

Abstract
A nonsense mutation is a substitutive mutation in a DNA sequence that causes a premature termination during translation and produces stalled proteins, resulting in dysfunction of a gene. Although it usually induces severe genetic disorders, there are no definite methods for inducing read through of premature termination codons (PTCs). Here, we present a targeted tool for bypassing PTCs, named CRISPR-pass, that uses CRISPR-mediated adenine base editors. CRISPR-pass, which should be applicable to 95.5% of clinically significant nonsense mutations in the ClinVar database, rescues protein synthesis in patient-derived fibroblasts, suggesting potential clinical utility.
Keywords: CRISPR-Cas9, base editing, nonsense mutation, stop codon read through, premature termination codon
Graphical Abstract
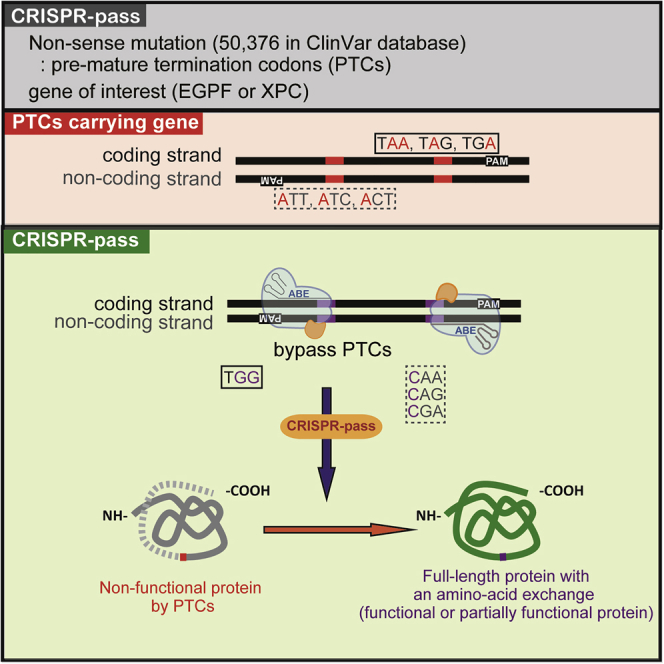
Lee et al. showed that CRISPR-pass, based on adenine base editors, would be a targeted tool for bypassing premature termination codons. This system could be applicable to ∼95% of clinically significant nonsense mutations, related to genetic diseases, in the ClinVar database.
Introduction
Nonsense mutations, in which premature termination codons (PTCs) are formed by base-pair substitution, truncate protein synthesis during translation. Such gene dysfunction is a source of severe pathological phenotypes in genetic diseases. Hence, compelling ribosomal read through of the full coding sequence is a reasonable strategy for treating such genetic disorders. To address this issue, previous studies showed to induce the skipping of exons containing PTCs by using antisense oligonucleotides (AONs).1, 2 In other ways, a few small-molecule drugs such as ataluren3, 4 and aminoglycosides5 (e.g., gentamicin) have been utilized to bypass nonsense mutations by introducing near-cognate tRNAs at the site of the PTC.6, 7 However, those approaches act transiently and have nonspecific effects for the drugs. Alternatively, CRISPR-mediated homology-directed repair (HDR) can be used for gene correction but is limited by low correction efficiency, especially in differentiated nonreplicating cells from higher eukaryotes including humans.8, 9, 10
It was reported that CRISPR-mediated base editing technologies enable highly efficient direct conversion of DNA bases without producing double-strand breaks (DSBs). Cytidine deaminase-based base editors (CBEs) produce C-to-T or G-to-A substitutions between the fourth and eighth bases in the nonbinding strand of single-guide RNA (sgRNA) at protospacer DNA.11, 12 On the other hand, A-to-G or T-to-C transitions in the same DNA positions can be achieved by adenine base editors (ABEs).13 In addition to the initial versions of CBEs and ABEs, including ABE7.10, Koblan et al.14 improved the base-editing activities by expression optimization and ancestral reconstruction, which were named BEmax and ABEmax, respectively. Moreover, Hu et al.15 and Nishimasu et al.16 independently developed new Cas9 variants, named xCas9 and SpCas9-NG, that recognize 5′-NG-3′ and 5′-NAR-3′ sequences, expanding the targetable sites. By combining xCas9 3.7 with ABE7.10 (called xABE here), Hu et al.15 further demonstrated how to expand the targetable sites for adenine base editing.
To date, a few groups have reported to successfully correct target genes by restoring the open reading frame in PTCs by using the ABEs, such as a TAG-PTC of EGFP gene in rice17 and both TAA-PTC of the Tyr gene and TAG-PTC of the DMD gene in mice.18 However, although a few meaningful examples were shown, the systematic gene rescue for all possible cases is not demonstrated yet. It is expected that by targeting the coding strand with ABEs, the three possible PTCs, 5′-TAA-3′, 5′-TAG-3′, and 5′-TGA-3′, can be converted to 5′-TGG-3′, which will be translated to tryptophan (Trp). Alternatively, by targeting the noncoding strand, the three PTCs can be converted to 5′-CAA-3′ (translated to glutamine; Gln), 5′-CAG-3′ (Gln), or 5′-CGA-3′ (arginine; Arg), respectively (Figure 1A). In this study, we established an ABE-mediated read-through method, named CRISPR-pass, to bypass PTCs by converting adenine to guanine or thymine to cytosine. We constructed all type of PTCs knockin (KI) cell lines and then showed the read through for all cases. Ultimately, we showed gene rescue at a patient-derived fibroblast containing PTC.
Figure 1.

CRISPR-Pass for Restoring Abbreviated Gene Expression
(A) Schematic of ABE-mediated CRISPR-pass. Targetable adenines are located in the coding or noncoding strand depending on the PAM’s orientation. All possible PTCs are shown in the upper boxes (coding strand targeting-TAA, TAG, TGA; noncoding strand targeting-TAA, TAG, TGA). The orange-colored shapes represent adenosine deaminase. (B) The percentages of different types of mutations causing pathological phenotypes in the ClinVar database. (C) The percentages of PTCs that are targetable by CRISPR-pass with various PAMs of variant ABEs and the recoverable rate of intact amino acids and bypassing alternative amino acids are depicted.
Results
In Silico Investigation of Applicable Targets for CRISPR-Pass in the ClinVar Database
We first inspected all targetable variations registered in the ClinVar database in silico to investigate how many genetic diseases with nonsense mutations could potentially be treated with CRISPR-pass. Of the 50,376 mutations causing pathological phenotypes in the database, nonsense mutations account for 16.2% (Figure 1B); among these, 41.1% are targetable by conventional ABEs that recognize a canonical protospacer adjacent motif (PAM), 5′-NGG-3′, and 95.5% are covered by xABEs, which recognizes a noncanonical set of PAMs, 5′-NG-3′, and 5′-NAR-3′ (Figure 1C). Only 31.5% of the nonsense mutations in the database can be exactly corrected to amino acids found in the nonmutant protein by xABEs, implying that the set of mutations that can be modified for read through by CRISPR-pass is much larger than the set for which exact gene correction in DNA is possible (Figure 1C; Data S1).
Construction of Six KI HeLa Cell Lines Carrying Various Types of PTCs in EGFP Gene
To demonstrate the efficiency of CRISPR-pass in human cells, as a proof of concept, we tried to construct six KI HeLa cell lines, each carrying a different mutated version of the EGFP gene. We first prepared six DNA plasmids having different types of PTCs in the EGFP gene. The mutant EGFP genes as a set contain each type of PTC at two locations: the three PTCs in a position that can be converted by targeting the coding strand and the three PTCs in a position that can be converted by targeting the noncoding strand. The first position corresponds to a codon for lysine (Lys53) and the second to a codon for aspartate (Asp217); the encoded residues are located in connecting loop domains of EGFP (Figures 2A and S1). After preparing plasmids containing the six mutated EGFP genes, each plasmid was inserted into the genome in an endogenous safe-harbor region, the AAVS1 site,19 using CRISPR-Cas9 via a nonhomologous end-joining (NHEJ) pathway20 (Figure 2B). The cell lines were named c-TAA, c-TAG, c-TGA, nc-TAA, nc-TAG, and nc-TGA, respectively.
Figure 2.

Restoring the Function of EGFP Gene Expression in Six KI HeLa Cell Lines Carrying Various Types of PTCs
(A) Scheme for restoration of EGFP expression by CRISPR-pass. The first set of PTCs, which can be converted by targeting the coding strand, affect a residue that is located on a loop between the third and fourth beta strands; the second set of PTCs, which can be converted by targeting the noncoding strand, affect a residue that is located on a loop between the 10th and 11th beta strands. c-PTC, coding strand PTC; nc-PTC, noncoding strand PTC. GFP structures were originated from Wikimedia Commons created by Zephyris. (B) Schematic of NHEJ-mediated KI of the EGFP-PTC constructs into the AAVS1 site. Mutated EGFP KI cell lines were established for the three types of PTCs (TAA, TAG, and TGA). EGFP-PTC constructs were inserted into the AAVS1 site by NHEJ-mediated KI methods. The hygromysin B-resistant gene was also inserted for cell selection. (C) Fluorescence image of rescued EGFP expression in the c-TAA cell line after CRISPR-pass treatment. Three different versions of ABEs (ABE7.10, xABE, and ABEmax) were used for bypassing the PTCs in the EGFP gene. All scale bars are 100 μm. (D) Flow cytometry data after the different versions of ABEs (ABE7.10, xABE, and ABEmax) were treated in the c-TAA cell line. (E) Targeted deep-sequencing data showing the percentages of each of the four nucleotides at each position in the target DNA sequences as a substitution table, which was obtained from the c-TAA cell line after the ABEmax treatment. Bar graphs showing recovered EGFP expression levels as determined by flow cytometry (F) and showing A-to-G substitution rates at PTC sites as determined by targeted deep sequencing (G) for each EGFP-PTC KI cell line, after treatment with ABEs (ABE7.10, xABE, or ABEmax). Each dot represents the three independent experiments. Error bars represent SEM.
CRISPR-Pass Rescues the Function of the EGFP Gene in Six KI HeLa Cell Lines
To test whether ABE treatment would allow bypass of these nonsense mutations, we transfected plasmids expressing sgRNAs designed to target both locations harboring PTCs, together with ABE-encoding plasmids, into the prepared HeLa cell lines. After ABE treatment by lipofection, we found that functional EGFP was expressed, as seen by green fluorescence, in all cell lines. For example, in the case of c-TAA cells, the function of EGFP would be rescued when two adenines are changed to guanines simultaneously for bypassing the PTC. As shown in Figure 2C, the functional EGFPs were observed after various ABEs (ABE7.10, xABE, and ABEmax) were treated. We quantified the ratios of rescued to mutated EGFPs by flow cytometry (Figure 2D). We also confirmed the A-to-G conversion at target DNA regions by targeted deep sequencing in bulk cell populations; the conversion rate of two adenines (A7A8) to two guanines (G7G8) at once was 11.2% here (Figure 2E).
Similar to the c-TAA cells, we repeatedly carried out CRISPR-pass for all other types of KI cell lines (Figure S2). The quantitative ratios of the functional EGFP expression were also measured by flow cytometry and targeted deep sequencing. As a result, the flow cytometry analysis demonstrated that nonsense mutations were bypassed in 0.7%–17.8% of cells (Figures 2F and S3; Table S1). And targeted deep sequencing analysis confirmed that 0.2%–15.2% of the cells showed A-to-G conversions at target regions with a comparable tendency (Figure 2G; Table S2). It is noteworthy to mention that ABEmax was the most effective one in all cases, resulting in up to 59.6% rescue of the mutant EGFP gene, compared to the ABE7.10 and xABE. And there seems to be some mismatches between EGFP expression levels and substitution rates especially in the case of c-TAG cells, which might be caused by the different expression capacity of each KI cell line due to cell-to-cell variations.
CRISPR-Pass Rescues the Function of the XPC Gene in Patient-Derived Fibroblasts
We next applied CRISPR-pass to the rescue of a nonsense mutation in fibroblasts (GM14867) derived from a patient with xeroderma pigmentosum, complementation group C (XPC). XPC, which affects the skin, is a genetic disorder caused by nonsense mutations in the XPC gene. The XPC protein is an initiator of global nucleotide excision repair.21 Thus, XPC-deficient cells accumulate DNA damage when they are exposed to chemical or physical stimuli including ultraviolet irradiation.22 GM14867 cells have a homozygous C > T nonsense mutation at nucleotide 1840 in the XPC gene, which creates a 5′-TGA-3′ stop codon that replaces a codon for Arg (Arg579) (1840C > T, Arg-579-UGA stop codon) (Figure 3A). After treating GM14867 cells with ABE7.10-encoding plasmid and sgRNA-encoding plasmid by electroporation, the adenine base in the 5′-TGA-3′ stop codon was converted to guanine to create 5′-TGG-3′ at a rate of 3.4%, as measured by targeted deep sequencing (Figure S4), indicating partial rescue of the XPC gene. Similar to the previous experiments, xABE and ABEmax resulted in higher base-editing rates more than 10% (Figures 3B and S4), respectively. Western blot analyses demonstrated that both xABE and ABEmax treatment led to recovery of expression of the full-length XPC protein, with a molecular weight similar to that in wild-type (WT) cells (ARPE-19), at considerably higher levels than induced by ataluren or gentamicin (Figure 3C).
Figure 3.

Restoring Abbreviated XPC Gene Expression in Patient-Derived Fibroblasts
(A) Scheme for ABE-induced read through of an XPC-associated PTC. (B) Targeted deep-sequencing data showing the A-to-G substitution rate induced by ABEmax treatment at the PTC site in the XPC gene. (C) Expression level of the XPC protein in XPC mutant cells rescued by treatment with ABEs (ABEmax or xABE), compared with the expression level in untreated cells and cells treated with ataluren or gentamicin for 48 h. (D) Cell viability of WT skin fibroblasts (BJ-5ta), XPC mutant cells (GM14867), and XPC mutant cells treated with ABEs (ABEmax or xABE), ataluren, or gentamicin at 3 days after exposure to 254 nm ultraviolet radiation at a dose of 25 J/m2. p values were calculated by one-way ANOVA with post-hoc Bonferroni’s multiple comparison tests (n = 6). p value indicators from a comparison with GM14867 cell viability are shown above each treatment group. NS, not significant (p > 0.05); *p < 0.05; ***p < 0.001. (E) Prolonged expression of the XPC protein after CRISPR-pass treatment. Significant and stable XPC protein expression was observed until at least 4 weeks after ABEmax treatment. However, XPC protein expression declined after removal of ataluren and gentamycin. Proteins were also prepared from ABEmax-treated XPC mutant cells at 2 and 4 weeks (subculturing twice per week) for comparison. Blue and red arrowheads indicate the positions of XPC protein.
Next, to determine the functional activity of the recovered XPC protein, we evaluated the viability of GM14867 cells at 72 h after exposure to 254 nm ultraviolet light. To our surprise, both xABE- and ABEmax-treated GM14867 cells had significantly regained resistance to ultraviolet irradiation-induced DNA damage, causing an increase in cellular viability (Student’s t test, p < 0.001; Figure 3D). More importantly, ABEmax-treated GM14867 cells sustained such XPC protein expression for at least 4 weeks, whereas the cells treated with ataluren or gentamicin gradually lost XPC protein expression (Figures 3E and S5), implying that CRISPR-pass induces persistent expression for the nonsense-mediated disease therapies.
Finally, to examine the off-target effects of CRISPR-pass in GM14867 fibroblasts, we searched for potential off-target sites using Cas-OFFinder23 and carried out targeted deep sequencing for 12 candidate target sites (Figure S6; Table S3). As a result, we found no noticeable off-target sites likewise to the previous ABE-based gene-editing studies,24, 25, 26 suggesting potential clinical utility.
Discussion
Previously, Kuscu et al.27 and Billon et al.28 demonstrated gene-silencing methods, named CRISPR-STOP and iSTOP, respectively, through CBE-induced nonsense mutations. In this study, we analogously demonstrated that CRISPR-pass is a straightforward method for inducing read through of PTCs by ABEs, covering most (95%) nonsense mutations in the ClinVar database that cause pathological phenotypes. We first demonstrated the CRISPR-pass activities in six types of EGPF-PTCs-KI human cells, as a proof of concept. In these experiments, CRISPR-pass efficiently rescued functional EGFP expression by bypassing all PTCs. Then we successfully confirmed the activity of CRISPR-pass in a patient-derived fibroblast, GM14867, which contains a nonsense mutation at the XPC gene.
Until now, researchers have tried to correct the PTC in the XPC coding gene by various approaches. One suggested a viral delivery method of intact XPC-coding plasmids,29 but it has potential problems such as a random integration of the transgene in viral delivery30 and overexpression effects of the exogenous XPC gene. Alternatively, another approach to correct the endogenous XPC gene was demonstrated by using meganucleases and TALENs.22 In this study, the authors tried to correct the XPC gene via a HDR pathway after producing double-strand breaks (DSBs) of DNA, which might induce DSB-mediated cell apoptosis,31 whereas the CRISPR-pass does not generate DSBs of DNA. Furthermore, we showed that the A-to-G conversions at a rate of about 10% can rescue the expression of functional XPC protein (Figures 3B–3D) without detectable off-target effects, strongly indicating that the CRISPR-pass is a relevant approach for rescuing the nonsense-associated diseases with higher editing efficiencies than using HDR32 and without the loss of large portion of protein via the exon removal33 or skipping strategies.34 More importantly, CRISPR-pass induced prolonged XPC protein expression, unlike ataluren and gentamycin that are known as current nonsense mutation disease therapies.3, 5
Recently, it is reported that DNA cleavages at on-target sites frequently cause undesired large deletions or complex genomic rearrangements.35 In this aspect, CRISPR-pass has important safety advantages relative to approaches that do rely on DNA cleavage. Furthermore, recent off-target profiling experiments on ABEs supported the high specificity of ABEs,24, 25, 26 increasing the potential clinical utility of it. These characteristics suggest that CRISPR-pass might be useful for gene rescue in a clinical setting, as an alternative to existing drugs.
Materials and Methods
General Methods and Cloning
All ABEs were purchased from Addgene (pCMV-ABE7.10, #102919; xCas9(3.7)-ABE(7.10), #108382; pCMV-ABEmax, #112095). The pXY-ECFP-AAVS1-NHEJ-KI donor vector (plasmid36, 37 provided by professor J.S. Woo at Korea University, South Korea, and modified by J.Y.) was digested with SacI and BsrGI, and Gibson assembly was then used to generate plasmids containing mutated versions of EGFP. The linearized vector was incubated with amplified EGFP DNA sequences, respectively containing each PTC at the appropriate location, with 20 nucleotides of overhanging homologous sequence at either end (5′-GGTCTATATAAGCAGAGCTC-3′ and 5′-TGTGCGGCTCACTTGTACAG-3′), in a solution containing 2× Gibson master mixture at 50°C for 1 h.38 After then, pXY-EGFP-AAVS1-NHEJ-KI vector was digested again with SacI. The linearized vector was incubated with additional amplified DNA sequences (5′-ggtctatataagcagagctctcgtcgacgagctcgtttagtgaaccgtcagatcgtttaaacaagttggtcgtgaggcactgggcaggtaagtatcaaggttacaagacaggtttaaggagaccaatagaaactgggcttgtcgagacagagaagactcttgcgtttctgataggcacctattggtcttactgacatccactttgcctttctctccacaggtgtccagggtaccgagctcgccgccatggtgag-3′) and 2× Gibson master mixture in 50°C. Each sequence of oligos encoding sgRNA was purchased from Macrogen (South Korea). Oligos were heated and cooled down by a thermocycler for complementary annealing. Double-strand oligos were ligated into linearized pRG2 plasmid linearized by BsaI restriction enzyme (Addgene, #104274). List of oligomers for sgRNAs and primer sequences for cloning are in Tables S4 and S5.
ClinVar Database Analysis
Bioinformatic analysis of the ClinVar database of human disease-associated mutations was conducted using Python. The ClinVar database (Common_and_clinical_20170905) was used for this analysis. The Python script used to analyze mutation patterns in human diseases and to identify mutations that could be CRISPR-pass targets can be accessed at https://github.com/Gue-ho/CRISPR-pass. In brief, the steps of the analysis were as follows.
-
1.
Among entries in the ClinVar database, we identified mutation patterns in the following categories: indels (insertions or deletions), silent mutations, nonsense mutations, and missense mutations.
-
a.
For precise analysis, entries in each mutation pattern category were subdivided depending on their nucleotide sequence using information about the surrounding genomic sequence and coding sequence (CDS). CDSs were extracted from the SNP database at NCBI. If no CDS was found in NCBI, than data were taken from GRCh38 and hg19.
-
2.
Among the sorted entries from the ClinVar database, the number of nonsense mutations that were potential CRISPR-pass targets were counted. The targetable Cas9 sites were grouped by their associated PAM sequences, such as GG, AG, GA, GC, GT, GAN, and AA.
-
a.
Each Cas9-targetable site was filtered depending on the ABE target range (positions 4 to 8 in the protospacer from the end distal to the PAM) it contained.
-
b.
To prevent counting sequences more than once, in the case of SpCas9, a sequence was counted when at least either GG or AG was possible; the number was counted as a targetable PAM for xABE when at least one of the PAM was possible.
Cell Culture and Transfection
HeLa (ATCC, CCL-2) cells were grown in DMEM with 10% fetal bovine serum (FBS) and a penicillin/streptomycin mix (100 units/mL and 100 mg/mL, respectively). 2.5 × 105 HeLa cells were transfected with each ABE (ABE, xABE, or ABEmax)-encoding plasmid (0.7 μg) and each sgRNA expression plasmid (0.3 μg) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were collected 5 days after transfection, and the cell’s genomic DNA was prepared using NucleoSpin Tissue (MACHEREY-NAGEL & Co.).
GM14867 (XPC mutant fibroblasts) were purchased from Coriell Institute and maintained in Eagle’s minimum essential medium (EMEM) with 15% FBS and a penicillin/streptomycin mix. BJ-5ta cells (cat. no. CRL-4001, ATCC) were maintained in a 4:1 mixture of DMEM and medium 199 with 10 μg/mL hygromycin B and 10% FBS. ARPE-19 cells (cat. no. CRL-2302, ATCC) were maintained in DMEM:F12 with 10% FBS and a penicillin/streptomycin mix. For plasmid-mediated expression of ABEs and sgRNAs, 6 × 105 fibroblasts were co-transfected with 14 μg of ABE-encoding plasmid and 6 μg of sgRNA-expressing plasmid. Fibroblasts were transfected with the Amaxa P3 primary cell 4D-nucleofector kit using program DS-137, according to the manufacturer’s protocol. A-to-G substitutions were analyzed 5 days after transfection.
EGFP-PTC-KI Cell Lines
2.5 × 105 HeLa cells were transfected with Cas9-encoding plasmid (0.35 μg), AAVS1-sgRNA-encoding plasmid (0.15 μg), and EGFP-PTC encoding plasmid (0.5 μg) using the Neon transfection system (Invitrogen) with the following parameters: pulse voltage, 1,005; pulse width, 35 ms; pulse number, 2. Seven days after transfection, the culture medium was changed to 150 μg/mL hygromycin B (Thermo Fisher Scientific, cat. no. 10687010)-containing HeLa cell culture medium. Seven days after hygromycin B treatment, single cells were selected and cultured. Single cell-derived clones were analyzed and used for further experiments.
Flow Cytometry
Five days after transfection, ABE-treated cells were trypsinized and resuspended in PBS. Single-cell suspensions were analyzed using a FACSCanto II (BD Biosciences) installed at Hanyang LINC+ Equipment Center (Seoul, South Korea).
Targeted Deep Sequencing
Genomic DNA segments that encompass the nuclease target sites were amplified using Phusion polymerase (New England Biolabs). Equal amounts of the PCR amplicons were subjected to paired-end read sequencing using Illumina MiSeq at Bio-Medical Science (South Korea). Rare sequence reads that constituted less than 0.005% of the total reads were excluded. Off-targets were selected by Cas-OFFinder (http://www.rgenome.net/cas-offinder/),23 and base substitutions were analyzed by BE-Analyzer (http://www.rgenome.net/be-analyzer/).39 Primer sequences and list of off-targets are in Tables S5 and S6.
Treatment with Ataluren and Gentamicin
GM-14867 cells and the cells treated with xABE and ABEmax were maintained in EMEM with 15% FBS. When the confluency was 60%–70%, the cells were treated with ataluren (10 μM; cat. no. S6003, Selleck) or gentamicin (1 mg/mL; cat. no. G1397, Sigma) for 48 h.
Western Blotting
Cell lysates were homogenized in 1× cell lysis buffer (cat. no. #9803, Cell Signaling Technology), and the supernatants were collected after centrifugation for 10 min at 14,000 × g. An equal amount (35 μg) of the protein was separated by SDS-PAGE in 4%–15% mini-PROTEAN TGX precast protein gels (cat. no. 4561084, Bio-Rad) and transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies overnight at 4°C. The primary antibodies utilized in this study were as follows: anti-XPC antibody (cat. no. MA1-23328, Thermo) and anti-β-actin antibody (catalog no. A2668, Sigma). Then, the membranes were treated with the appropriate species-specific secondary antibodies (cat. no. sc-2357 and sc-516102, Santa Cruz) for 1 h at room temperature. After treatment of the membranes with reagents from the EZ-Western Lumi pico kit (cat. no. DG-WP100, DoGEN), the protein bands were visualized using the ImageQuant LAS4000 system with the accompanying software program (GE).
Functional Assessment
To assess the functional recovery of GM14867 cells, which carry a homozygous mutation in the XPC gene, after treatment with xABE, ABEmax, ataluren, or gentamicin, these cells, together with BJ-5ta WT cells, were exposed to ultraviolet irradiation at 254 nm at 1 J/m2/s (cat. no. CL-1000, Analytik Jena) and left to grow for 72 h. Cells treated with ataluren or gentamicin underwent treatment for 48 h before ultraviolet exposure. Cell survival was evaluated with a water-soluble tetrazolium salt assay using an EZ-Cytox kit (cat. no. EZ-1000, DoGEN).
Statistics
All statistical analyses were performed using the GraphPad Prism 5 program (GraphPad), and results are indicated in the figure legends. The values of each mean and SEM were visualized as horizontal lines and error bars, respectively, in graphs.
Data Availability
Sequencing data has been uploaded to the Sequence Read Archive under Bioproject: PRJNA518883. All data are available from the authors upon reasonable request.
Author Contributions
Jeong H. Kim and S.B. conceived this project; C.L., D.H.J., J.Y., and S.P. performed the experiments; G.-H.H. performed bioinformatics analyses; C.L., D.H.J., J.Y., and Jin H. Kim analyzed the data; J.-S.K. gave critical comments; C.L., D.H.J., Jeong H. Kim, and S.B. wrote the manuscript with the approval of all other authors.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by National Research Foundation of Korea (NRF) grants (2017R1A6A3A04004741 to D.H.J.; 2015M3A9E6028949, 2017M3A9B4062654, and 2018M3D1A1058826 to Jeong H. Kim; and 2018M3A9H3022412 to S.B.); a grant from the Korea Research Institute of Standards and Science (KRISS – 2018 – GP2018-0018 to Jeong H. Kim); and grants from the Next Generation BioGreen 21 Program (PJ01319301), Korea Healthcare Technology R&D Project (HI16C1012), and Technology Innovation Program (20000158) (to S.B.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.05.013.
Contributor Information
Jeong Hun Kim, Email: steph25@snu.ac.kr.
Sangsu Bae, Email: sangsubae@hanyang.ac.kr.
Supplemental Information
References
- 1.Alter J., Lou F., Rabinowitz A., Yin H., Rosenfeld J., Wilton S.D., Partridge T.A., Lu Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006;12:175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 2.Aartsma-Rus A., van Ommen G.J. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roy B., Friesen W.J., Tomizawa Y., Leszyk J.D., Zhuo J., Johnson B., Dakka J., Trotta C.R., Xue X., Mutyam V. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA. 2016;113:12508–12513. doi: 10.1073/pnas.1605336113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiqui N., Sonenberg N. Proposing a mechanism of action for ataluren. Proc. Natl. Acad. Sci. USA. 2016;113:12353–12355. doi: 10.1073/pnas.1615548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuschal C., DiGiovanna J.J., Khan S.G., Gatti R.A., Kraemer K.H. Repair of UV photolesions in xeroderma pigmentosum group C cells induced by translational readthrough of premature termination codons. Proc. Natl. Acad. Sci. USA. 2013;110:19483–19488. doi: 10.1073/pnas.1312088110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A., Ursic D., Davies J. Phenotypic suppression and misreading Saccharomyces cerevisiae. Nature. 1979;277:146–148. doi: 10.1038/277146a0. [DOI] [PubMed] [Google Scholar]
- 7.Pichavant C., Aartsma-Rus A., Clemens P.R., Davies K.E., Dickson G., Takeda S., Wilton S.D., Wolff J.A., Wooddell C.I., Xiao X., Tremblay J.P. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 2011;19:830–840. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim J.S. Precision genome engineering through adenine and cytosine base editing. Nat. Plants. 2018;4:148–151. doi: 10.1038/s41477-018-0115-z. [DOI] [PubMed] [Google Scholar]
- 9.Komor A.C., Badran A.H., Liu D.R. Editing the Genome Without Double-Stranded DNA Breaks. ACS Chem. Biol. 2018;13:383–388. doi: 10.1021/acschembio.7b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nami F., Basiri M., Satarian L., Curtiss C., Baharvand H., Verfaillie C. Strategies for In Vivo Genome Editing in Nondividing Cells. Trends Biotechnol. 2018;36:770–786. doi: 10.1016/j.tibtech.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 13.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C., Zong Y., Wang Y., Jin S., Zhang D., Song Q., Zhang R., Gao C. Expanded base editing in rice and wheat using a Cas9-adenosine deaminase fusion. Genome Biol. 2018;19:59. doi: 10.1186/s13059-018-1443-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 19.DeKelver R.C., Choi V.M., Moehle E.A., Paschon D.E., Hockemeyer D., Meijsing S.H., Sancak Y., Cui X., Steine E.J., Miller J.C. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maresca M., Lin V.G., Guo N., Yang Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013;23:539–546. doi: 10.1101/gr.145441.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugasawa K., Ng J.M.Y., Masutani C., Iwai S., van der Spek P.J., Eker A.P.M., Hanaoka F., Bootsma D., Hoeijmakers J.H. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 22.Dupuy A., Valton J., Leduc S., Armier J., Galetto R., Gouble A., Lebuhotel C., Stary A., Pâques F., Duchateau P. Targeted gene therapy of xeroderma pigmentosum cells using meganuclease and TALEN™. PLoS ONE. 2013;8:e78678. doi: 10.1371/journal.pone.0078678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z., Lu Z., Yang G., Huang S., Li G., Feng S., Liu Y., Li J., Yu W., Zhang Y. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun. 2018;9:2338. doi: 10.1038/s41467-018-04768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang P., Xie X., Zhi S., Sun H., Zhang X., Chen Y., Chen Y., Xiong Y., Ma W., Liu D. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat. Commun. 2019;10:67. doi: 10.1038/s41467-018-07988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H.K., Willi M., Miller S.M., Kim S., Liu C., Liu D.R., Hennighausen L. Targeting fidelity of adenine and cytosine base editors in mouse embryos. Nat. Commun. 2018;9:4804. doi: 10.1038/s41467-018-07322-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuscu C., Parlak M., Tufan T., Yang J., Szlachta K., Wei X., Mammadov R., Adli M. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods. 2017;14:710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 28.Billon P., Bryant E.E., Joseph S.A., Nambiar T.S., Hayward S.B., Rothstein R., Ciccia A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell. 2017;67:1068–1079.e4. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warrick E., Garcia M., Chagnoleau C., Chevallier O., Bergoglio V., Sartori D., Mavilio F., Angulo J.F., Avril M.F., Sarasin A. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells. Mol. Ther. 2012;20:798–807. doi: 10.1038/mt.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 31.Roos W.P., Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Hess G.T., Tycko J., Yao D., Bassik M.C. Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol. Cell. 2017;68:26–43. doi: 10.1016/j.molcel.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benchaouir R., Meregalli M., Farini A., D’Antona G., Belicchi M., Goyenvalle A., Battistelli M., Bresolin N., Bottinelli R., Garcia L., Torrente Y. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell. 2007;1:646–657. doi: 10.1016/j.stem.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 35.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Backliwal G., Hildinger M., Chenuet S., Wulhfard S., De Jesus M., Wurm F.M. Rational vector design and multi-pathway modulation of HEK 293E cells yield recombinant antibody titers exceeding 1 g/l by transient transfection under serum-free conditions. Nucleic Acids Res. 2008;36:e96. doi: 10.1093/nar/gkn423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen T.A., Jo M.H., Choi Y.G., Park J., Kwon S.C., Hohng S., Kim V.N., Woo J.S. Functional Anatomy of the Human Microprocessor. Cell. 2015;161:1374–1387. doi: 10.1016/j.cell.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 38.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 39.Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E., Kim S.T., Eils R., Kim J.S., Bae S. Web-based design and analysis tools for CRISPR base editing. BMC Bioinformatics. 2018;19:542. doi: 10.1186/s12859-018-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data has been uploaded to the Sequence Read Archive under Bioproject: PRJNA518883. All data are available from the authors upon reasonable request.