

Abstract
Here we investigate the photophysics and photochemistry of Ni(II) aryl halide complexes common to cross-coupling and Ni/photoredox reactions. Computational and ultrafast spectroscopic studies reveal that these complexes feature long-lived 3MLCT excited states, implicating Ni as an underexplored alternative to precious metal photocatalysts. Moreover, we show that 3MLCT Ni(II) engages in bimolecular electron transfer with ground-state Ni(II), which enables access to Ni(III) in the absence of external oxidants or photoredox catalysts. As such, it is possible to facilitate Ni-catalyzed C–O bond formation solely by visible light irradiation, thus representing an alternative strategy for catalyst activation in Ni cross-coupling reactions.
Graphical abstract
INTRODUCTION
Transition metal complexes play critical roles as photocatalysts for solar-to-electrical energy conversion,1 solar fuel synthesis,2 and chemical synthesis.3 Photocatalysts typically work by absorbing light to generate electronically excited charge-transfer (CT) states that engage in electron and energy transfer with substrates, catalysts, and materials.4 Transition metal-based photocatalysts are generally derived from expensive precious metals such as Ru and Ir that impose significant environmental footprints due to terrestrial scarcity. Consequently, there is broad interest in the development of sustainable photocatalysts based on earth-abundant first-row transition metals.5 For example, researchers have extensively investigated photocatalysts derived from Fe(II) as d6 homologues of Ru(II) and Ir(III) complexes.5b However, most first-row transition metal complexes display exceptionally short CT lifetimes arising from deactivation through low-lying ligand-field excited states (Figure 1A).5a In contrast, precious metal complexes typically possess more favorable photophysics due to strong ligand field splitting,6 as evidenced by the long triplet metal-to-ligand charge-transfer (3MLCT) lifetime for Ru(bpy)32+ (0.63 μs) compared to Fe(bpy)32+ (~100 fs).7
Figure 1.

(A) Electronic structure of d6 photocatalysts compared to Ni(II) aryl halide complexes. (B) Structure of Ni complexes and X-ray crystal structure of 1-Cl. (C) Comparison of component absorption spectra. (D) Absorption spectra for aryl halide complexes.
In organic chemistry, photocatalysts have been employed together with nickel catalysts to accomplish novel cross-coupling reactions.8 In this area, one of our groups recently reported Csp3–H cross-coupling reactions proceeding via unimolecular photoinduced bond dissociation from Ni aryl halide complexes.9,10 Accordingly, we questioned what properties of Ni enabled its favorable photochemical behavior and whether it would be possible to develop other Ni-based photocatalytic reactions. To date, little is known about the photophysics of organometallic Ni complexes relevant to cross-coupling,11 despite their prevalence as intermediates in Ni/photoredox applications. Herein we present our initial findings on the fundamental photophysics and photochemistry of these complexes, including the discovery that Ni(II) aryl halide complexes feature long-lived 3MLCT excited states that can engage in bimolecular electron-transfer reactions. This study implicates Ni as an underexplored alternative to precious metal photocatalysts, demonstrates a novel pathway for photoinduced activation of Ni complexes toward cross-coupling, and offers new mechanistic insights into recently reported Ni/photoredox transformations.
RESULTS AND DISCUSSION
We selected Ni(II)(dtbbpy)(o-tolyl) halide complexes (dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl) for photophysical study due to the prevalence of dtbbpy as a ligand in Ni/photoredox transformations and the thermal stability afforded by the o-tolyl substituent. Our photophysical studies began with ultraviolet–visible absorption spectroscopy. The spectra of complexes 1-Cl, -Br, and -I are characterized by two prominent features in the visible (λmax = 475 nm) and ultraviolet (λmax = 297 nm), each displaying multiple bands flanking the absorption maximum (Figure 1D). Interestingly, the UV features are similar in shape and position to the spectrum of free dtbbpy, suggesting they may arise from dtbbpy-oriented π–π* transitions (Figure 1C). In addition, the visible features are only present in the Ni(II) aryl halide spectra, suggesting that these transitions may be unique to the square planar complex, as evidenced by comparison to the absorption spectrum of tetrahedral Ni(II)-(dtbbpy)Cl2 2. Consistent with this observation, the computed electronic structures of 1-Cl, -Br, and -I indicate that the visible features arise from multiple CT transitions. Highest occupied orbitals (113–120) are metal-, tolyl-, or halide-centered and lowest unoccupied orbitals (121–123) are dtbbpy-π*-centered (Figure 2A).
Figure 2.
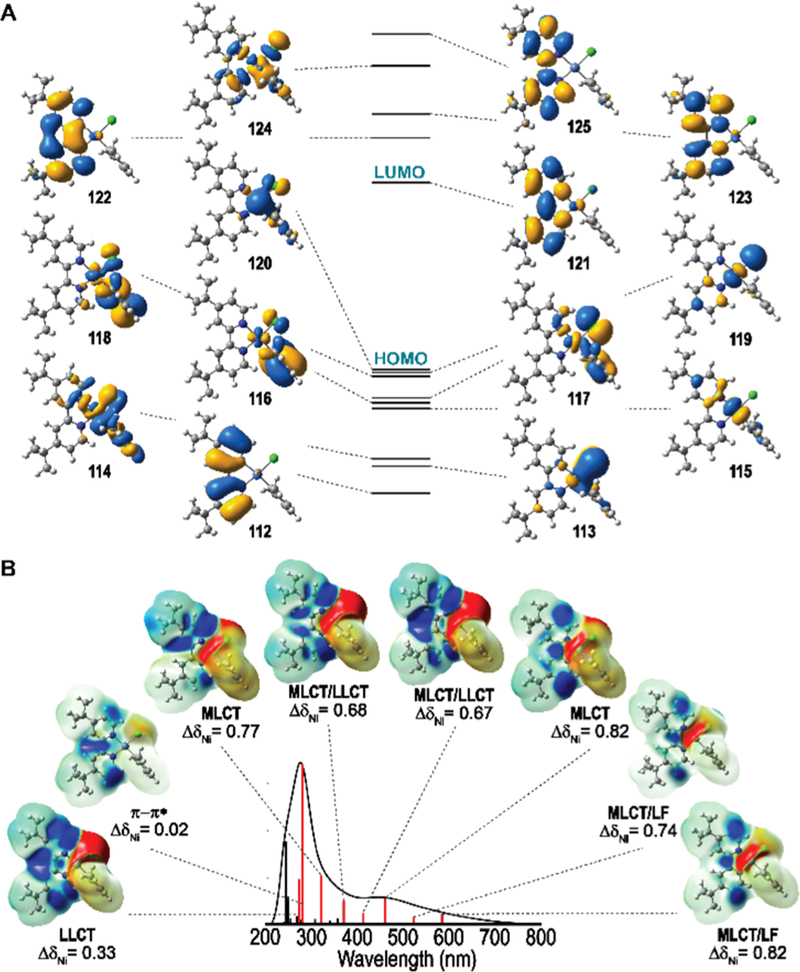
Summary of computational studies. (A) DFT computed electronic structure of 1-Cl displayed together with Kohn–Sham orbitals. (B) TD-DFT computed absorption spectrum, peak assignments, and population analysis. Difference density isosurfaces demonstrate the loss (red) and build-up (blue) of electron density. Changes in charge are normalized such that Δδ Ni(III)–Ni(II) = 1.
To assess the interplay between the computed electronic structure and state structure of the complexes, time-dependent density functional theory (TD-DFT) band assignments were carried out. Importantly the computed absorption spectra are in good agreement with experimental spectra (Figure S12). Difference density maps and orbital contributions for dominant transitions suggest that the visible manifold is predominantly 1MLCT with some halide-to-dtbbpy ligand-to-ligand charge-transfer (1LLCT) character, while the ultraviolet manifold is best described as a ligand-centered π–π* transition (Figure 2B). The MLCT assignment of the visible manifold is in good agreement with an electrochemical estimate based on the cyclic voltammetry peak potentials for Ni(III)/Ni(II) and dtbbpy/dtbbpy, redox couples of complex 1-Cl, giving λMLCT = 614 nm from a band gap of 2.02 eV (Figure S151). By comparison, TD-DFT gives a HOMO–LUMO 1MLCT1 transition at 634 nm (Table S1, Figure S23). Moreover, natural population analysis revealed that relative to ionization of Ni(II), all transitions in the visible result in significant positive charge buildup on Ni, while the band at 280 nm shows little atomic CT, consistent with the π–π* transition assignment.
The computed electronic structures of 1-Cl, -Br, and -I qualitatively resemble that of a precious metal polypyridyl photocatalyst, with unoccupied metal-centered orbitals higher in energy than ligand-π* orbitals.6 Accordingly, we posited that the halide series may have significantly longer excited MLCT lifetimes than typical first-row transition metal complexes. An ultrafast transient absorption (TA) spectroscopy study was undertaken to evaluate the excited-state dynamics of the complexes. Photoexcitation of 1-Cl into the π–π* band (λpump= 295 nm) produced an initial transient spectrum resembling that of dtbbpy co-evolved with the Ni(II) difference spectrum, while photoexcitation of 1-Cl into the 1MLCT manifold (λpump = 400 nm) produced an entirely different early time TA spectrum (Figures 3B and 4B). Notably, transient signals from both pump wavelengths coalesced in a long-lived state (τ = 4.1 ns) after 10 ps; this state is characterized by a distinct excited-state absorption at 390 nm and ground-state bleach at 470 nm.
Figure 3.
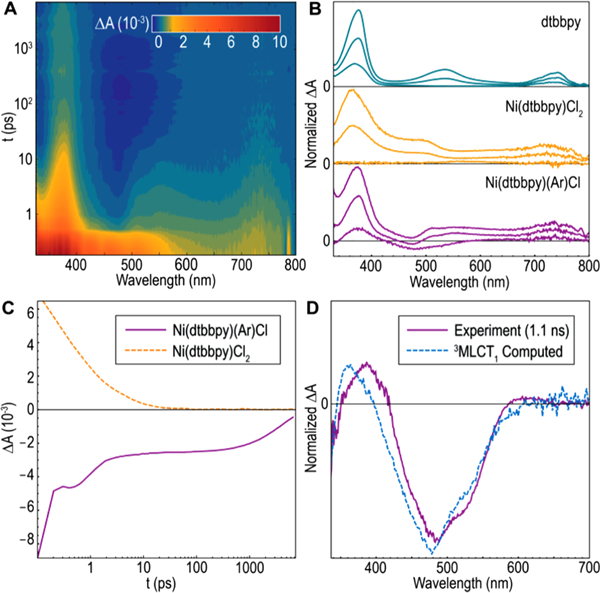
Initial transient absorption experiments. (A) Full contour plot for TA spectrum of 1-Cl (pump 295 nm). (B) Comparison of TA spectra of free dtbbpy (1–1000 ps, pump 305 nm), Ni(dtbbpy)Cl2 (1–60 ps, pump 295 nm), and 1-Cl (1–1000 ps, pump 295 nm ).(C) Single-wavelength kinetics for Ni(dtbbpy)Cl2 (367 nm, pump 295 nm) and 1-Cl (476 nm, pump 400 nm). (D) Experimental spectrum versus simulated 3MLCT1 spectrum of 1-Cl.
Figure 4.
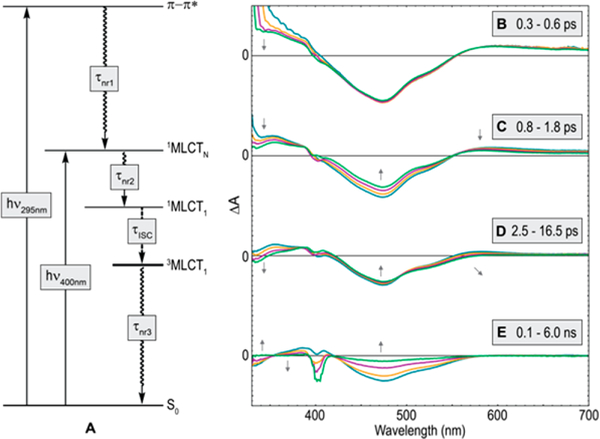
Summary of photophysics Ni(II) aryl halide complexes and evolution of TA spectra for 1-Cl upon visible excitation (λpump = 400 nm). (A) Jablonski diagram for all aryl halide complexes. (B) Initial cooling. (C) ISC associated spectral evolution (τISC = 1.1 ps). (D) Bleach broadening. (E) Decay to ground state (τnr3 = 4.1 ns). The negative feature at 400 nm is the result of pump scattering.
The long-lived component of the transient spectrum of 1-Cl is especially striking when compared to that of 2, which produces transient spectra (λpump = 295 nm) that decay completely within 60 ps. Since the frontier orbital structure of2 (Figures S21 and S22) is similar to 1-Cl, the presence of metal-centered states cannot account for the disparate rates of de-excitation. However, in its ground state, complex 2 is expected to be a triplet with tetrahedral geometry whereas 1-Cl is a singlet with square planar geometry, a function of ligand field strength (Figure S32).12 If the long-lived transient species of 1-Cl is an excited triplet state, de-excitation via a spin-forbidden pathway (versus spin-allowed for 2) could account for the differential dynamics. To probe this hypothesis, the transient spectrum for triplet 1-Cl was simulated by subtraction of the steady-state absorption spectrum of Ni(II) from the computed triplet absorption spectrum (Figure 3D). The observed difference absorption spectrum is in good agreement with the simulated spectrum, which suggests that the long-time dynamics are that of T1. Furthermore, the normalized change in charge on Ni associated with transition from S0 to T1 was computed to be 0.94, consistent with a 3MLCT1 assignment.
Two generalized kinetic scenarios resulting from excitation of the MLCT or π–π* manifold are summarized in Figure 4A. Closer inspection of the ultrafast dynamics of 1-Cl, after excitation in the MLCT manifold and initial cooling (Figure 4B), revealed the simultaneous decay of the excited-state absorption feature at 600 nm and the negative signal from 400 to 550 nm over the first few picoseconds (Figure 4C). This is followed closely by a broadening of the remaining negative signal to give the 3MLCT spectrum (Figure 4D). The observed early time spectral evolution, in conjunction with the similarity of the resultant 3MLCT spectrum to the UV pumped case (Figure 3B), points to the assignment of the initial decay as intersystem crossing (ISC, Figure 4C). Surprisingly, both the ultrafast and nanosecond dynamics are largely retained within the halide series of 1 (Table S11). The conservation of ISC dynamics (τISC: Cl = 1.1 ps, Br = 1.3 ps, I = 2.0 ps) is consistent with either spin–orbit coupling originating from Ni or energetically similar 1MLCT and 3MLCT excited states facilitating hot ISC.11a,13 The latter possibility is noteworthy because it suggests that deliberate energy level matching of triplet states could be a new strategy to facilitate ISC and compensate for the lack of spin–orbit coupling in base metals.
Having established that photoexcitation of complexes 1-Cl, -Br, and -I results in the generation of long-lived 3MLCT Ni(II), we questioned whether these species could engage in photoinduced electron-transfer (PET) reactions. Interestingly, over the course of our spectroscopic studies, degradation of complex 1-Cl was detected at both pump wavelengths (Figures S41 and S49). To distinguish between thermal degradation via heating from irradiation or nickel-based photochemistry, the stability of 1-Cl was studied. Irradiation of a THF solution of 1-Cl with a 34 W blue light emitting diode resulted in the gradual decay of UV–vis features associated with the complex and simultaneous growth of a new peak centered at 650 nm (Figure S125). Monitoring degradation by 1H NMR, various organic products derived from the complex were detected (Figures 5A and S122). It was also observed that irradiation resulted in heating of the reaction medium up to 34 °C. In the absence of light, little degradation was observed at the same temperature (Figure S123).
Figure 5.
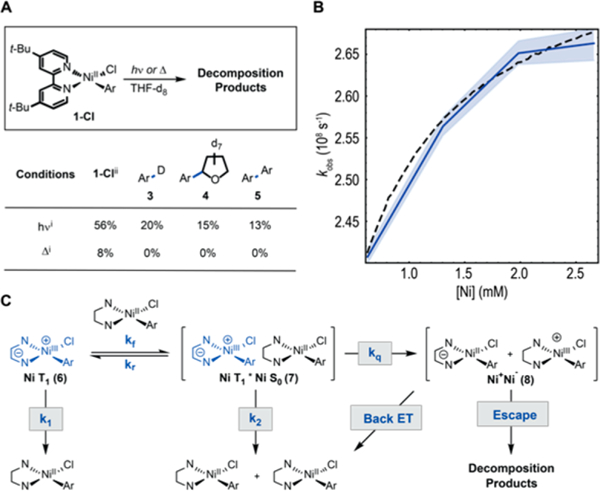
Criteria for bimolecular photoinduced electron transfer. (A) Visible light photolysis and thermolysis experiments. (i) Yield determined by 1H NMR. (ii) Conversion is reported. (B) Experimental concentration dependent kinetics (solid blue) plotted together with 95% confidence interval (blue shading) and simulated saturation kinetics (black dashed). (C) Kinetic hypothesis.
To account for the observed degradation, we hypothesized that 3MLCT 1-Cl may engage in bimolecular PET with another Ni(II). To assess the energetic feasibility of the PET pathway, the excited-state redox potentials of 3MLCT1 1-Cl were estimated from the computed S0–3MLCT1 gap (1.64 eV, 37.8 kcal mol−1), due to absence of observable photoluminescence. The excited-state reduction potentials were estimated to be 0.47 V (Ni(II)*/Ni(I), versus SCE in THF) and −0.79 V (Ni(III)/Ni(II)*, versus SCE in THF) (Figure S31). Together, with the measured peak potentials (Ni(III)/ Ni(II) = 0.85 V versus SCE in THF, Ni(II)/Ni(I) = −1.17 V versus SCE in THF)9 these data suggest that PET may be slightly endergonic. Therefore, we turned to stoichiometric redox bracketing to determine the feasibility of PET. Notably, oxidation of 1-Cl occurred rapidly upon addition of a THF solution of FcBArF4 (Fc+/Fc = 0.38 V vs SCE in THF)14 suggesting that 3MLCT1 Ni is capable of oxidizing ground-state Ni(II) via PET; only in the presence of light did this reaction give similar products to the Ni-based photochemical reaction (Figure S128).
We next sought spectroscopic evidence to differentiate the proposed electron transfer and potential unimolecular photochemistry. For bimolecular PET, the observed rate of decay of 3MLCT Ni (kobs), is expected to display a Ni concentration dependence. For collisional quenching, this dependence should take the form of a line with slope and intercept equal to the quenching rate constant and nonradiative decay rate respectively (Figure S138). Importantly, for a unimolecular decomposition reaction or reaction with solvent, no concentration dependence is expected. To differentiate these possibilities, the concentration-dependent dynamics of complex 1-Cl were explored. When kobs is plotted against the concentration of Ni, a clear concentration dependence is observed (Figure 5B). However, the concentration dependence takes the form of a saturation curve rather than the line necessitated by pure collisional quenching. The observed negative deviation from linearity could be explained by a Michaelis–Menten-like saturation scheme as shown in Figure 5C. Indeed, modeling of this kinetic scheme reproduces the observed saturation curve when a quasi-steady-state condition is met, suggesting the observed bimolecular reaction may proceed via excimer 7. Moreover, the bleach broadening observed at early times (Figure 4D) is consistent with the formation of an excimer.15
The demonstration that Ni(II) complexes can participate in photoinduced disproportionation holds important implications for the interpretation and design of photochemical reactions with Ni.16 For example, Ni catalyzed C–O cross-coupling is thermodynamically infeasible via Ni(II) reductive elimination (Figure 6).17 In contrast, cross-coupling reactions which proceed via a Ni(III) intermediate are facile, suggesting that photoinduced disproportionation could offer a mechanism for initiating Ni(I)/(III) catalytic cycles for C–O cross-coupling. In keeping with this hypothesis, a combination of Ni(cod)2 and dtbbpy was capable of efficiently catalyzing the cross-coupling of n-butanol and 4-bromoacetophenone under visible light irradiation.18 Preliminary controls and computational studies suggest that this transformation could take place via Ni(II)-oriented photoinduced electron transfer to initiate a Ni(I)/(III) C–O coupling cycle (see Supporting Information). Notably, the observed reactivity provides proof of principle that Ni(II) can act as a photocatalyst.
Figure 6.
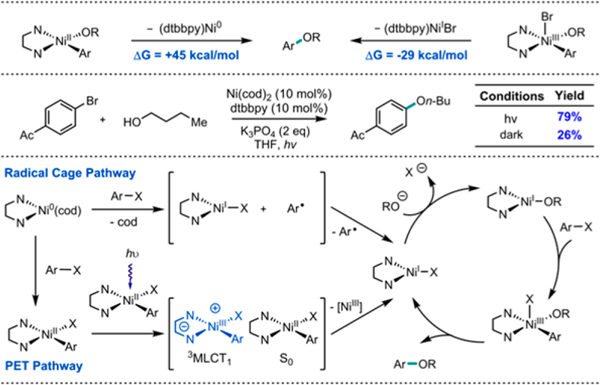
Ni-catalyzed C–O coupling and proposed mechanism. Yields determined by GC-FID using 1-fluoronaphthalene as an external standard.
CONCLUSION
Computational and ultrafast spectroscopic studies have shown that long-lived 3MLCT states of Ni(II) aryl halide complexes are accessible through both ligand localized UV and MLCT visible transitions. Moreover, it was demonstrated that 3MLCT Ni(II) aryl halide complexes engage in electron transfer, suggesting that Ni is a promising candidate for the development of base-metal photocatalysts. Formation of long-lived 3MLCT Ni(II) was strikingly unaffected by halide substitution, pointing to possible contributions from a high density of energetically similar 3MLCT excited states. Together with mimicking the frontier electronic structure of Ru(II) and Ir(III) polypyridyl complexes, this could offer a new design principle for developing photocatalysts based on earth-abundant first-row transition metals. Finally, we showed that photoinduced disproportionation of Ni(II) can promote C–O cross-coupling in the absence of a precious metal photoredox catalyst.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by a Princeton Innovation Fund grant and NIGMS (R01 GM100985) to A.G.D. This material is based on work supported by the National Science Foundation Graduate Research Fellowship under Grant Number DGE-1656466 (to B.K.). G.D.S. gratefully acknowledges the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through Grant No. DE-SC0015429. We thank Phil Jeffrey of Princeton University for X-ray crystallo-graphic structure determination.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b13281.
Procedures, data, analysis, and statistics, including Figures S1–S151 and Tables S1–S32 (PDF)
X-ray crystallographic data for 1-Cl, disorder model (CIF)
X-ray crystallographic data for 1-Cl, major conformer (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Nozik AJ; Miller J Chem. Rev. 2010, 110, 6443. [DOI] [PubMed] [Google Scholar]
- (2).(a)Grätzel M Inorg. Chem. 2005, 44, 6841. [DOI] [PubMed] [Google Scholar]; (b)Morris AJ; Meyer GJ; Fujita E Acc. Chem. Res. 2009, 42, 1983. [DOI] [PubMed] [Google Scholar]; (c)Esswein AJ; Nocera DG Chem. Rev. 2007, 107, 4022. [DOI] [PubMed] [Google Scholar]; (d)Willkomm J; Orchard KL; Reynal A; Pastor E; Durrant JR; Reisner E Chem. Soc. Rev. 2016, 45, 9. [DOI] [PubMed] [Google Scholar]
- (3).(a)Romero NA; Nicewicz DA Chem. Rev. 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]; (b)Prier CK; Rankic DA; MacMillan DW Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c)Tucker JW; Stephenson CR J. Org. Chem. 2012, 77, 1617. [DOI] [PubMed] [Google Scholar]; (d)Schultz DM; Yoon TP Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a)Kavarnos GJ; Turro NJ Chem. Rev. 1986, 86, 401. [Google Scholar]; (b)Arias-Rotondo DM; McCusker JK Chem. Soc. Rev. 2016, 45, 5803. [DOI] [PubMed] [Google Scholar]; (c)Kalyanasundaram K Coord. Chem. Rev. 1982, 46, 159. [Google Scholar]
- (5).(a)Larsen CB; Wenger OS Chem. -Eur. J. 2017, 23, xxx. [Google Scholar]; (b)Liu Y; Persson P; Sundstrom V; Warnmark K Acc. Chem. Res. 2016, 49, 1477. [DOI] [PubMed] [Google Scholar]; (c)Scaltrito DV; Thompson DW; O’Callaghan JA; Meyer GJ Coord. Chem. Rev. 2000, 208, 243. [Google Scholar]; (d)Büldt LA; Guo X; Vogel R; Prescimone A; Wenger OS J. Am. Chem. Soc. 2017, 139, 985. [DOI] [PubMed] [Google Scholar]; (e)Creutz SE; Lotito KJ; Fu GC; Peters JC Science 2012, 338, 647. [DOI] [PubMed] [Google Scholar]
- (6).Balzani V; Bergamini G; Campagna S; Puntoriero F Top. Curr. Chem. 2007, 280, 1. [Google Scholar]
- (7).(a)Caspar JV; Meyer TJ J. Am. Chem. Soc. 1983, 105, 5583. [Google Scholar]; (b)Cannizzo A; Milne CJ; Consani C; Gawelda W; Bressler C; van Mourik F; Chergui M Coord. Chem. Rev. 2010, 254, 2677. [Google Scholar]
- (8).(a)Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DW C. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b)Tellis JC; Primer DN; Molander GA Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shields BJ; Doyle AG J. Am. Chem. Soc. 2016, 138, 12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Heitz DR; Tellis JC; Molander GA J. Am. Chem. Soc. 2016, 138, 12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a)Chergui M Acc. Chem. Res. 2015, 48, 801. [DOI] [PubMed] [Google Scholar]; (b)Powers DC; Anderson BL; Nocera DG J. Am. Chem. Soc. 2013, 135, 18876. [DOI] [PubMed] [Google Scholar]; (c)Lockwood G; McGarvey JJ; Devonshire R Chem. Phys. Lett. 1982, 86, 127. [Google Scholar]; (d)Hwang SJ; Powers DC; Maher AG; Anderson BL; Hadt RG; Zheng SL; Chen YS; Nocera DG J. Am. Chem. Soc. 2015, 137, 6472. [DOI] [PubMed] [Google Scholar]; (e)Klein A; Kaiser A; Wielandt W; Belaj F; Wendel E; Bertagnolli H; Zalis S Inorg. Chem. 2008, 47, 11324. [DOI] [PubMed] [Google Scholar]
- (12).Biswas S; Weix DJ J. Am. Chem. Soc. 2013, 135, 16192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Frei F; Rondi A; Espa D; Mercuri ML; Pilia L; Serpe A; Odeh A; Van Mourik F; Chergui M; Feurer T; Deplano P; Vlcek A; Cannizzo A Dalton Trans. 2014, 43, 17666. [DOI] [PubMed] [Google Scholar]
- (14).Noviandri I; Brown KN; Fleming DS; Gulyas PT; Lay PA; Masters AF; Phillips LJ Phys. Chem. B 1999, 103, 6713. [Google Scholar]
- (15).Brown KE; Salamant WA; Shoer LE; Young RM; Wasielewski M R J. Phys. Chem. Lett. 2014, 5, 2588. [DOI] [PubMed] [Google Scholar]
- (16).Ishida N; Masuda Y; Ishikawa N; Murakami M Asian J. Org. Chem. 2017, 6, 669. [Google Scholar]
- (17).(a)Terrett JA; Cuthbertson JD; Shurtleff V; MacMillan DW C. Nature 2015, 524, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b)Welin ER; Le C; Rotondo DM; McCusker JK; MacMillan, D. W. C. Science 2017, 355, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). The efficiency of photocatalytic C–O coupling with Ni is dependent on the reaction conditions (see Supporting Information), consistent with a report by the MacMillan group (ref 17a) wherein no background reactivity was observed for a Ni/Ir dual catalyst system under distinct conditions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.