round-bottomed flask equipped with a magnetic stir bar were added 20a-f (1.0 eq), 6 (1.2 eq), DIPEA (2.0 eq), and DMA (1.0 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column to afford 21a–f.
4.1.13.1. 4-(2-Ethylbutyl)-4-hydroxy-N-(3-(4-methoxyphenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (21a).
Coupling of 20a (229 mg, 0.5 mmol) with 6 (86 mg, 0.6 mmol) in the presence of DIPEA afforded 21a (200 mg, 81%), eluted with hexane/ethyl acetate (1/1, V/V). White solid, M.P. 79 – 81 °C. 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.15 (s, 1H), 6.99 (d, J = 9.0 Hz, 2H), 6.90 (d, J = 9.0 Hz, 2H), 6.82 (s, 1H), 6.46 (s, 1H), 3.90 – 3.83 (m, 2H), 3.81 (s, 3H), 3.32 – 3.22 (m, 2H), 1.65 – 1.58 (m, 5H), 1.35 – 1.18 (m, 6H), 1.04 (s, 1H), 0.93 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.35, 156.33, 154.18, 149.02, 141.31, 132.07 (d, J = 33.3 Hz), 123.60 (d, J = 266.6 Hz), 121.13, 115.01, 111.32, 110.19, 108.23, 71.48, 55.57, 45.60, 40.34, 38.02, 33.75, 28.06, 11.34. HRMS (ESI) m/z [M + Na]+ calcd. for C26H33F3N2NaO4 517.2290, found 517.2299.
4.1.13.2. N-(3-(4-Carbamoylphenoxy)-5-(trifluoromethyl)phenyl)-4-(2-ethylbutyl)-4-hydroxypiperidine-1-carboxamide (21b).
Coupling of 20b (236 mg, 0.5 mmol) with 6 (95 mg, 0.6 mmol) in the presence of DIPEA afforded 21b (100 mg, 39%), eluted with ethyl acetate. White solid, M.P. 98 – 101 °C. 1H NMR (400 MHz, DMSO) δ 8.84 (s, 1H), 7.94 (d, J = 8.3 Hz, 3H), 7.76 (s, 1H), 7.49 (s, 1H), 7.34 (s, 1H), 7.11 (d, J = 8.2 Hz, 2H), 6.92 (s, 1H), 3.85 – 3.70 (m, 2H), 3.14 (t, J = 11.6 Hz, 2H), 1.55 – 1.40 (m, 5H), 1.36 – 1.22 (m, 7H), 0.90 (d, J = 6.3 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 167.05, 158.32, 156.82, 154.08, 143.62, 130.52 (d, J = 32.3 Hz), 129.92, 129.83, 123.81 (d, J = 273.7 Hz), 118.38, 111.99, 110.55, 107.92, 68.38, 45.25, 42.06, 38.87, 34.95, 28.75, 11.30. HRMS (ESI) m/z [M + Na]+ calcd. for C26H32F3N3NaO4 530.2243, found 530.2251.
4.1.13.3. 4-(2-Ethylbutyl)-4-hydroxy-N-(3-(4-(methylcarbamoyl)-phenoxy)-5 (trifluoromethyl)phenyl)piperidine-1-carboxamide (21c).
Coupling of 20c (243 mg, 0.5 mmol) with 6 (112 mg, 0.6 mmol) in the presence of DIPEA afforded 21c (117 mg, 45%), eluted with hexane/ethyl acetate (1/7, V/V). White solid, M.P. 98 – 100 °C. 1H NMR (400 MHz, DMSO) δ 8.84 (s, 1H), 8.41 (d, J = 4.4 Hz, 1H), 7.89 (d, J = 8.6 Hz, 2H), 7.76 (s, 1H), 7.50 (s, 1H), 7.12 (d, J = 8.6 Hz, 2H), 6.91 (s, 1H), 3.79 (d, J = 13.1 Hz, 2H), 3.13 (t, J = 11.4 Hz, 2H), 2.78 (d, J = 4.4 Hz, 3H), 1.52 – 1.33 (m, 5H), 1.32 – 1.21 (m, 7H), 0.80 (t, J = 7.3 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 166.22, 158.58, 157.24, 154.48, 144.05, 130.94 (d, J = 31.3 Hz), 130.58, 129.77, 124.24 (d, J = 270.7 Hz), 118.85, 112.44, 110.97, 108.35, 68.87, 46.53, 37.13, 35.06, 31.09, 27.18, 26.67, 11.15. HRMS (ESI) m/z [M + Na]+ calcd. for C27H34F3N3NaO4 544.2394, found 544.2391.
4.1.13.4. N-(3-(4-(Dimethylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)-4-(2-ethylbutyl)-4-hydroxypiperidine-1-carboxamide (21d).
Coupling of 20d (250 mg, 0.5 mmol) with 6 (95 mg, 0.6 mmol) in the presence of DIPEA afforded 21d (180 mg, 67%), eluted with hexane/ethyl acetate (1/10, V/V). White solid, M.P. 82 – 84 °C. 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.56 (s, 1H), 7.33 (d, J = 6.0 Hz, 2H), 7.13 (s, 1H), 6.97 (d, J = 6.1 Hz, 2H), 6.89 (s, 1H), 3.90 – 3.65 (m, 2H), 3.30 – 2.86 (m, 8H), 1.56 – 1.44 (m, 6H), 1.40 – 1.30 (m, 5H), 1.25 (s, 1H), 0.93 (d, J = 5.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 171.43, 157.69, 157.31, 154.67, 142.48, 132.35 (d, J = 33.3 Hz), 131.41, 129.12, 123.80 (d, J = 273.7 Hz), 118.95, 112.28, 111.52, 109.57, 70.14, 45.07, 42.54, 37.27, 35.01, 34.62, 28.38, 11.62. HRMS (ESI) m/z [M + Na]+ calcd. for C28H36F3N3NaO4 558.2556, found 558.2661.
4.1.13.5. Methyl 4-(3-(4-(2-ethylbutyl)-4-hydroxypiperidine-1-carboxamido)-5-(trifluoromethyl)phenoxy)benzoate (21e).
Coupling of 20e (487 mg, 1.0 mmol) with 6 (189 mg, 1.2 mmol) in the presence of DIPEA afforded 21e (280 mg, 54%), eluted with hexane/ethyl acetate (1/1, V/V). White solid, M.P. 80 – 82 °C. 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 8.3 Hz, 2H), 7.42 (s, 1H), 7.00 (d, J = 8.3 Hz, 2H), 6.93 (s, 2H), 6.31 (s, 1H), 3.93 – 3.83 (m, 2H), 3.30 – 3.18 (m, 2H), 2.98 (s, 3H), 1.60 – 1.54 (m, 5H), 1.30 – 1.24 (m, 6H), 1.10 (s, 1H), 0.92 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 166.51, 160.64, 156.63, 154.08, 141.72, 132.49 (d, J = 33.3 Hz), 131.79, 125.27, 123.44 (d, J = 272.7 Hz), 117.92, 113.65, 111.88, 110.46, 70.13, 52.07, 45.97, 42.47, 37.10, 34.83, 27.28, 11.02. HRMS (ESI) m/z [M + Na]+ calcd. for C27H33F3N2NaO5 545.2239, found 545.2230.
4.1.13.6. 4-(2-Ethylbutyl)-4-hydroxy-N-(3-(4-(methylsulfonyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (21f).
Coupling of 20f (253 mg, 0.5 mmol) with 6e (112 mg, 0.6 mmol) in the presence of DIPEA afforded 21f (160 mg, 59%), eluted with hexane/ethyl acetate (1/2, V/V). White solid, M.P. 86 – 88 °C. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 7.9 Hz, 2H), 7.47 – 7.36 (m, 2H), 7.22 – 7.16 (m, 1H), 7.01 (d, J = 7.9 Hz, 2H), 6.85 (s, 1H), 3.83 – 3.65 (m, 2H), 3.25 – 3.10 (m, 2H), 2.98 (s, 3H), 1.56 – 1.37 (m, 5H), 1.36 – 1.19 (m, 7H), 0.76 (t, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 161.52, 155.62, 154.22, 142.24, 134.54, 132.48 (d, J = 32.3 Hz), 129.73, 123.39 (d, J = 273.7 Hz), 118.23, 114.22, 112.59, 110.56, 70.07, 46.71, 44.71, 40.42, 37.00, 35.29, 27.25, 10.79. HRMS (ESI) m/z [M + Na]+ calcd. for C26H33F3N2NaO5S 565.1954, found 565.1944.
4.1.14. General procedure to synthesize 22a-d and 24a-b.
To a dried round two neck bottomed flask equipped with a magnetic stir bar were added 0.6 M LaCl3-2LiCl in THF (1.5 eq) under nitrogen, Grignard reagents in THF (1.5 eq) was added slowly through syringe at 0 °C. After stirring at room temperature for 3 h, a solution of ketones (5 or 23a-b) (1.0 eq) in THF (1.0 M) was added into the mixture. The reaction was stirred for another18 h until the reaction was completed as determined by TLC and then quenched with 25% acetic acid. The mixture was extracted with ethyl acetate, the ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was used directly for the next step without purification.
To a round-bottomed flask equipped with a magnetic stir bar were added above crude product, 10% Pd/C and methanol (0.5 M). The reaction was bubbled with hydrogen gas for 6 h at room temperature until the reaction was completed as determined by TLC and then filtered through celite. The filtrate was concentrated under reduced pressure to afford yellow oil product 22a-d and 24a-b.
4.1.14.1. 4-Methylpiperidin-4-ol (22a).
Yield: 93%. 1H NMR (400 MHz, CDCl3) δ 5.86 (s, 1H), 3.03 – 2.87 (m, 2H), 2.48 – 2.34 (m, 2H), 1.68 – 1.55 (m, 4H), 1.26 (s, 3H).
4.1.14.2. 4-Ethylpiperidin-4-ol (22b).
Yield: 98%. 1H NMR (400 MHz, CDCl3) δ 5.82 (s, 1H), 3.00 – 2.86 (m, 2H), 2.44 – 2.23 (m, 2H), 1.72 – 1.39 (m, 7H), 0.95 – 0.80 (m, 3H).
4.1.14.3. 4-Isopropylpiperidin-4-ol (22c).
Yield: 97%. 1H NMR (400 MHz, CDCl3) δ 3.06 – 2.84 (m, 4H), 1.67 – 1.51 (m, 5H), 0.90 (d, J = 6.9 Hz, 6H).
4.1.14.4. 4-Isobutylpiperidin-4-ol (22d).
Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 3.44 (s, 1H), 3.38 – 3.17 (m, 4H), 1.88 – 1.76 (m, 4H), 1.48 – 1.40 (m, 2H), 1.24 – 1.17 (m, 2H), 0.95 (d, J = 6.1 Hz, 6H).
4.1.14.5. 3-Isobutylazetidin-3-ol (24a).
Yield: 93%. 1H NMR (400 MHz, CDCl3) δ 3.95 – 3.87 (m, 2H), 3.75 – 3.65 (m, 2H), 1.96 – 1.82 (m, 1H), 1.75 – 1.58 (m, 2H), 0.91 (d, J = 5.8 Hz, 6H).
4.1.14.6. 3-Isobutylpyrrolidin-3-ol (24b).
Yield: 92%. 1H NMR (400 MHz, CDCl3) δ 3.75 – 3.41 (m, 4H), 2.12 – 1.78 (m, 4H), 1.71 – 1.49 (m, 3H), 1.02 – 0.91 (m, 6H).
4.1.15. General procedure to synthesize 25a-j.
To a round-bottomed flask equipped with a magnetic stir bar were added 20c or 20f (1.0 eq), 22a-d or 24a-b (1.2 eq), DIPEA (2.0 eq), and DMA (1.0 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column to afford 25a–j.
4.1.15.1. 4-Hydroxy-4-methyl-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25a).
Coupling of 20c (243 mg, 0.5 mmol) with 22a (69 mg, 0.6 mmol) in the presence of DIPEA afforded 25a (160 mg, 62%), eluted with hexane/ethyl acetate (1/5, V/V). White solid, M.P. 103 – 106 °C. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.8 Hz, 2H), 7.50 (s, 1H), 7.40 (s, 1H), 7.27 (s, 1H), 7.12 (d, J = 8.7 Hz, 2H), 6.98 (s, 1H), 6.61 (s, 1H), 3.79 – 3.72 (m, 2H), 3.42 – 3.32 (m, 2H), 3.06 (s, 3H), 1.67 – 1.61 (m, 4H), 1.31 (s, 3H). 13C
NMR (101 MHz, DMSO) δ 165.80, 158.15, 156.81, 154.07, 143.60, 130.51 (d, J = 32.3 Hz), 130.16, 129.33, 123.79 (d, J = 273.7 Hz), 118.42, 112.05, 110.59, 107.92, 66.02, 40.34, 38.21, 29.70, 26.23. HRMS (ESI) m/z [M + Na]+ calcd. for C22H25F3N3O4 452.1792, found 452.1787.
4.1.15.2. 4-Hydroxy-4-methyl-N-(3-(4-(methylsulfonyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25b).
Coupling of 20f (253 mg, 0.5 mmol) with 22a (69 mg, 0.6 mmol) in the presence of DIPEA afforded 25b (160 mg, 68%), eluted with hexane/ethyl acetate (1/2, V/V). White solid, M.P. 94 – 96 °C. 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 7.4 Hz, 2H), 7.42 (s, 1H), 7.03 – 6.91 (m, 4H), 6.32 (s, 1H), 3.84 – 3.69 (m, 2H), 3.41 – 3.25 (m, 2H), 2.98 (s, 3H), 1.63 – 1.56 (m, 5H), 1.28 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.50, 155.71, 154.15, 142.11, 134.66, 132.59 (d, J = 32.3 Hz), 129.74, 123.37 (d, J = 273.7 Hz), 118.26, 114.14, 112.54, 110.71, 67.66, 44.67, 40.71, 38.25, 30.18. HRMS (ESI) m/z [M + H]+ calcd. for C21H24F3N2O5S 473.1353, found 473.1343.
4.1.15.3. 4-Ethyl-4-hydroxy-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25c).
Coupling of 20c (243 mg, 0.5 mmol) with 22b (78 mg, 0.6 mmol) in the presence of DIPEA afforded 25c (160 mg, 69%), eluted with ethyl acetate. White solid, M.P. 104 – 107 °C. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 8.5 Hz, 2H), 7.42 (s, 1H), 7.28 (s, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.94 (s, 1H), 6.84 (s, 1H), 6.24 (s, 1H), 3.86 – 3.76 (m, 2H), 3.35 – 3.24 (m, 2H), 2.99 (d, J = 4.8 Hz, 3H), 1.61 – 1.53 (m, 6H), 1.12 (s, 1H), 0.93 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 165.79, 158.14, 156.81, 154.06, 143.61, 130.50 (d, J = 32.3 Hz), 130.16, 129.33, 123.79 (d, J = 273.7 Hz), 118.42, 112.04, 110.61, 107.89, 67.83, 40.05, 35.85, 34.78, 26.22, 7.14. HRMS (ESI) m/z [M + H]+ calcd. for C23H27F3N3O4 466.1948, found 466.1940.
4.1.15.4. 4-Ethyl-4-hydroxy-N-(3-(4-(methylsulfonyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25d).
Coupling of 20f (253 mg, 0.5 mmol) with 22b (78 mg, 0.6 mmol) in the presence of DIPEA afforded 25d (240 mg, 99%), eluted with hexane/ethyl acetate (1/2, V/V). White solid, M.P. 89 – 91 °C. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.7 Hz, 2H), 7.50 (s, 1H), 7.41 (s, 1H), 7.12 (d, J = 8.7 Hz, 2H), 6.97 (s, 1H), 6.66 (s, 1H), 3.86 – 3.75 (m, 2H), 3.38 – 3.27 (m, 2H), 3.06 (s, 3H), 1.65 – 1.54 (m, 6H), 1.11 (s, 1H), 0.94 (t, J = 7.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 161.51, 155.68, 154.16, 142.17, 134.64, 132.56 (d, J = 33.3 Hz), 129.73, 123.38 (d, J = 273.7 Hz), 118.23, 114.15, 112.56, 110.68, 110.67, 44.67, 40.45, 36.04, 35.52, 6.96. HRMS (ESI) m/z [M + H]+ calcd. for C22H26F3N2O5S 487.1509, found 487.1502.
4.1.15.5. 4-Hydroxy-4-isopropyl-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25e).
Coupling of 20c (243 mg, 0.5 mmol) with 22c (86 mg, 0.6 mmol) in the presence of DIPEA afforded 25e (108 mg, 44%), eluted with ethyl acetate. White solid, M.P. 100 – 102 °C. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 11.8 Hz, 2H), 7.26 (s, 1H), 7.02 (d, J = 8.5 Hz, 2H), 6.95 (s, 1H), 6.52 (s, 1H), 3.91 (s, 3H), 3.83 – 3.76 (m, 2H), 3.38 – 3.28 (m, 2H), 1.90 – 1.80 (m, 1H), 1.65 – 1.62 (m, 2H), 1.45 – 1.41 (m, 2H), 1.06 (s, 1H), 0.98 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.00, 158.89, 157.12, 154.51, 141.94, 132.28 (d, J = 33.3 Hz), 130.07, 128.89, 123.53 (d, J = 374.7 Hz), 118.55, 112.60, 111.58, 109.88, 71.42, 40.25, 38.00, 33.77, 26.75, 16.33. HRMS (ESI) m/z [M + H]+ calcd. for C24H29F3N3O4 480.2105, found 480.2097.
4.1.15.6. 4-Hydroxy-4-isopropyl-N-(3-(4-(methylsulfonyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25f).
Coupling of 20f (253 mg, 0.5 mmol) with 22c (86 mg, 0.6 mmol) in the presence of DIPEA afforded 25f (200 mg, 80%), eluted with hexane/ethyl acetate (1/2;, V/V). White solid, M.P. 92 – 104 °C. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.8 Hz, 2H), 7.50 (s, 1H), 7.41 (s, 1H), 7.12 (d, J = 8.8 Hz, 2H), 6.97 (s, 1H), 6.63 (s, 1H), 3.91 – 3.84 (m, 2H), 3.32 – 3.23 (m, 2H), 3.06 (s, 3H), 1.66 – 1.58 (m, 5H), 1.06 (s, 1H), 0.93 (d, J = 6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 161.50, 155.71, 154.08, 142.14, 134.68, 132.59 (d, J = 33.3 Hz), 129.74, 123.38 (d, J = 271.7 Hz), 118.23, 114.12, 112.48, 110.69, 71.43, 44.67, 40.34, 38.02, 33.75, 16.36. HRMS (ESI) m/z [M + H]+ calcd. for C23H28F3N2O5S 501.1659, found 501.1666.
4.1.15.7. 4-Hydroxy-4-isobutyl-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25g).
Coupling of 20c (243 mg, 0.5 mmol) with 22d (94 mg, 0.6 mmol) in the presence of DIPEA afforded 25g (159 mg, 64%), eluted with hexane/ethyl acetate (1/7, V/V). White solid, M.P. 190 – 192 °C. 1H NMR (400 MHz, DMSO) δ 8.80 (s, 1H), 8.38 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.72 (s, 1H), 7.47 (s, 1H), 7.09 (d, J = 7.4 Hz, 2H), 6.88 (s, 1H), 3.85 – 3.65 (m, 2H), 3.20 – 3.00 (m, 2H), 2.75 (s, 3H), 2.47 (s, 1H), 1.85 – 1.70 (m, 1H), 1.52 – 1.21 (m, 6H), 0.87 (d, J = 5.5 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 166.21, 158.57, 157.24, 154.50, 147.82, 144.04, 140.77, 131.09, 130.58, 129.77, 118.86, 112.45, 109.69 (d, J = 267.7 Hz), 68.79, 51.67, 40.48, 37.29, 26.67, 25.37, 23.17. HRMS (ESI) m/z [M + Na]+ calcd. for C25H30F3N3NaO4 516.2081, found 516.2069.
4.1.15.8. 4-Hydroxy-4-isobutyl-N-(3-(4-(methylsulfonyl)phenoxy)-5-(trifluoromethyl)phenyl)piperidine-1-carboxamide (25h).
Coupling of 20f (253 mg, 0.5 mmol) with 22d (94 mg, 0.6 mmol) in the presence of DIPEA afforded 25h (210 mg, 82%), eluted with hexane/ethyl acetate (1/2;, V/V). White solid, M.P. 87 – 90 °C. 1H NMR (400 MHz, CDCl3) δ 7.90 (s, 2H), 7.50 (s, 1H), 7.42 (s, 1H), 7.12 (s, 2H), 6.97 (s, 1H), 6.72 (s, 1H), 3.87 – 3.72 (m, 2H), 3.41 – 3.24 (m, 2H), 3.06 (s, 3H), 1.94 – 1.70 (m, 1H), 1.71 – 1.40 (m, 7H), 1.07 – 0.90 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 161.52, 155.64, 154.20, 142.21, 134.58, 132.51 (d, J = 32.3 Hz), 129.73, 123.38 (d, J = 275.7 Hz), 118.22, 114.17, 112.55, 110.60, 70.03, 51.89, 44.67, 40.42, 37.05, 24.82, 23.24. HRMS (ESI) m/z [M + Na]+ calcd. for C24H29F3N2NaO5S 537.1641, found 537.1641.
4.1.15.9. 3-Hydroxy-3-isobutyl-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)azetidine-1-carboxamide (25i).
Coupling of 20c (243 mg, 0.5 mmol) with 24a (78 mg, 0.6 mmol) in the presence of DIPEA afforded 25i (110 mg, 47%), eluted with hexane/ethyl acetate (1/5, V/V). White solid, M.P. 107 – 109 °C. 1H NMR (400 MHz, DMSO) δ 8.80 (s, 1H), 8.42 (s, 1H), 7.90 (d, J = 7.4 Hz, 2H), 7.77 (s, 1H), 7.52 (s, 1H), 7.12 (d, J = 7.5 Hz, 2H), 6.92 (s, 1H), 3.95 – 3.65 (m, 4H), 2.78 (s, 3H), 1.91 – 1.77 (m, 1H), 1.65 – 1.45 (m, 2H), 1.22 (s, 1H), 0.88 (d, J = 5.3 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 166.22, 158.46, 157.45, 156.37, 143.47, 131.11 (d, J = 32.3 Hz), 130.68, 129.79, 124.19 (d, J = 273.7 Hz), 118.98, 111.63, 110.24, 108.36, 69.11, 63.86, 47.30, 26.67, 24.52, 23.50. HRMS (ESI) m/z [M + H]+ calcd. for C23H27F3N3O4 466.1948, found 466.1939.
4.1.15.10. 3-Hydroxy-3-isobutyl-N-(3-(4-(methylcarbamoyl)phenoxy)-5-(trifluoromethyl)phenyl)pyrrolidine-1-carboxamide (25j).
Coupling of 20c (243 mg, 0.5 mmol) with 24b (86 mg, 0.6 mmol) in the presence of DIPEA afforded 25j (100 mg, 42%), eluted with hexane/ethyl acetate (1/5, V/V). White solid, M.P. 113 – 116 °C. 1H NMR (400 MHz, DMSO) δ 8.48 (d, J = 45.8 Hz, 2H), 8.11 – 7.43 (m, 4H), 7.13 (s, 2H), 6.93 (s, 1H), 3.55 – 3.40 (m, 2H), 2.90 – 2.67 (m, 2H), 2.10 – 1.68 (m, 3H), 1.60 – 1.37 (m, 2H), 1.24 (s, 1H), 0.93 (s, 6H). 13C NMR (101 MHz, DMSO) δ 165.81, 158.17, 156.81, 153.43, 143.45, 130.52 (d, J = 32.3 Hz), 130.15, 129.35, 123.82 (d, J = 271.7 Hz), 118.41, 111.94, 110.49, 107.89, 58.16, 47.16, 45.37, 44.48, 26.25, 24.37, 24.18, 24.13. HRMS (ESI) m/z [M + H]+ calcd. for C24H29F3N3O4 480.2105, found 480.2098.
4.1.16. General procedure to synthesize 26a-e.
To a round-bottomed flask equipped with a magnetic stir bar was added 14 (1.0 eq), corresponding phenols (1.0 eq), K3PO4 (2.0 eq) and DMA (1.0 M). The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 26a–e.
4.1.16.1. 1-(4-Fluorophenoxy)-3-(4-methoxyphenoxy)-5-nitrobenzene (26a).
Compound 26a was eluted with hexane/ethyl acetate (10/1, V/V). Yield: 93%. 1H NMR (400 MHz, CDCl3) δ 7.34 (dd, J = 4.8, 2.1 Hz, 2H), 7.12 – 6.96 (m, 6H), 6.96 – 6.89 (m, 2H), 6.88 – 6.84 (m, 1H), 3.80 (s, 3H).
4.1.16.2. 6-(3-(4-Fluorophenoxy)-5-nitrophenoxy)-2-methyl-3,4-dihydroisoquinolin-1(2H)-one (26b).
Compound 26b was pure enough for next step. Yield: 99%. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.6 Hz, 1H), 7.49 (d, J = 12.3 Hz, 1H), 7.24 (s, 1H), 7.17 – 6.90 (m, 6H), 6.83 (s, 1H), 3.59 (t, J = 6.6 Hz, 2H), 3.16 (s, 3H), 3.00 (t, J = 6.6 Hz, 2H).
4.1.16.3. 6-(3-(4-Fluorophenoxy)-5-nitrophenoxy)-3,4-dihydroisoquinolin-1(2H)-one (26c).
Compound 26c was pure enough for next step. Yield: 40%. 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.74 – 7.35 (m, 2H), 7.19 – 6.78 (m, 6H), 6.65 (s, 1H), 3.74 – 3.47 (m, 2H), 3.11 – 2.84 (m, 2H).
4.1.16.4. N-(2-Fluoroethyl)-4-(3-(4-fluorophenoxy)-5-nitrophenoxy)benzamide (26d).
Compound 26d was eluted with hexane/ethyl acetate (3/2, V/V). Yield: 31%. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.7 Hz, 2H), 7.51 – 7.45 (m, 2H), 7.14 – 7.10 (m, 4H), 7.08 – 7.04 (m, 2H), 6.95 (t, J = 2.2 Hz, 1H), 6.48 (s, 1H), 4.68 (t, J = 4.7 Hz, 1H), 4.56 (t, J = 4.7 Hz, 1H), 3.83 (dd, J = 10.2, 5.1 Hz, 1H), 3.76 (dd, J = 10.2, 5.1 Hz, 1H).
4.1.16.5. (3-(4-Fluorophenoxy)-5-nitrophenyl)(4-(methylsulfonyl-)phenyl)sulfane (26e).
Compound 26e was eluted with hexane/ethyl acetate (2/1, V/V). Yield: 15%. 1H NMR (400 MHz, CDCl3) δ 7.92 – 7.88 (m, 3H), 7.65 (s, 1H), 7.47 (d, J = 8.3 Hz, 2H), 7.28 (s, 1H), 7.16 – 7.02 (m, 5H), 3.08 (s, 3H).
4.1.17. General procedure to synthesize 27a-e.
To a round-bottomed flask equipped with a magnetic stir bar were added 26a–e (1.0 eq), 10% Pd/C and ethyl acetate. The reaction was bubbled with hydrogen gas for 16 h at room temperature until the reaction was completed as determined by TLC. The mixture then was filtered through celite. To the filtrate was added NaHCO3 (2.0 eq) followed by adding 2,2,2-trichloroethyl chloroformate (1.0 eq) slowly through syringe under nitrogen at 0 °C. The reaction was warmed to room temperature and stirred for 3 h until the reaction was completed as determined by TLC. Then, the mixture was washed with water, saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was directly used for next step without purification.
4.1.17.1. 2,2,2-Trichloroethyl (3-(4-fluorophenoxy)-5-(4-methoxyphenoxy)phenyl)carbamate (27a).
Yield: 96%. 1H NMR (400 MHz, CDCl3) δ 7.05 – 6.97 (m, 6H), 6.90 – 6.87 (m, 2H), 6.79 (s, 1H), 6.76 (s, 1H), 6.67 (s, 1H), 6.32 (t, J = 2.2 Hz, 1H), 4.76 (s, 2H), 3.80 (s, 3H).
4.1.17.2. 2,2,2-Trichloroethyl (3-(4-fluorophenoxy)-5-((2-methyl-1-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)oxy)phenyl)carbamate (27b).
Yield: 96%, 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.5 Hz, 1H), 7.24 (s, 1H), 7.14 – 6.96 (m, 4H), 6.96 – 6.82 (m, 3H), 6.81 – 6.75 (m, 1H), 6.39 (s, 1H), 4.77 (s, 2H), 3.55 (t, J = 6.6 Hz, 2H), 3.14 (s, 3H), 2.96 (t, J = 6.5 Hz, 2H).
4.1.17.3. 2,2,2-Trichloroethyl (3-(4-fluorophenoxy)-5-((1-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)oxy)phenyl)carbamate (27c).
Yield: 95%, 1H NMR (400 MHz, CDCl3) δ 8.05 – 7.99 (m, 1H), 7.16 (s, 1H), 7.09 – 7.00 (m, 4H), 6.96 – 6.91 (m, 1H), 6.91 – 6.86 (m, 2H), 6.84 – 6.79 (m, 1H), 6.41 (s, 1H), 6.13 (s, 1H), 4.78 (s, 2H), 3.60 – 3.51 (m, 2H), 2.96 (t, J = 6.1 Hz, 2H).
4.1.17.4. 2,2,2-Trichloroethyl (3-(4-((2-fluoroethyl)carbamoyl)phenoxy)-5-(4-fluorophenoxy)phenyl)carbamate (27d).
Yield: 90%. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.7 Hz, 2H), 7.10 – 6.97 (m, 7H), 6.88 (s, 1H), 6.83 (s, 1H), 6.45 (s, 1H), 6.39 (t, J = 2.1 Hz, 1H), 4.77 (s, 2H), 4.66 (t, J = 4.7 Hz, 1H), 4.54 (t, J = 4.7 Hz, 1H), 3.81 (dd, J = 10.3, 5.0 Hz, 1H), 3.74 (dd, J = 10.2, 5.1 Hz, 1H).
4.1.17.5. 2,2,2-Trichloroethyl (3-(4-fluorophenoxy)-5-((4-(methylsulfonyl)phenyl)thio)phenyl)carbamate (27e).
Yield: 99%. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 7.12 (s, 1H), 7.08 – 6.98 (m, 6H), 6.90 (s, 1H), 4.79 (s, 2H), 3.04 (s, 3H).
4.1.18. General procedure to synthesize 28a-g.
To a round-bottomed flask equipped with a magnetic stir bar were added 27a-e (1.0 eq), 6 or 22d (1.2 eq), DIPEA (2.0 eq) and DMA. The reaction vessel was immersed in a 100 °C preheated oil bath for 12 h until the reaction was completed as determined by TLC. After cooling, the reaction was diluted with water and extracted with ethyl acetate. The ethyl acetate layer was washed with saturated brine and dried over anhydrous MgSO4. After filtration and concentration, the crude product was purified on a silica gel column, eluted with hexane/ethyl acetate to afford 28a–g.
4.1.18.1. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-(4-methoxyphenoxy)phenyl)-4-hydroxypiperidine-1-carboxamide (28a).
Compound 28a was eluted with hexane/ethyl acetate (1/1, V/V) as white solid. Yield: 52%, M.P. 79 – 80 °C. 1H NMR (400 MHz, CDCl3) δ 6.98 (tt, J = 10.2, 3.0 Hz, 6H), 6.88 – 6.83 (m, 2H), 6.75 (t, J = 2.0 Hz, 1H), 6.64 (t, J = 2.0 Hz, 1H), 6.57 (s, 1H), 6.25 (t, J = 2.2 Hz, 1H), 3.79 (s, 3H), 3.72 (d, J = 12.9 Hz, 2H), 3.31 – 3.21 (m, 2H), 1.59 (dd, J = 8.8, 3.8 Hz, 4H), 1.36 (dd, J = 15.0, 8.8 Hz, 7H), 0.84 (t, J = 7.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 160.07, 159.12, 158.91 (d, J = 243.4), 156.09, 154.05, 152.28 (d, J = 3.0), 149.38, 141.13, 121.07, 120.76 (d, J = 8.1), 116.26 (d, J = 23.2), 114.84, 103.44, 103.22, 102.33, 70.12, 55.60, 46.71, 40.74, 36.90, 35.36, 27.30, 10.82. HRMS (ESI) m/z [M + H]+ calcd. for C31H38FN2O5 537.2759, found 537.2751.
4.1.18.2. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-((2-methyl-1-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)oxy)phenyl)-4-hydroxypiperidine-1-carboxamide (28b).
Compound 28b was eluted with hexane/ethyl acetate (1/3, V/V) as white solid. Yield: 24%, M.P. 80 – 82 °C. 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 1H), 7.07 – 6.95 (m, 3H), 6.94 – 6.84 (m, 2H), 6.81 (s, 1H), 6.75 (s, 1H), 6.57 (s, 1H), 6.32 (s, 1H), 3.85 – 3.71 (m, 2H), 3.53 (t, J = 6.5 Hz, 2H), 3.34 – 3.20 (m, 2H), 3.12 (s, 3H), 2.94 (t, J = 6.5 Hz, 2H), 1.62 – 1.52 (m, 4H), 1.42 – 1.30 (m, 7H), 1.16 (s, 1H), 0.84 (t, J = 6.6 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 164.49, 159.90, 159.14, 158.92 (d, J = 243.4 Hz), 157.12, 154.47, 152.17 (d, J = 2.0 Hz), 142.20, 140.19, 130.13, 124.12, 120.82 (d, J = 8.1 Hz), 116.48, 116.28 (d, J = 23.2 Hz), 116.00, 105.62, 105.11, 103.78, 70.06, 47.98, 46.73, 40.38, 37.05, 35.28, 35.04, 27.91, 27.26, 10.80. HRMS (ESI) m/z [M + Na]+ calcd. for C34H40FN3NaO5 612.2844, found 612.2846.
4.1.18.3. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-((1-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)oxy)phenyl)-4-hydroxypiperi-dine-1-carboxamide (28c).
Compound 28c was eluted with ethyl acetate as white solid. Yield: 26%, M.P. 81 – 83 °C. 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.5 Hz, 1H), 7.07 – 6.96 (m, 3H), 6.96 – 6.89 (m, 1H), 6.88 – 6.77 (m, 2H), 6.56 (s, 1H), 6.33 (s, 1H), 5.95 (s, 1H), 3.85 – 3.71 (m, 2H), 3.61 – 3.48 (m, 2H), 3.34 – 3.18 (m, 2H), 2.99 – 2.87 (m, 2H), 1.65 – 1.52 (m, 5H), 1.44 – 1.31 (m, 7H), 1.15 (s, 1H), 0.84 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 166.00, 160.41, 159.24, 158.97 (d, J = 243.4 Hz), 157.03, 154.45, 152.13 (d, J = 2.0 Hz), 142.15, 141.14, 130.07, 123.60, 120.88 (d, J = 8.1 Hz), 116.52, 116.32 (d, J = 23.2 Hz), 116.30, 105.61, 105.10, 103.90, 70.13, 46.72, 40.41, 40.09, 37.06, 35.32, 28.43, 27.27, 10.80. HRMS (ESI) m/z [M + Na]+ calcd. for C33H38FN3NaO5 598.2688, found 598.2680.
4.1.18.4. 4-(2-Ethylbutyl)-N-(3-(4-((2-fluoroethyl)carbamoyl)phenoxy)-5-(4-fluorophenoxy)phenyl)-4-hydroxypiperidine-1-carboxamide (28d).
Compound 28d was eluted with hexane/ethyl acetate (7/3, V/V) as white solid. Yield: 88%, M.P. 79 – 82 °C. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 7.6 Hz, 2H), 7.14 – 6.93 (m, 6H), 6.90 (s, 1H), 6.86 – 6.76 (m, 3H), 6.31 (s, 1H), 4.68 – 4.46 (m, 2H), 3.84 – 3.62 (m, 4H), 3.26 – 3.12 (m, 2H), 1.58 – 1.44 (m, 4H), 1.38 – 1.22 (m, 8H), 0.88 – 0.78 (m, 6H). 13C NMR (101 MHz, DMSO) δ 166.16, 159.35, 158.84 (d, J = 241.39 Hz), 158.94, 157.41, 154.60, 152.46, 144.02, 129.81, 129.59, 121.50 (d, J = 8.1 Hz), 118.39, 117.00 (d, J = 24.2 Hz), 104.51, 104.03, 102.45, 82.60 (d, J = 166.7 Hz), 68.88, 46.52, 40.38, 37.18, 35.07, 27.18, 11.15. HRMS (ESI) m/z [M + Na]+ calcd. for C33H39F2N3NaO5 618.2750, found 618.2727.
4.1.18.5. 4-(2-Ethylbutyl)-N-(3-(4-fluorophenoxy)-5-((4-(methylsulfonyl)phenyl)thio)phenyl)-4-hydroxypiperidine-1-carboxamide (28e).
Compound 28e was eluted with hexane/ethyl acetate (2/3, V/V) as white solid. Yield: 27%, M.P. 78 – 80 °C. 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 7.21 (s, 1H), 7.14 (s, 1H), 7.05 – 6.93 (m, 4H), 6.72 (s, 1H), 6.59 (s, 1H), 3.85 – 3.70 (m, 2H), 3.24 – 3.15 (m, 2H), 3.02 (s, 3H), 1.65 – 1.53 (m, 4H), 1.44 – 1.28 (m, 7H), 1.14 (s, 1H), 0.84 (t, J = 6.7 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 159.06 (d, J = 243.4 Hz), 159.00, 154.25, 151.94 (d, J = 2.0 Hz), 145.85, 142.15, 137.33, 132.65, 127.90, 127.79, 120.92 (d, J = 9.1 Hz), 119.08, 116.86, 116.45 (d, J = 24.2 Hz), 110.06, 70.11, 46.72, 44.50, 40.39, 37.03, 35.31, 27.28, 10.82. HRMS (ESI) m/z [M + Na]+ calcd. for C31H37FN2NaO5S2 623.2020, found 623.2018.
4.1.18.6. N-(3-(4-((2-fluoroethyl)carbamoyl)phenoxy)-5-(4-fluorophenoxy)phenyl)-4-hydroxy-4-isobutylpiperidine-1-carboxamide (28f).
Compound 28f was eluted with hexane/ethyl acetate (1/2;, V/V) as white solid. Yield: 70%, M.P. 81 – 83 °C. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.2 Hz, 2H), 7.06 – 6.90 (m, 7H), 6.86 – 6.75 (m, 3H), 6.31 (s, 1H), 4.68 – 4.46 (m, 1H), 4.55 – 4.46 (m, 1H), 3.82 – 3.60 (m, 4H), 3.19 (t, J = 11.5 Hz, 2H), 1.87 – 1.74 (m, 1H), 1.60 – 1.46 (m, 4H), 1.39 – 1.28 (m, 3H), 0.93 (d, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 167.44, 159.72, 159.28, 158.99 (d, J = 243.4 Hz), 157.35, 154.55, 152.04 (d, J = 2.0 Hz), 142.05, 128.97, 128.89, 120.96 (d, J = 8.1 Hz), 118.10, 116.34 (d, J = 23.2 Hz), 105.01, 104.83, 103.55, 82.54 (d, J = 167.7 Hz), 70.03, 51.86, 40.54, 40.31, 37.06, 24.84, 23.22. HRMS (ESI) m/z [M + Na]+ calcd. for C31H35F2N3NaO5 590.2437, found 590.2430.
4.1.18.7. N-(3-(4-Fluorophenoxy)-5-((4-(methylsulfonyl)phenyl)-thio)phenyl)-4-hydroxy-4-isobutylpiperidine-1-carboxamide (28g).
Compound 28g was eluted with hexane/ethyl acetate (2/3, V/V) as white solid. Yield: 35%, M.P. 79 – 81 °C. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 5.6 Hz, 2H), 7.41 – 7.15 (m, 4H), 7.12 – 6.95 (m, 4H), 6.88 (s, 1H), 6.75 (s, 1H), 3.90 – 3.68 (m, 2H), 3.40 – 3.18 (m, 2H), 3.04 (s, 3H), 1.95 – 1.75 (m, 1H), 1.70 – 1.50 (m, 4H), 1.46 – 1.28 (m, 3H), 0.99 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 159.10 (d, J = 244.4 Hz), 159.07, 154.15, 151.93 (d, J = 3.0 Hz), 145.75, 141.99, 137.46, 132.85, 127.99, 127.83, 120.95 (d, J = 8.1 Hz), 118.96, 116.95, 116.48 (d, J = 23.2 Hz), 109.95, 70.13, 51.94, 44.53, 40.43, 37.10, 24.84, 23.27. HRMS (ESI) m/z [M + Na]+ calcd. for C29H33FN2NaO5S2 595.1707, found 595.1693.
4.2. Sphingosine-1-phosphate receptors binding assay
[32P]S1P was freshly prepared by following our previously published protocol26 and dissolved in DMSO, which was further diluted to 0.3–0.6 nM with assay buffer (50 mM HEPES-Na, pH 7.5, 5 mM MgCl2, 1 mM CaCl2, 0.5% fatty acid-free BSA). The test compounds were dissolved in DMSO and diluted into six different concentrations (0.03, 0.3, 3.0, 30, 300, and 3000 nM) with assay buffer. The commercial cell membranes expressing recombinant human S1PRs were diluted with assay buffer to make a 20–40 μg/mL of solution. To a 96-well plate was added 50 μL of cell membranes, 50 μL of test compounds, and 50 μL of [32P]S1P. Each well has a final volume of 150 μL containing 0.1–0.2 nM of [32P]S1P, 1–2 μg of membrane protein (S1PRs), and different concentrations (0.01–1000 nM) of test compounds. The plate was incubated for 60 min at room temperature with shaking and terminated by collecting the membranes onto 96-well glass fiber (GF/B) filtration plates (Millipore, Billerica, MA). Each filter was washed with 200 μL of assay buffer for a total of five washes. The filter bound radionuclide was measured by a Beckman LS 3801 scintillation counter using Cherenkov counting. The IC50 values were fitted from GraphPad Prism 6 using one site Nonlinear Regression.
Supplementary Material
Acknowledgements
This study was supported by the National Multiple Sclerosis Society [RG150705331]; National Institutes of Health [NS103988, NS075527, and EB025815]
Abbreviations
- S1P
sphingosine 1-phosphate
- S1PR
sphingosine 1-phosphate receptor
- CNS
central nervous system
- HTS
high throughput screening
- BBB
blood-brain barrier
- PET
positron emission tomography
- DMF
N,N-dimethylformamide
- DMA
N,N-dimethylacetamide
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide
- THF
tetrahydrofuran
- RT
room temperature
- TLC
thin-layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hla T, Physiological and pathological actions of sphingosine 1-phosphate, Semin Cell Dev Biol. 2004;15:513–520. 10.1016/j.semcdb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Hla T, Signaling and biological actions of sphingosine 1-phosphate, Pharmacol Res. 2003;47:401–407. 10.1016/S1043-6618(03)00046-X. [DOI] [PubMed] [Google Scholar]
- 3.Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL, The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis, J Biol Chem. 2004;279:29367–29373. 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 4.Spiegel S, Milstien S, Functions of a new family of sphingosine-1-phosphate receptors, Biochim Biophys Acta, Mol Cell Biol Lipids. 2000;1484:107–116. 10.1016/S1388-1981(00)00010-X. [DOI] [PubMed] [Google Scholar]
- 5.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S, Sphingosine 1-phosphate receptor signaling, Annu Rev Biochem. 2009;78:743–768. 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 6.Chun J, Goetzl EJ, Hla T, Igarashi Y, Lynch KR, Moolenaar W, Pyne S, Tigyi G, International Union of Pharmacology. XXXIV. Lysophospholipid Receptor Nomenclature, Pharmacol Rev. 2002;54:265–269. [DOI] [PubMed] [Google Scholar]
- 7.Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH, International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid Receptor Nomenclature, Pharmacol Rev. 2010;62:579–587. 10.1124/pr.110.003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T, Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN, Arterioscler Thromb Vasc Biol. 2007;27:1312–1318. 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 9.Imasawa T, Koike K, Ishii I, Chun J, Yatomi Y, Blockade of sphingosine 1-phosphate receptor 2 signaling attenuates streptozotocin-induced apoptosis of pancreatic beta-cells, Biochem Biophys Res Commun. 2010;392:207–211. 10.1016/j.bbrc.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, Dent P, Spiegel S, Shi R, Xu W, Liu X, Bohdan P, Zhang L, Zhou H, Hylemon PB, Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes, Hepatology. 2012;55:267–276. 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takashima S, Sugimoto N, Takuwa N, Okamoto Y, Yoshioka K, Takamura M, Takata S, Kaneko S, Takuwa Y, G12/13 and Gq mediate S1P2-induced inhibition of Rac and migration in vascular smooth muscle in a manner dependent on Rho but not Rho kinase, Cardiovasc Res. 2008;79:689–697. 10.1093/cvr/cvn118. [DOI] [PubMed] [Google Scholar]
- 12.Kimura A, Ohmori T, Kashiwakura Y, Ohkawa R, Madoiwa S, Mimuro J, Shimazaki K, Hoshino Y, Yatomi Y, Sakata Y, Antagonism of sphingosine 1-phosphate receptor-2 enhances migration of neural progenitor cells toward an area of brain, Stroke. 2008;39:3411–3417. 10.1161/STROKEAHA.108.514612. [DOI] [PubMed] [Google Scholar]
- 13.Akahoshi N, Ishizaki Y, Yasuda H, Murashima YL, Shinba T, Goto K, Himi T, Chun J, Ishii I, Frequent spontaneous seizures followed by spatial working memory/anxiety deficits in mice lacking sphingosine 1-phosphate receptor 2, Epilepsy Behav. 2011;22:659–665. 10.1016/j.yebeh.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT, Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury, J Am Soc Nephrol. 2012;23:266–280. 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, Qiang X, Sun L, Gurley EC, Lai G, Zhang L, Liang G, Nagahashi M, Takabe K, Pandak WM, Hylemon PB, Zhou H, The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice, Hepatology. 2017;65:2005–2018. 10.1002/hep.29076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R, Li X, Hylemon PB, Zhou H, Conjugated bile acids promote invasive growth of esophageal adenocarcinoma cells and cancer stem cell expansion via sphingosine 1-phosphate receptor 2–mediated Yes-associated protein activation, Am J Pathol. 2018;188:2042–2058. 10.1016/j.ajpath.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, Piccio L, Schmidt RE, Cross AH, Crosby SD, Klein RS, Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility, J Clin Invest. 2014;124:2571–2584. 10.1172/JCI73408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Lan T, Xie X, Huang K, Peng J, Huang J, Shen X, Liu P, Huang H, S1P2 receptor mediates sphingosine-1-phosphate-induced fibronectin expression via MAPK signaling pathway in mesangial cells under high glucose condition, Exp Cell Res. 2012;318:936–943. 10.1016/j.yexcr.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Osada M, Yatomi Y, Ohmori T, Ikeda H, Ozaki Y, Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist, Biochem Biophys Res Commun. 2002;299:483–487. 10.1016/S0006-291X(02)02671-2. [DOI] [PubMed] [Google Scholar]
- 20.Satsu H, Schaeffer M- T, Guerrero M, Saldana A, Eberhart C, Hodder P, Cayanan C, Schurer S, Bhhatarai B, Roberts E, Rosen H, Brown SJ, A sphingosine 1-phosphate receptor 2 selective allosteric agonist, Bioorg Med Chem. 2013;21:5373–5382. 10.1016/j.bmc.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kusumi K, Shinozaki K, Kanaji T, Kurata H, Naganawa A, Otsuki K, Matsushita T, Sekiguchi T, Kakuuchi A, Seko T, Discovery of novel S1P2 antagonists. Part 1: Discovery of 1,3-bis(aryloxy)benzene derivatives, Bioorg Med Chem Lett. 2015;25:1479–1482. 10.1016/j.bmcl.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 22.Kusumi K, Shinozaki K, Yamaura Y, Hashimoto A, Kurata H, Naganawa A, Ueda H, Otsuki K, Matsushita T, Sekiguchi T, Kakuuchi A, Seko T, Discovery of novel S1P2 antagonists. Part 2: Improving the profile of a series of 1,3-bis(aryloxy)benzene derivatives, Bioorg Med Chem Lett. 2015;25:4387–4392. 10.1016/j.bmcl.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 23.Kusumi K, Shinozaki K, Yamaura Y, Hashimoto A, Kurata H, Naganawa A, Otsuki K, Matsushita T, Sekiguchi T, Kakuuchi A, Yamamoto H, Seko T, Discovery of novel S1P2 antagonists, part 3: Improving the oral bioavailability of a series of 1,3-bis(aryloxy)benzene derivatives, Bioorg Med Chem Lett. 2016;26:1209–1213. 10.1016/j.bmcl.2016.01.031. [DOI] [PubMed] [Google Scholar]
- 24.Yue X, Jin H, Liu H, Rosenberg AJ, Klein RS, Tu Z, A potent and selective C-11 labeled PET tracer for imaging sphingosine-1-phosphate receptor 2 in the CNS demonstrates sexually dimorphic expression, Org Biomol Chem. 2015;13:7928–7939. 10.1039/c5ob00951k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Z, Yue X, Yang H, Liu H, Klein RS, Tu Z, Design and synthesis of pyrazolopyridine derivatives as sphingosine 1-phosphate receptor 2 ligands, Bioorg Med Chem Lett. 2018;28:488–496. 10.1016/j.bmcl.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg AJ, Liu H, Tu Z, A practical process for the preparation of [32P]S1P and binding assay for S1P receptor ligands, Appl Radiat Isot. 2015;102:5–9. 10.1016/j.apradiso.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.