

Abstract
Background:
Oxidative stress exacerbates lower airway diseases including asthma and chronic obstructive pulmonary disease (COPD); however, its role in upper airway (sinonasal) chronic inflammatory disorders is less clear. Nuclear erythroid 2 p45-related factor (Nrf2) is an endogenous mechanism that upon activation invokes an antioxidant response pathway via nuclear translocation and upregulation of cytoprotective genes. We sought to determine whether deletion of Nrf2 enhances susceptibility to allergic sinonasal inflammation in vivo.
Methods:
Nrf2−/− mice were subjected to the ovalbumin (Ova)-induced murine model of rhinosinusitis and indices of sinonasal inflammation and epithelial barrier dysfunction were assessed.
Results:
We show that deletion of Nrf2 results in enhances indices of allergen-induced sinonasal inflammation including aggravated eosinophil accumulation and goblet cell hyperplasia. An exaggerated increase in epithelial derived inflammatory cytokines including interleukin 33 (IL-33) and thymic stromal lymphopoietin (TSLP) was observed in the nasal lavage fluid and sinonasal mucosal tissue of Nrf2−/− mice. Furthermore, Nrf2−/− mice showed heightened Ova-induced barrier dysfunction as measured by serum albumin accumulation in nasal lavage fluid of mice.
Conclusion:
These data show that the endogenous Nrf2 pathway limits Ova-induced sinonasal inflammation, epithelial derived inflammatory cytokine production, and epithelial barrier dysfunction in vivo and identify a potential therapeutic target in the management of allergic sinonasal inflammatory disorders. This is the first study to our knowledge which shows that Nrf2 regulates allergic inflammation in the sinonasal cavity in vivo.
Keywords: epithelial barrier dysfunction, interleukin-33, Nrf2, oxidative stress, sinonasal inflammation
The human airway plays an essential role in oxygen exchange and is subjected to airborne environmental, allergen, and infectious exposures. These agents incite oxidative stress either directly or indirectly through recruitment of inflammatory cells which release reactive oxygen species (ROS).1,2 Oxidative stress has been linked to disease severity of lower airway chronic inflammatory disorders such as asthma and COPD; however, its role in upper airway (sinonasal) chronic inflammatory disorders is less clear.1-3 Interestingly, increased expression of cytoprotective enzymes has been reported in chronic rhinosinusitis (CRS), suggesting that oxidative stress may play a role in CRS pathophysiology.4,5
The epithelial barrier plays an essential role in the airway as the interface between the external environment and underlying tissues of the body. This barrier is composed of a variety of cell junction proteins including adherens junctions such as epithelial cadherin (E-cadherin), and tight junctions including zonula occludens-1 (ZO-1), junctional adhesion molecule-A (JAM-A), and claudin family members.6,7 Adherens and tight junction proteins either directly comprise or aid in the reinforcement of intercellular epithelial interactions. Junction proteins thereby generate an intercellular barrier that limits paracellular transit of noxious or allergenic stimuli.
Epithelial barrier dysfunction is a proposed pathogenic mechanism in chronic inflammatory disorders involving multiple organ systems including asthma, Crohn’s disease, atopic dermatitis, and psoriasis.8-10 Barrier dysfunction has been hypothesized to lead to chronic exposure of the underlying interstitial tissue to environmental and allergic stimuli. This may in turn result in increased inflammatory mediator release, inflammatory cell accumulation, and tissue damage. Defective barrier integrity has been reported in allergic inflammation of the sinonasal tract and decreased epithelial barrier function has been hypothesized to exacerbate sinonasal inflammation.11,12
Nrf2 is an endogenous mechanism that upon activation invokes an antioxidant response pathway via nuclear translocation and upregulation of cytoprotective genes.6 Disruption of the Nrf2 pathway in mice has been reported to enhance susceptibility to ovalbumin (Ova)-induced asthma.13 Recent evidence suggests that activation of the Nrf2 pathway stabilizes the sinonasal epithelial barrier against multiple stimuli including house dust mite antigen, particulate matter, and cigarette smoke extract in vitro.6,14,15 Indeed, Nrf2 activation was found in these studies to reduce sinonasal barrier dysfunction as assessed by paracellular fluorescein isothiocyanate (FITC)-dextran transit as well as by maintenance of cell surface epithelial tight and adherens junction proteins.6,14,15 We therefore hypothesized that Nrf2 deletion may worsen Ova-induced sinonasal inflammation and epithelial barrier dysfunction in vivo and act as an endogenous regulator of upper airway sinonasal inflammation.
Materials and methods
Animals
All animal procedures were approved by the Johns Hopkins Institutional Animal Care and Use Committee. Mice with Nrf2-targeted gene ablation were purchased from Jackson Laboratory (Bar Harbor, ME) and were backcrossed to C57BL6/J Nrf2 wild-type mice for at least 5 generations. Four-week-old to 6-week-old male homozygous Nrf2−/− mice and littermate Nrf2+/+ mice were used in this study. Animals were housed in a specific pathogen-free facility with single ventilated cages and received an Ova-free diet.
Ova sensitization
Allergic sinonasal inflammation was induced as described.16 Briefly, mice were sensitized by intraperitoneal (IP) injection of 900 μg of Ova (grade V; Sigma Chemical Co., Saint Louis, MO) and 77 μL of Imject alum adjuvant (Thermo Scientific, Waltham, MA) in 103 μL of phosphate buffered saline (PBS). On day 7, mice received additional IP injections of 450 μg of Ova in 77 μL of alum adjuvant and 103 μL of PBS. Beginning at day 14, mice were anesthetized with isoflurane and challenged with 500 μg of Ova in 20 μL of PBS distributed equally between the 2 nostrils. Intranasal challenges were administered daily for 14 days.
Histopathology
After perfusion with 4% paraformaldehyde (PFA) mouse heads were fixed with 4% PFA, decalcified overnight in TBD2 (Thermo Scientific, Waltham, MA), equilibrated in sucrose, and cryoembedded in Tissue-Tek OCT (Sakura, Torrance, CA). Sectioned tissue was used for Alcian blue-nuclear fast red staining or immunofluorescence analysis. Immunofluorescence staining was performed on 12-μm sections. Sections were blocked and permeabilized overnight in 10% Normal donkey serum and 0.1% Triton X-100 in PBS. Antigen retrieval was performed by boiling sections in 10mM sodium citrate and 0.05% Tween-20. After incubation with primary antibodies overnight at 4°C. Claudin-18 (sc-17687) and eosinophil major basic protein (EMBP; sc-33938) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). E-cadherin was from Novus (NB120-11512, Littleton, CO), Krt5 was from Covance (PRB-160P, San Diego, CA). The slides were washed in PBS and then incubated with Alexa Fluor® 488/568 conjugated secondary antibodies (Invitrogen, Waltham, MA). Sections were then washed, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Waltham, MA). Confocal images were collected with a LSM700 confocal laser scanning microscope (Zeiss, Oberkochen, Germany). EMBP-positive cells in sections of nasal respiratory tissue were counted. Data were presented as mean ± standard error of the mean (SEM) from 5 images from 3 independent experiments.
RNA extraction and reverse transcription
Sinonasal mucosa was extracted from mice as described.17 Briefly, to harvest sinonasal tissue for messenger RNA (mRNA) extraction, the skull was bisected sagittally and septal and turbinate mucosa were removed and placed in 1 mL of RNAlater solution (Ambion, Inc, Foster City, CA). Total RNA was isolated from nasal mucosa homogenates with the RNeasy Mini kit (Qiagen, Hilden, Germany) using the manufacturer’s protocol. RNA was quantified spectrophotometrically and absorbance ratios at 260/280 nm were >1.80 for all samples further evaluated. Five-hundred nanograms (500 ng) of total RNA was reverse transcribed in a 20-μL volume with random hexamer primers (Invitrogen, Waltham, MA) and 20 U of RNase inhibitor (Applied Biosystems, Foster City, CA) using the Omniscript RT kit (Qiagen, Hilden, Germany) under conditions provided by the manufacturer. Real-time polymerase chain reaction (PCR) was performed using the Applied Biosystems StepOnePlus under standard cycling parameters (Foster City, CA). Expression levels of 18S (Mm04277571_s1), interleukin 33 (IL-33; Mm00505403_m1), and thymic stromal lymphopoietin (TSLP; Mm01157588_m1) were quantified in each sample in duplicate using the TaqMan gene expression system (Applied Biosystems, Foster City, CA). Amplicon expression of each target gene was normalized to its 18S RNA content and the level of expression of target mRNA was measured as the ΔCt, the difference in threshold cycles for each target and housekeeping gene. Relative changes in gene expression compared to wild-type vehicle control was calculated as 2−ΔΔCt.
Quantification of nasal lavage fluid proteins
Nasal lavages were collected by insertion of a flexible catheter into the opening of the pharynx and 500 μL of PBS was flushed through both nares as described.17 Nasal lavages were centrifuged at 6000 rpm (~3400g) for 10 minutes. Supernatants were then analyzed for concentrations of IL-33 and TSLP by ELISA (E-Biosciences, Waltham, MA). Lavage fluids were analyzed for serum albumin by Albumin Blue Fluorescent Assay Kit (Active Motif, Carlsbad, CA).
Statistical analysis
All values are shown as mean ± SEM. Eosinophil and goblet cell counts were compared by 1-way analysis of variance (ANOVA) followed by Tukey post hoc test. The expression of inflammatory mediator mRNA and protein levels in nasal lavage fluids between genetically matched groups were compared by employing the nonparametric Kruskal-Wallis H test followed by Dunn’s multiple comparisons test. Statistical significance was considered to be p < 0.05. Results were analyzed with the use of the statistical software Prism 6 (GraphPad, La Jolla, CA).
Results
Nrf2−/− mice show enhanced eosinophil accumulation in response to chronic Ova exposure
To test this hypothesis, we subjected Nrf2−/− mice to the Ova-induced model of allergic chronic sinonasal inflammation.16 While repeated intranasal Ova administration increased eosinophil accumulation in sinonasal tissue in Nrf2+/+ mice, the recruitment of eosinophils was significantly amplified in Nrf2−/− littermate mice (Fig. 1A,B). Nrf2−/− mice also showed an exacerbated increase in goblet cell hyperplasia in response to Ova exposure (Fig. 2A,B). Sinonasal epithelial remodeling was assessed via epithelial cell thickness and although increased in Nrf2+/+ and Nrf2−/− mice upon Ova administration, there was no significant difference between Nrf2+/+ and Nrf2−/− mice (Fig. 3).
FIGURE 1.

Nrf2−/− mice demonstrate exaggerated eosinophil accumulation in response to chronic Ova exposure (A) Immunofluorescence for EMBP (red) and Krt5 (green). White arrows indicate areas of increased eosinophil accumulation. (B) Eosinophil counts per millimeter of basal lamina from imaging of the entire nasal septum (n = 4 per group). ***p < 0.001, ****p < 0.0001. Data are represented as mean ± SEM. EMBP = eosinophil major basic protein; Krt5 = Keratin-5; Ova = ovalbumin; SEM = standard error of the mean.
FIGURE 2.

Nrf2−/− mice demonstrate exaggerated goblet cell hyperplasia in response to chronic Ova exposure. (A, B) Quantification of goblet cells per millimeter of basal lamina from Alcian blue-nuclear fast red stain for mucus glycoproteins (n = 4 per group). **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are represented as mean ± SEM. NS = not significant; SEM = standard error of the mean.
FIGURE 3.
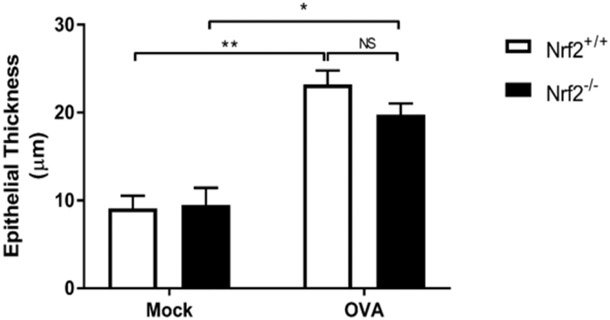
Epithelial thickness is significantly increased after Ova challenge to a similar degree in Nrf2+/+ and Nrf2−/− mice. Measurements were taken from an entire nasal septum from the basal lamina to the cilia (n = 4 per group). Ova = ovalbumin.
Epithelial-derived cytokines are expressed at an increased level in Ova-treated Nrf2−/− mice
Inflammatory cytokines and chemokines have been shown to drive chronic allergen-induced airway inflammation.18,19 Indeed, an activated and unstable epithelial barrier may also respond to allergic inflammation through inflammatory cytokine release. There is emerging evidence implicating epithelial derived inflammatory cytokines including IL-33 and TSLP in the pathogenesis of chronic sinonasal inflammation.20 We therefore analyzed nasal lavage samples for IL-33 and TSLP protein levels and found an exaggerated 12.76-fold increase in IL-33 and 32.38-fold increase in TSLP in Nrf2−/− compared to Nrf2+/+ mice (Fig. 4A,B). An increase in inflammatory cytokine expression levels was also observed in sinonasal mucosal tissue by qPCR (Fig. 4C,D).
FIGURE 4.

Nrf2−/− mice express increased epithelial derived cytokines. (A, B) Analysis of nasal lavage fluids for IL-33 and TSLP by ELISA (n = 4 to 6 per group). (C, D) mRNA expression levels of IL-33 and TSLP are increased in Nrf2−/− mice upon Ova stimulation (n = 6 to 8 per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are represented as mean ± SEM. ELISA = enzyme-linked immunosorbent assay; IL-33 = interleukin 33; mRNA = messenger RNA; NS = not significant; Ova = ovalbumin; SEM = standard error of the mean; TSLP = thymic stromal lymphopoietin.
Nrf2−/− mice show increased barrier dysfunction in response to chronic Ova exposure
Nasal lavage fluid samples were analyzed for serum albumin content, a measurement of chronic sinonasal barrier dysfunction. Although repeated intranasal Ova administration increased serum albumin content in nasal lavage fluid from Nrf2+/+ mice, a 1.75-fold increase in albumin content was observed upon chronic Ova administration in Nrf2−/− mice (Fig. 5).
FIGURE 5.
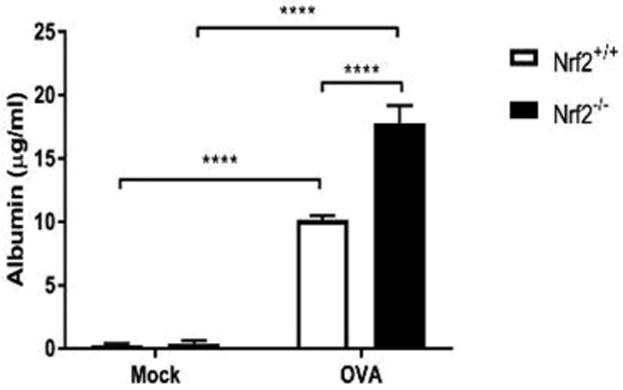
Nrf2−/− mice demonstrate exaggerated barrier dysfunction in response to chronic Ova exposure. Analysis of nasal lavage fluids for serum albumin via ELISA (n=4 to 5 per group). ELISA=enzyme-linked immunosorbent assay; Ova = ovalbumin.
Discussion
The human sinonasal tract is under constant exposure to airborne environmental, allergen, and infectious exposures.21,22 Oxidative stress has been linked to disease severity of lower airway chronic inflammatory disorders such as asthma and COPD; however, its role in upper airway (sinonasal) chronic inflammatory disorders is less clear.1,3 Furthermore, oxidative stress has been implicated in disruption of sinonasal barrier function.6,14,21 Nrf2 is known for its role in invoking an antioxidant response. The data presented here show that the Nrf2 pathway regulates Ova-induced sinonasal epithelial barrier dysfunction in vivo as loss of Nrf2 resulted in increased eosinophil accumulation, epithelial derived cytokine expression, goblet cell hyperplasia, and epithelial barrier breakdown (Fig. 6).
FIGURE 6.

Schematic representation. (A) Under normal conditions, Keap1 sequesters Nrf2 in the cytoplasm that can be released to invoke and antioxidant response. (B) When Nrf2 is deleted, exposure to allergens leads to multiple downstream effects including excessive sinonasal barrier dysfunction, epithelial inflammatory cytokine release, sinonasal inflammatory cell accumulation, and goblet cell hyperplasia.
Epithelial barrier dysfunction is a proposed pathogenic mechanism in chronic inflammatory disorders of multiple organ systems including asthma, CRS, Crohn’s disease, atopic dermatitis, and psoriasis.8-10 Barrier dysfunction may lead to chronic exposure of the underlying interstitial tissue to environmental and allergic stimuli, which may in turn result in increased inflammatory mediator release, inflammatory cell accumulation, and tissue damage. Although barrier dysfunction has been proposed to play a pathogenic role in sinonasal inflammation, the identification of pathways such as Nrf2 that can stabilize this barrier and test this hypothesis have been lacking. In addition to simply forming a barrier, sinonasal epithelial cells respond to inflammatory stimuli through inflammatory cytokine release. Our studies reported here in Nrf2−/− mice identified specifically a heightened increase in inflammatory cytokines regarded as epithelial-derived, namely IL-33 and TSLP, in response to Ova exposure. These results combined with the exaggerated barrier dysfunction likely place the sinonasal epithelium as the key area of dysregulation in the Nrf2 pathway. However, the mechanism of barrier destabilization has yet to be ascertained. One possibility, given the known role of Nrf2 in generating an antioxidant response, is that an increase in reactive oxygen species may attenuate tight junctions or lead to altered epithelial tight junction protein dynamics.23,24 Alternatively, the increased levels of inflammatory cytokines reported here may act to directly disrupt barrier function. Future studies using tissue-specific deletion of the Nrf2 pathway will be helpful in elucidating the specific contribution of the sinonasal epithelium.
Interestingly, Nrf2−/− mice showed an exaggerated increase in goblet cells in response to repeated Ova exposure (Fig. 2A,B). An increase in goblet cell accumulation has been noted in chronic allergic airway disease as well as mouse models of chronic sinonasal inflammation.25,26 A recent study reported increased goblet cell hyperplasia in human nasal epithelial cultures treated with IL-33 in vitro.27 Thus, the exaggerated increase of goblet cells in Nrf2−/− mice may be in response to the elevated expression of IL-33 (Fig. 4A,C). Goblet cell metaplasia has also been a suggested mechanism to increased goblet cell accumulation in chronic airway inflammation.28 Further studies will help to clarify the mechanism of goblet cell accumulation and the link to sinonasal epithelial cell dysfunction.
Conclusion
Collectively, the data presented here show that the endogenous Nrf2 pathway limits Ova-induced sinonasal inflammation, epithelial-derived inflammatory cytokine production, and epithelial barrier dysfunction in vivo and identify a potential therapeutic target in the management of CRS. This is the first study to our knowledge which shows that Nrf2 regulates allergic inflammation in the sinonasal cavity in vivo. The exaggerated epithelial-derived inflammatory cytokine production combined with the exaggerated barrier dysfunction likely place the sinonasal epithelium as the key area of dysregulation in the Nrf2 pathway.
Acknowledgements
We thank C. Kiefe for graphical assistance.
Funding sources for the study: NIH (National Institute of Environmental Health Sciences [NIEHS] ES020859 to M.R.); Flight Attendant Medical Research Institute (FAMRI) Clinician Innovator Award (to M.R.).
Footnotes
Potential conflict of interest: N.R.L. is a patent co-inventor for methods treating vascular barrier dysfunction licensed to Navigen Pharmaceuticals, which is unrelated to the Nrf2 pathway. N.R.L. also holds a small amount of stock in Navigen Pharmaceuticals, which is currently of no value. The remaining authors declare no competing financial interests.
Presented presented at the European Rhinologic Society on July 3–7, 2016, in Stockholm, Sweden.
References
- 1.Bowler R Oxidative stress in the pathogenesis of asthma. Curr Allergy Asthma Rep. 2004;4:116–122. [DOI] [PubMed] [Google Scholar]
- 2.Jiang L, Diaz PT, Best TM, Stimpfl JN, He F, Zuo L. Molecular characterization of redox mechanisms in allergic asthma. Ann Allergy Asthma Immunol. 2014;113:137–142. [DOI] [PubMed] [Google Scholar]
- 3.Loukides S, Bakakos P, Kostikas K. Oxidative stress in patients with COPD. Curr Drug Targets. 2011;12:469–477. [DOI] [PubMed] [Google Scholar]
- 4.Mrowicka M, Zielinska-Blizniewska H, Milonski J, Olszewski J, Majsterek I. Evaluation of oxidative DNA damage and antioxidant defense in patients with nasal polyps. Redox Rep. 2015;20:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Z, Wang Y, Zhang J, et al. Expression of heme oxygenase-1 in eosinophilic and non-eosinophilic chronic rhinosinusitis with nasal polyps: modulation by cytokines. Int Forum Allergy Rhinol. 2015;5:734–740. [DOI] [PubMed] [Google Scholar]
- 6.London NR, Tharakan A, Rule AM, Lane AP, Biswal S, Ramanathan M. Air pollutant mediated disruption of sinonasal epithelial cell barrier function is reversed by activation of the Nrf2 pathway. J Allergy Clin Immunol. 2016;138:1736–1738.e4. [DOI] [PubMed] [Google Scholar]
- 7.Steelant B, Farré R, Wawrzyniak P, et al. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J Allergy Clin Immunol. 2016;137:1043–1053.e5. [DOI] [PubMed] [Google Scholar]
- 8.Wise S, Laury AM, Katz EH, Den Beste KA, Parkos CA, Nusrat A. Interleukin-4 and interleukin-13 compromise the sinonasal epithelial barrier and perturb intercellular junction protein expression. Int Forum Allergy Rhinol. 2014;4:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronaghan NJ, Shang J, Iablokov V, et al. The serine protease-mediated increase in intestinal epithelial barrier function is dependent on occludin and requires an intact tight junction. Am J Physiol Gastrointest Liver Physiol. 2016;311:G466–G479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sweerus K, Lachowicz-Scroggins M, Gordon E, et al. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J Allergy Clin Immunol. 2017; 139:72–81.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soyka MB, Wawrzyniak P, Eiwegger T, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol. 2012;130:1087–1096.e10. [DOI] [PubMed] [Google Scholar]
- 12.Steelant B, Seys SF, Boeckxstaens G, Akdis CA, Ceuppens JL, Hellings PW. Restoring airway epithelial barrier dysfunction: a new therapeutic challenge in allergic airway disease. Rhinology. 2016;54:195–205. [DOI] [PubMed] [Google Scholar]
- 13.Rangasamy T, Guo J, Mitzner WA, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tharakan A, Halderman A, Lane A, Biswal S, Ramanathan M. Reversal of cigarette smoke extract induced sinonasal epithelial cell barrier function through Nrf2 activation. Int Forum Allergy Rhinol. 2016;6:1145–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.London NR, Tharakan A, Lane AP, Biswal S, Ramanathan M. Nuclear erythroid 2-related factor 2 activation inhibits house dust mite-induced sinonasal epithelial cell barrier dysfunction. Int Forum Allergy Rhinol. 2017;7:536–541. [DOI] [PubMed] [Google Scholar]
- 16.Mendiola M, Chen M, Asempa T, Lane AP, Ramanathan M. Characterization of a novel high-dose ovalbumin-induced murine model of allergic sinonasal inflammation. Int Forum Allergy Rhinol. 2016;6:964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho S, Oh SY, Zhu Z, Lee J, Lane AP. Spontaneous eosinophilic nasal inflammation in a genetically-mutant mouse: comparative study with an allergic inflammation model. PLoS One. 2012;7:e35114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corren J, Lemansek RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. [DOI] [PubMed] [Google Scholar]
- 19.London NR, Lane AP. Innate immunity and chronic rhinosinusitis: what we have learned from animal models. Laryngoscope Investig Otolaryngol. 2016;1:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw J, Fakhri S, Citardi MJ, et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188:432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.London NR, Tharakan A, Ramanathan M. The role of innate immunity and aeroallergens in chronic rhinosinusitis. Adv Otorhinolaryngol. 2016;79:69–77. [DOI] [PubMed] [Google Scholar]
- 22.Ramanathan M, London NR, Tharakan A, et al. Airborne particulate matter induces nonallergic eosinophilic sinonasal inflammation in mice. Am J Respir Cell Mol Biol. 2017;57:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007; 21:3666–3676. [DOI] [PubMed] [Google Scholar]
- 24.Xu R, Li Q, Zhou XD, Perelman JM, Kolosov VP. Oxidative stress mediates the disruption of airway epithelial tight junctions through a TRPM2-PLCγ 1-PKCα signaling pathway. Int J Mol Sci. 2013;14:9475–9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn BH, Park YH, Shin SH. Mouse model of aspergillus and Alternaria induced rhinosinusitis. Auris Nasus Larynx. 2009;36:422–426. [DOI] [PubMed] [Google Scholar]
- 26.Du J, Ba L, Li B, et al. Distinct expression of NK2 homeobox 1 (NKX2-1) and goblet cell hyperplasia in nasal polyps with different endotypes. Int Forum Allergy Rhinol. 2017;7:690–698. [DOI] [PubMed] [Google Scholar]
- 27.Ishinaga H, Kitano M, Today M, et al. Interleukin-33 induces mucin gene expression and goblet cell hyperplasia in human nasal epithelial cells. Cytokine. 2017;90:60–65. [DOI] [PubMed] [Google Scholar]
- 28.Kouzaki H, Matsumoto K, Kikuoka H, et al. Endogenous protease inhibitors in airway epithelial cells contribute to eosinophilic chronic rhinosinusitis. Am J Respir Crit Care Med. 2017;195:737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]