

Abstract
The mammalian heart undergoes complex structural and functional remodeling to compensate for stresses such as pressure overload. While studies suggest that, at best, the adult mammalian heart is capable of very limited regeneration arising from the proliferation of existing cardiomyocytes, how myocardial stress affects endogenous cardiac regeneration or repair is unknown. To define the relationship between left ventricular afterload and cardiac repair, we induced left ventricle pressure overload in adult mice by constriction of the ascending aorta (AAC). One week following AAC, we normalized ventricular afterload in a subset of animals through removal of the aortic constriction (de-AAC). Subsequent monitoring of cardiomyocyte cell cycle activity via thymidine analog labeling revealed that an acute increase in ventricular afterload induced cardiomyocyte proliferation. Intriguingly, a release in ventricular overload (de-AAC) further increases cardiomyocyte proliferation. Following both AAC and de-AAC, thymidine analog-positive cardiomyocytes exhibited characteristics of newly generated cardiomyocytes, including single diploid nuclei and reduced cell size as compared to age-matched, sham-operated adult mouse myocytes. Notably, those smaller cardiomyocytes frequently resided alongside one another, consistent with local stimulation of cellular proliferation. Collectively, our data demonstrate that adult cardiomyocyte proliferation can be locally stimulated by an acute increase or decrease of ventricular pressure, and this mode of stimulation can be harnessed to promote cardiac repair.
Keywords: Cardiac repair, cardiac remodeling, LV pressure overload, hypertrophy, cardiomyocyte proliferation, aortic constriction
Graphical Abstract
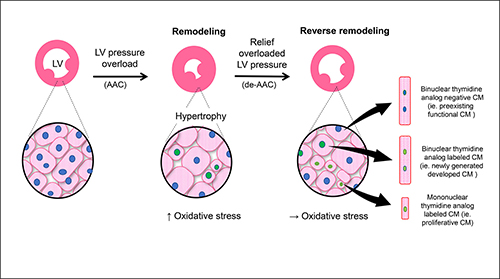
Ascending aortic constriction (AAC) triggers new cardiomyocyte formation due to LV pressure overload (middle panel). Relieving this pressure overload by de-banding or de-AAC (far right panel) further stimulates myocyte formation. Redox stress, secondary to LV pressure alteration, plays a critical role in this process.
1. Introduction
Over the last decade, increasing evidence have suggested that the adult mammalian heart possesses appreciable repair and some regeneration ability, arising in part from the proliferation of existing adult cardiomyocytes.1,2 However, reported cardiomyocyte turnover frequencies fluctuate considerably depending on the degree of injury and exact pathologic condition.1,3,4,5,6 For example, emerging evidence demonstrates that newly formed cardiomyocytes may replace lost cardiomyocytes, albeit at a very low level, in animal model of myocardial infarction (MI).7, 8 Stimulation of new cardiomyocyte formation is thought to be triggered by “left ventricular (LV) loading”, an effort made by the heart to normalize the ventricular stresses that accompanies pathologic states like MI. However, the direct relationship between ventricular afterload and cardiomyocyte proliferation is unclear.
In the present study, we find that an acute increase in ventricular afterload pressures via ascending aortic constriction (AAC) promotes new cardiomyocyte formation as evidenced by cardiomyocyte entry into the cell cycle and labeled thymidine analog incorporation. Excitingly, our data show that relief of elevated afterload and return to the normal condition through removal of the aortic constriction further stimulates cardiomyocyte cell cycle re-entry. Based on gene array analysis, we observed that oxidative stress might play a critical role in regulating cardiomyocyte proliferative capacity in response to changes in ventricular loading pressure. Collectively, our data suggest that in addition to hypertrophic response, cardiomyocyte proliferation also serves to adapt to the alteration in ventricular loading. Importantly, cellular redox regulation plays a critical role in such processes and may be targeted to augment cardiac repair and regeneration.
2. Materials and Methods – (See supplement for expanded methods)
2.1. Experimental design and surgical procedures
All protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Brigham and Women’s Hospital. Eight weeks old C57/BL6J male mice were purchased from Jackson Labs (Bar Harbor, ME). After 1 week of housing in our animal facility, mice were subjected to ascending aortic constriction, as previously described.9 Briefly, mice were fully anesthetized with isoflurane and intubated to maintain proper ventilation. The ascending aorta was exposed via left side parasternal incision, and a 26G needle was attached against the ascending aorta and tightly banded using an 8.0 silk suture. Immediately after banding, the needle was removed to regain aortic blood flow.10 One week after the operation, the chest was again opened, and aortic banding was removed in a subset of animals. Control sham operated animals underwent initial left side parasternal incision with handling of the aorta without constriction of the aorta.
2.2. Echocardiography
Echocardiography was performed at the serial time points using a high-resolution, high-frequency digital imaging system (Vevo 2100, VisualSonics).9 For details, see Supplemental Material.
2.3. TUNEL staining
TUNEL staining was performed for detecting apoptotic cardiomyocytes at postoperative day 21, following the manufacture provided protocol (Sigma-Aldrich, product No.11684795910).
2.4. Thymidine analog labeling
Two thymidine analogs, 5-bromo-2’-deoxyuridine (BrdU, Sigma-Aldrich) and 5-ethynyl-2’-deoxyuridine (EdU, Invitrogen), markers for S phase cell cycle entry, were used for in vivo cell fate tracking. Both thymidine analogs were administered by osmotic mini pumps (Alzet model 2100, 1007D or 1002), according to manufacturer’s instructions. For details, see Supplemental Material.
2.5. Y chromosome labeling by FISH probe
Isolated adult cardiomyocytes were fixed with 70% paraformaldehyde for 30 seconds. Fixed cardiomyocytes were seeded on glass slides and further processed with 0.25% trypsin EDTA for 6 minutes. They were then washed with PBS followed by distilled water; FISH probe was applied for detecting mouse Y chromosome (Chromosome Science Labo. Sapporo, Japan). After denaturing for 5 minutes at 80°C, samples were incubated overnight at 37°C.On the following day, samples were washed with 50% formamide in 2× saline sodium citrate buffer.
2.6. RNA sequencing analysis
Total RNA was extracted from isolated cardiomyocytes using a RNeasy Mini Kit (QIAGEN, Hilden, Germany), according to the instructions of the manufacturer. Complementary DNA synthesis and library preparation was performed using a REPLI-g WTA Single Cell Kit (QIAGEN) and a Nextera XT DNA Library Preparation kit (Illumina, San Diego, CA) according to the instructions of the manufacturer. Sequencing was performed using a HiSeq2500 next-generation sequencing platform with a 2×75 bp end reads (Illumina). Obtained reads were aligned to the mm9 reference genome using Tophat2.11 Counting of reads on each gene was performed by HiSeq.12 Differential expression was calculated using DESeq213 with default parameters, where ranking metrics were calculated as a product of sign of fold change and −log10(adjusted p value). Genes with insufficient read coverage for calculating p-values by DESeq2 were excluded from subsequent analyses. Gene set enrichment was performed using the whole part of pre-ranked list including all remaining genes.14 We considered gene sets with false discovery rate (FDR) <0.25 as statistically significant.
2.7. 8-Isoprostane assay for evaluating oxidative stress
Left ventricular tissues were harvested from day 14 and 21 post-operated animals. Oxidative stress was determined using 8-Isoprostane ELISA Kit (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer instructions. Briefly, 0.01 mL/mg of homogenization buffer (0.1 M phosphate buffer, containing 1 mM EDTA and 0.005% BHT) was added to tissue samples. Samples were homogenized thoroughly using a homogenizer followed by sonication. The homogenized samples were centrifuged at 8,000 × g for 10 minutes and transferred the supernatant to a new tube. An aliquot of this supernatant was used to determine protein concentration. Following the assay protocol, 100 μL of ELISA buffer followed by 50 μL of sample and 50 μL of 8-Isoprostane AChE Tracer were added to 96-well plate. The plate was covered with plastic film and incubated 18 hours at 4°C. The wells were rinsed five times with wash buffer. 200 μL of Ellman’s reagent was added to each well. Samples were incubated in the dark at room temperature for 120 minutes and the readout was collected at 405–420 nm.
2.8. Statistical analysis
Data are presented as mean ± SEM. The difference of means between two groups was tested for using the unpaired two-tailed Student’s t-test. To test for differences across multiple groups, the one factor ANOVA was used with group class as the factor followed by use of the TukeyHSD test to determine if the difference of means between each possible combination of groups were significant. P values less than 0.05 considered as significant.
3. Results
3.1. Ventricular loading and unloading
To study the relationship between cardiomyocyte proliferation and ventricular loading, we used a well-established mouse model of pressure overload secondary to AAC. One-week post AAC, mice were randomized into two groups. The de-AAC group underwent surgery to unload/release the aortic constriction-mediated increased loading pressure through removal of the aortic constriction, while in AAC group the animals received a sham operation without release of the aortic constriction (Supplement 1A). Given that cardiac remodeling is highly dependent on the degree of pressure gradient induced15–17, we assayed and precisely controlled for the degree of ventricular loading. Only mice showing a pressure gradient of 40–60 mm Hg following AAC as measured by Doppler flow imaging were included in this study (Supplement 1B and 1C). Following removal of the suture to relieve aortic constriction, aortic outflow velocity was again measured to ensure that the pressure gradient in de-AAC mice was released (0 mm Hg as shown in Supplement 1C), whereas the pressure gradient in AAC mice remained at 40–60 mm Hg.
As shown in Figure 1A–C, AAC-induced increased LV pressure resulted in ventricular remodeling, as evidenced by increased LV wall thickness and LV chamber dimension, and a corresponding hypercontractile phenotype with augmented fractional shortening, as determined by serial, non-invasive transthoracic echocardiography. In de-AAC animals, ventricular chamber dimensions and function returned to baseline values with relief of the ventricular stress. Consistent with the observed changes in ventricular structure and function, heart weight and tissue fibrosis were elevated in AAC animals but not in de-AAC animals (Figure 1D–E, Supplement 2). Similar to these findings, apoptotic cardiomyocytes were increased in AAC compared to sham or de-AAC animals at postoperative day 21 (Supplement 3). As expected, AAC-induced cardiomyocyte hypertrophy was evidenced by increased cardiomyocyte cross-sectional area, while in de-AAC animals’ cell size returned to baseline levels (Figure 1F). Interestingly, at day 21, the cross-sectional area of cardiomyocytes isolated from de-AAC animals demonstrated a bimodal distribution with peaks at ~200 μm2 (small cardiomyocytes) and ~400 μm2 (mature cardiomyocytes) (Figure 1G, Supplement 4), which merged into a single peak centered at ~300 μm2 of mature cardiomyocytes by day 49 (Figure 1H).
Figure 1. Structural, morphological and cellular remodeling secondary to LV pressure overload and release.

A serial transthoracic echocardiographic measurement indicated that changes in (A) LV diastolic wall thickness, (B) LV diastolic chamber dimension, and (C) fractional shortening after AAC (AAC, black line, *p< 0.05 vs sham, †p <0.05 vs de-AAC). Such alterations were restored to the level similar to sham operated animals (Sham, grey dashed line) at 14 days post removal of the aortic constriction (de-AAC, red line). Similarly, the AAC increased (D) heart weight/body ratio (*p< 0.05 vs sham, †p <0.05 vs AAC) and (E) tissue fibrosis (*p< 0.05 vs sham, †p <0.05 vs AAC) were also diminished in de-AAC groups (red bar) as compared to the AAC group (grey bar) to the level of the sham-operated (light grey bar) mice. N= 4 each for AAC, de-AAC and sham animals. Immunohistochemical analysis using WGA staining showed that (F) cardiomyocyte cross sectional area increased following aortic constriction (AAC: dark grey bar; de-AAC: red bar; Sham: light grey bar). However, two weeks and six weeks following removal of the aortic constriction, the myocyte cross sectional area return to the area similar to sham operated group (light grey bar, *p< 0.05 vs sham, †p <0.05 vs AAC). Further analysis of the distribution of cross-sectional area the WGA stained section, the data revealed that (G) a double peaked distribution of cardiomyocyte cross-sectional area was observed at day 21. (H) This distribution was normalized at day 49 (sham; n=4, de-AAC; n=4). Five hundred cardiomyocyte from cross sectional areas in each mouse, for a total of 2,000 cardiomyocytes, were measured in each group to generate this histogram.
3.2. LV loading or unloading triggers DNA synthesis in cardiomyocytes.
Given the increased number of mononuclear diploid cardiomyocytes noted in ACC hearts, and their increased presence in post-ACC hearts, we decided to examine cell proliferative capacity more directly. As DNA synthesis always precedes cell proliferation, to determine if acute increases (ACC) or decreases (de-ACC) in ventricular pressure stimulate DNA synthesis, we used two different thymidine analogs, 5-bromo-2’-deoxyuridine (BrdU) or ethynyl deoxyuridine (EdU), in pulse-chase assays to report on DNA synthesis in AAC and de-AAC mouse models.
BrdU or EdU was administered via osmotic mini pumps for a period of two weeks following sham or AAC surgery (Figure 2A). The abundance of BrdU positive (BrdU+) cardiomyocytes increased with LV loading greater than 50-fold, compared to sham-operated mice (Figure 2B). At early time points (day 21), BrdU+ cardiomyocytes exhibited smaller myocyte cross-sectional area as compared to non-labeled cardiomyocytes but were comparable in size by day 49 (Figure 2C–D), the pattern expected for proliferating cells. To further validate these data, we also assessed the size of isolated cardiomyocytes using EdU labeling. Similar to BrdU+ cardiomyocytes, EdU+ cardiomyocytes were initially smaller in size, but were indistinguishable from EdU-cardiomyocytes after 7 weeks post AAC (Figure 2E).
Figure 2. Pressure overload triggers cardiomyocytes proliferation.

A. Study design and time line. B. AAC increases the number of BrdU+ cardiomyocytes at 21 days and 49 days post-AAC (*p <0.05 vs sham, sham, light grey bar, n=4, de-AAC, dark grey bar, n=5). C. Representative images of BrdU+ cardiomyocytes at 21 days and 49 days post-AAC (Scale bar; 5 μm. Hoechst, blue; BrdU, green, a-actin, red; WGA, white). D. Distribution of cross-sectional area between BrdU+ and BrdU− cardiomyocytes. BrdU+ cardiomyocytes exhibit smaller cross-sectional area at day 21 in comparison to the cross-sectional area at day 49. Cross sectional areas of forty to 80 BrdU+ cardiomyocytes per sample, for a total of 320 cardiomyocytes were measured. E. Size differences of EdU+ and EdU− cardiomyocyte (*p < 0.05 vs EdU−, †p < 0.05 vs EdU+ at day 21). Eighty EdU+ and 100 EdU- isolated cardiomyocytes’ length and width were measured.
To determine whether de-AAC affected the initiation of DNA synthesis in cardiomyocytes, we performed double thymidine labeling with administration of BrdU for a period of one week immediately after AAC and followed by administration of EdU for one more week immediately after de-AAC or sham operation (Figure 3A). Remarkably, the total number of BrdU+, EdU+, BrdU+/EdU+ cardiomyocytes was significantly increased in de-AAC relative to the already elevated levels seen in AAC animals, suggesting greater cardiomyocyte incorporation of thymidine analogue with alleviation of ventricular pressure (Figure 3B), and implicating perturbation in ventricular pressure in either direction, stimulate cardiomyocyte proliferative capacity. Among thymidine analog labeled cardiomyocytes, the greatest fractional increase in de-AAC relative to AAC animals were observed in Brdu+/Edu+ double-labeled cells (Figure 3C–E). de-ACC treatment led to a 3-fold increase in the number of BrdU+/EdU+ double-labeled cells.
Figure 3. Removal of aortic constriction one-week post-AAC induces further cardiomyocyte proliferation in LV area distant from injury.

A. Study design and timeline. B. Percentage of total thymidine analog positive cardiomyocytes (*p <0.05 vs sham, †p <0.05 vs AAC, n=6 in each group). de-AAC further increases thymidine analog positive cardiomyocytes. C. Representative images of thymidine analog labeling in de-AAC heart sections (Scale bar = 50 μm and 10 μm in the inset). Green and red arrows indicate BrdU+ and EdU+ cardiomyocytes, respectively. D. Double thymidine analog positive cardiomyocyte in heart section (left panel) and isolated cardiomyocyte (right panel). Scale bars indicate 10 μm and 20 μm in left and right panel, respectively. E. Percentage of double thymidine analog positive cardiomyocyte (indicated yellow) in de-AAC is significantly increased compared to that in AAC (*p <0.05 vs AAC). F. Representative images of de-AAC heart sections (upper panels). Red arrows indicate EdU+ cardiomyocytes. Dotted circles indicate EdU+ nuclei in cardiomyocytes (lower right). Scale bars indicate 20 μm. 27.4% (left) and 32.2% (right) of thymidine analog positive cardiomyocytes reside in close proximity in BrdU+ and EdU+ cardiomyocytes, respectively. (n = 6 in each group). G. Representative images of Ki67+ α-actin+ cells. Scale bars indicate 10 μm. Ki67+ α-actin+ cells are increased in de-AAC compared to AAC hearts at post-operative day 9 (2 days after de-AAC, *p <0.05).
Interestingly, within de-AAC hearts, approximately one third of thymidine analog-positive cardiomyocytes (BrdU+; 29.9%, EdU+; 32.0%) were localized in close proximity to each other (Figure 3F), consistent with what would be expected of newly divided cell clusters. For further verification, we determined the number of ACC and de-AAC cardiomyocytes expressing Ki67, a marker of cell proliferation. Our data (Figure 3G) showed that the number of Ki67+ cardiomyocytes was significantly increased in de-AAC relative to AAC at post-operative day 9, ie., 2 days post de-AAC. Collectively, these data support the hypothesis that a subpopulation of cardiomyocytes that initiated DNA synthesis due to the induced pressure overload (at week 1) continued to synthesize DNA and proliferate following normalization of ventricular pressure to release the injury/stress.
3.3. de-AAC hearts show increase in mononuclear diploid cardiomyocytes.
It has been shown that mononuclear diploid cardiomyocytes exhibit a greater capacity to enter the cell cycle than multinuclear cardiomyocytes.18, 19 Therefore, we were interested to assess how AAC and de-AAC may affect the ploidy and number of nuclei in isolated cardiomyocytes. Our results show that 21 days post de-AAC, 57.7% of thymidine analog-positive cardiomyocytes were mononuclear compared to 37.7% with AAC alone. However, levels of single nuclear thymidine analog-positive cardiomyocytes were similar at 49 days post operation (Figure 4A). Ploidy was further assessed by measuring Y chromosome abundance. Using this measure, we found that 61.8% and 69.7% of sham and AAC cardiomyocytes were diploid, respectively. Interestingly, following de-AAC diploid mononuclear cardiomyocyte abundance increased to 88.1% (Figure 4B upper panels). However, these differences in ploidy were not observed in multi-nuclear cardiomyocytes (Figure 4B lower panels). Taken together, our data indicate that the subpopulation of mononuclear diploid cardiomyocytes, which is considered more proliferative, increased at day 21 in de-AAC, and develop into mature multinucleated cardiomyocytes at day 49.
Figure 4. A fraction of single nuclear diploid cardiomyocytes is further increased in de-AAC compared to AAC at postoperative day 21.

A. Representative images of isolated EdU+ cardiomyocytes (left panels, scale bar = 50 μm). Percentage of thymidine analog positive single nuclear cardiomyocyte is increased in de-AAC compared to that in AAC at day 21 (*p <0.05 vs sham, †p <0.05 vs AAC at day 21), which is restored to AAC level at day 49 (‡p <0.05 vs sham at day49). B. Cardiomyocytes ploidy analysis at day 21, following AAC and de-AAC, respectively. Y chromosomes are detected by FISH. Each single spot indicates a single Y chromosome. One spot in single nuclei indicates diploid cell (upper left panel for example, scale bar = 10 μm). Percentage of single nuclear diploid cardiomyocyte is increased in de-AAC compared to sham (*p <0.05 vs sham), while there is no difference in multi nuclear cardiomyocyte.
3.4. Thymidine analog-positive cardiomyocytes arise from pre-existing cardiomyocytes
To investigate the origins of thymidine analog-positive cardiomyocytes in de-AAC hearts, we conducted genetic fate mapping experiments20. We generated a mouse line linking GFP expression to the myocyte specific Myh6 promotor to ensure cardiomyocyte specificity of GFP expression and performed thymidine analog labeling (Figure 5A–B). In order to determine whether or not the EdU+ proliferative cells were cardiomyocytes (EdU+, GFP+) or cells of a different lineage (EdU+, GFP−), we conducted consecutive intra-peritoneal injections of 2 mg tamoxifen for three days to induce GFP expression in approximately 80% of cardiomyocytes (Figure 5B–D). Subsequently, we performed AAC followed by de-AAC and pulse with EdU to label proliferative cells with active DNA-synthesis. We found that %GFP labeled EdU− and EdU+ cardiomyocytes isolated from de-AAC hearts were similar (EdU− 87.8%, EdU+ 85.7%), indicating that these EdU+ cardiomyocytes originated from pre-existing cardiomyocytes (Figure 5E–F).
Figure 5. Genetic fate mapping study for accessing the origin of thymidine analog positive cardiomyocytes.

A. Scheme of genetically engineered mouse for genetic fate mapping. Administration of tamoxifen can induce GFP specifically in cardiomyocytes. B. Study timeline. C. Representative images of GFP expressing cardiomyocytes after each tamoxifen injection. D. Total 6 mg of tamoxifen can induce GFP in about 80% of cardiomyocytes (n = 3 in each group). E. Representative images for GFP/EdU double positive cardiomyocytes. A total of 6 mg of tamoxifen was administered to induce GFP expression in cardiomyocytes. AAC operation was performed at 9-weeks followed by de-AAC operation one week after initial operation. F. The frequency of GFP positivity was equivalent between EdU negative and positive cardiomyocytes. This result indicates that EdU positive cardiomyocytes were generated from pre-existing cardiomyocytes
3.5. Mononuclear cardiomyocytes exhibit both proliferative and myogenic characteristics in gene expression.
Our results suggest that alteration of ventricular loading pressure may be sufficient to initiate DNA synthesis in smaller mono-nuclear diploid cardiomyocytes. To understand how de-ACC impacts gene expression patterns in the subpopulation of mononuclear cardiomyocytes we compared the RNA expression profiles between mononuclear and binuclear cardiomyocytes before and after de-ACC. We isolated and sequenced RNA from 80–100 independently collected mono or binuclear cardiomyocytes obtained from de-AAC and sham operated mice at day 14 post ACC, ie. 7 days after de-AAC. The gene expression profiles were analyzed using HiSeq. Consistent with the increased proliferation capacities as noted in the experimental data, gene set enrichment analysis (GSEA) identified a gene set associated with proliferating cell nuclear antigen (PCNA) as one of the most significantly enriched transcripts (MORF_PCNA gene set, p= 0.000, FDR= 0.078, Figure 6A–B) in de-ADD mononuclear cardiomyocytes compared to mononuclear cardiomyocytes isolated from sham-treated individuals. Furthermore, global transcriptomic analysis comparing binuclear and mononuclear de-ACC cardiomyocytes revealed an enrichment in the abundance of multiple hallmarks of myogenesis in mononuclear cells as compared to binuclear cells from identical mice (N= 2, HALLMARK_MYOGENESIS gene set, p= 0.004, FDR= 0.111, Figure 6C–D). Collectively, our data indicate that mononuclear cardiomyocytes specifically in the de-ACC group have a potential to reenter the cell cycle for proliferation and can develop into mature cardiomyocytes.
Figure 6. Global gene expression analysis on isolated cardiomyocytes.

A. Gene set enrichment analysis (GSEA). A global expression profile of mononuclear cells obtained from deAAC mice (N = 2) was compared with that obtained from sham mice (N = 2) at day 14. Enrichment profile for MORF_PCNA (a set of genes showing statistically significant co-expression with Pcna in MORF dataset) is shown with its normalized enrichment score (NES), nominal p value (P), and false discovery rate (FDR). B. Summary of gene expression in the MORF_PCNA dataset. The heatmap shows top 40 differentially expressed genes within the gene set. Expression level of each gene was normalized as a log2 fold change from the median read count of all samples. Asterisks indicate core enrichment genes. C. GSEA comparing mono- and bi-nuclear cells obtained from de-AAC mice (N = 2). Enrichment plot shows the result for the HALLMARK_MYOGENESIS gene set (genes involved in myogenesis). D. Differentially expressed genes in the gene set.
3.6. Pressure overload induced oxidative stress inhibits proliferative capacity of cardiomyocytes
Oxidative stress has been shown to be involved in pressure overload.21 Consistent with the literature, we also found that 8-Isoprostane levels were increased in the cells isolated from AAC animals as compared to sham treated as well as de-ACC mice (Figure 7A). To determine if elevated oxidative stress is normalized following de-AAC, we explored candidate genes controlling redox balance by microarray and performed a subsequent pathway enrichment analysis (Supplement 5 A, B). Our data revealed four genes (Nox4, Cyp1b1, Gcdh, Cyp27a1) known to be associated with the redox process differentially expressed between the AAC and de-AAC groups. Moreover, the elevated gene expression levels of Nox4 and its related gene, p22phox were restored to the levels of sham in the de-AAC group (Figure 7B, Supplement 5C, D).
Figure 7. Abscising from oxidative stress after de-AAC contributes to cardiomyocyte proliferation.

A. The levels of oxidative stress in cardiac specimens were analyzed by 8-isoprostane assay. The levels of 8-isoprostane were increased in AAC heart in both postoperative day 14 and day 21, which were decreased to sham operation group levels after de-banding (sham; n = 3, AAC; n = 4, de-AAC; n = 4, *p<0.05). B. mRNA expression levels of NADPH oxidase subunits Nox4 and p22phox were increased in AAC compared to sham at postoperative day 814, and were comparable in de-banded heart (sham; n = 3, AAC; n = 4, de-AAC; n = 4). C. Study timeline. D. Total number of cardiomyocyte to find 50 EdU+ cardiomyocytes was increased in AAC heart compare to de-AAC heart, which was canceled by anti-oxidative agent, NAC, injection (de-AAC; n = 5, AAC/Vehicle; n = 5, AAC/NAC; n = 6, *p<0.05).
Hypoxic environments have been shown to induce cardiomyocyte repair or regeneration even in adult mammalian hearts, albeit at a very low frequency.22 Therefore, we hypothesized that redox homeostasis is critical for regulating proliferative capacity of cardiomyocytes. To test whether reducing the elevated oxidative stress could alter the proliferative capacity of cardiomyocytes in AAC hearts, we injected 150 mg/kg/day antioxidant N-acetylcysteine (NAC) intraperitoneally for 14 days starting at post-AAC operative day 7 (Figure 7C). Two weeks after NAC treatment, cardiomyocytes were isolated and suspended in PBS. An equal volume of the suspension was dispensed into each well in a 96 well plate, and we determined what percentage of cardiomyocytes were EdU+. A detail method is described in the Supplemental Figure 6. As shown in Figure 7D, the NAC-injected AAC group showed a significant reduction in the total cardiomyocyte count needed to reach 50 EdU+ cardiomyocytes as compared to AAC only group. As such, NAC treated AAC group showed higher EdU+ cardiomyocytes compared to AAC group without NAC treatment. The data support the notion that oxidative stress occurred during pressure overload exerts a negative impact on cardiomyocyte proliferative capacity, which reduces the overall myocyte proliferative capacity. Collectively, our data support the hypothesis that ameliorating oxidative stress, induced by pressure overload, may trigger cardiomyocytes endogenous repair and regenerative capacities.
3.7. AAC and de-AAC injury models exhibit distinct proliferative microenvironments.
It was reported that cardiomyocyte proliferation is most commonly observed in the areas bordering the injury sites, where cardiomyocytes are replaced by fibrosis.2 In our studies, we also noted that most thymidine analog-positive cardiomyocytes were located near fibrotic regions of the heart. To quantify this effect, we measured the number of thymidine-positive cardiomyocytes within the fibrotic area and border zone (defined as within 200 μm from the edge of fibrosis) and the remote area (>200 μm from the border of fibrosis) in ACC and de-ACC hearts as illustrated in Figure 8A. One-week following the final surgical procedure i.e. aortic banding for AAC group and de-banding for de-AAC group, more than 70% of thymidine analog-positive cardiomyocytes were found immediately adjacent to the fibrotic and border area in each group. At two-weeks post surgical operation, the percentage of thymidine analog-positive cardiomyocytes within fibrotic or peri-fibrotic area decreased to around 40% in both groups. In contrast, only in the de-AAC group but not in AAC group, thymidine analog-positive cardiomyocytes in areas remote to injury increased to around 40% (Figure 8B–C). These data demonstrate that DNA synthesis in cardiomyocytes is initiated proximate to regional cardiomyocyte damage/death following AAC, whereas with relief of overloaded ventricular pressure following de-AAC, DNA synthesis is observed in areas remote to injury. These findings may suggest that cardiomyocyte proliferation is differentially regulated post ACC and de-AAC.
Figure 8. De-AAC induces further cardiomyocyte regeneration in LV area distant from injury.

A. Illustration of examined LV areas in AAC and de-AAC heart sections. B. Representative pictures from AAC (left and middle) and de-AAC (right) heart sections. Left panels show massive fibrotic and peri-fibrotic areas. C, Higher numbers of thymidine analog positive cardiomyocytes were observed in remote areas of de-AAC hearts compared to AAC. Error bars indicate SED for remote area, *p <0.05 vs AAC at 2nd week.
4. Discussion
Recent data using rodent models have challenged the paradigm that the adult heart is a post-mitotic terminally differentiated organ fully devoid of myogenetic regenerative capacity, and have instead suggested that, during physiological and pathological conditions, new cardiomyocyte formation may occur.23 Our findings suggest that alteration of ventricular pressure may affect cardiac myogenesis, albeit in a rather limited fashion. Moreover, we provide evidence that the AAC and de-AAC models differentially regulate the cardiac microenvironment, regulating dynamic changes in cardiomyocyte proliferation in a distinct manner. After a period of injury, i.e., increased ventricular loading, new cardiomyocyte formation is stimulated, particularly proximal to areas that may have experienced the greatest degree of local wall stress. However, following periods of increased stress, a return to the normal hemodynamic state, mimicking treatment, can also stimulate new cardiomyocyte formation, particularly among a select group of cells initially responsive to increased pressure load. According to our findings, LV pressure load provides cardiomyocytes, especially mononucleated diploid small cardiomyocytes, a cue for proliferation and increases oxidative stress at the same time, resulting in a limited incidence of cardiomyocyte proliferation. After removal of pressure loading, the level of oxidative stress is rapidly diminished, while the proliferation signal could be retained.
We fully recognize that thymidine analog incorporation into DNA is not exclusive to cardiomyocyte proliferation but also includes multi-nucleation or polyploidization.24 Our data showed that thymidine analog positive cardiomyocytes are initially small in size with a single nucleus and develop into larger multinucleated cardiomyocytes over time after aortic pressure loading/unloading. Importantly, ploidy analysis showed that the fraction of single nuclear diploid cardiomyocytes was increased especially after de-AAC. These findings, therefore, indicate that DNA synthesis initiated after LV pressure fluctuation is followed by karyokinesis and cytokinesis events. Additionally, histochemical analysis revealed that about one third of thymidine analog positive cardiomyocytes were found immediate adjacent to each other, which further supports the notion that those cells arose from a cytokinesis event.
Significant efforts have been made to identify whether the main source of newly generated cardiomyocytes is from proliferation of pre-existing cardiomyocytes or de novo cardiomyocytes stemmed from the myogenic differentiation of endogenous cardiac progenitor cells.2,20 Recent report by Senyo and colleagues suggests that proliferation of pre-existing cardiomyocytes represents the main source of newly generated cardiomyocytes in normal aging and following injury20. Consistent with their data, our data obtained from pulse-chase genetic fate mapping experiments also suggest that thymidine analog-positive cardiomyocytes in de-AAC heart are generated by the division of pre-existing cardiomyocytes, at least in cells already expressing αMHC. It is noteworthy that our methodology only labels cells that already express αMHC, and therefore we may be underestimating the number of newly generated cardiomyocytes. Moreover, we show that more than 50% of thymidine analog-positive cardiomyocytes are mononuclear at early time points after de-AAC, and that the population of bi-nucleated cardiomyocytes increases at later time point, a pattern consistent with the maturation of newly generated cardiomyocytes. Further support can be derived from our global gene expression profiling, which indicates that single nuclear cardiomyocytes express a set of genes associated with cell proliferation and myogenesis. While it has been reported that Neureglin1/Erb4 signaling can induce proliferation of mono-nucleated but not poly-nucleated cardiomyocytes in mice,25 it is important to highlight that there is no specific transcription factor has been identified for cardiac progenitor and post further challenge in understanding the potential endogenous cardiac regenerate/repair capacities. Nonetheless, our data further support the idea that some degree, albeit limited, of endogenous repair/regeneration capacity remains, at least, in rodent hearts.
Previous reports linked LV positive remodeling with cardiomyocyte regeneration.5–8 Angert and colleagues have shown that cardiac regeneration is accelerated in a mouse model of isoproterenol induced hypertrophy.7 Boström and colleagues reported that newly generated cardiomyocytes were observed during physiological LV hypertrophy.5 Although evidence of cardiac regeneration in humans has not been directly demonstrated, Wohlschlaeger and coworkers showed that the degree of polyploidy in cardiomyocytes increases in heart failure patients.26
Our data may reveal critical mechanisms associated with the functional benefit seen in patients with overall improvement in ventricular function, following unloading of ventricular stress/burdens with anti-hypertensive agents or relief of aortic stenosis. The clinical situation that is most relevant to our study is that of patients with the use of a left ventricular assist device (LVAD). A number of patients with severe heart failure exhibit improved LV function after LVAD therapy and are able to survive without LVAD or cardiac transplantation thereafter.28–30 Consistent with our experimental finding, the number of polyploid cardiomyocytes in these patients is reduced while diploid cardiomyocytes increased in HF patients with hemodynamic unloading with LVAD.26 Similarly, Canseco et al, recently showed evidence of cardiomyocyte mitosis and proliferation in adult human hearts following implantation of LVAD.27
In summary, while the rate of new cardiomyocyte formation in diseased hearts is quite low,31, 32 our current study sheds light on how cardiomyocyte mitosis and proliferation could be stimulated by LV pressure overloading and unloading (see graphical abstract). Understanding the specific stimuli that give rise to new cardiomyocyte formation under alteration of hemodynamic states may aid in the future development of therapeutic strategies to stimulate/augment the endogenous capacity of cardiac regeneration and repair.
Supplementary Material
Highlights.
LV pressure loading induces cardiomyocyte hypertrophy, and LV pressure unloading induces LV reverse remodeling with the return of hypertrophied cardiomyocytes toward normal size. In the process of LV reverse remodeling, the fraction of smaller cardiomyocytes increases.
Smaller cardiomyocytes observed during LV reverse remodeling are characterized by single diploid nucleus, entry into the cell cycle, and expression of proliferation-related genes.
Fate mapping shows that these cardiomyocytes originate from preexistent cardiomyocytes. Importantly, these newly generated cardiomyocytes can further develop into mature functional cardiomyocytes.
Although, LV pressure overload can cue cell division, sustained pressure overload triggers oxidative stress, which adversely affects cardiomyocyte proliferation overtime. Relieving the LV pressure overload establishes the redox balance and triggers additional cardiomyocyte proliferation.
Acknowledgments
We wish to sincerely thank the late Dr. Michael Bauer for his inspiration, creativity and intellectual contribution in the initiation of this research work. Dr. Bauer unfortunately passed during a marathon in Austria; his contrition to this project is eternally remembered and appreciated. In addition, we are grateful for language editorial support by Dr. Megan Mayerle (Stanford CVI). We also wish to thank the Cardiovascular Physiology Core at Brigham and Women’s Hospital and S. Ngoy for surgical assistance.
Sources of Funding
This work was supported in part by NIH HL R01HL093148 and R01HL099073 (to RL), a Grant-in-Aid for Scientific Research (C) from the Ministry of Education, Culture, Science and Technology of Japan and the Takeda Science Foundation (to KU). AO was supported by American Heart Association Scientist Development Grant 17SDG33660794.
Abbreviations
- LV
Left ventricle
- LVAD
LV assist device
- AAC
ascending aortic constriction
- de-AAC
ascending aortic de-constriction
- BrdU
5-bromo-2’-deoxyuridine
- EdU
5-ethynyl-2’-deoxyuridine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
References
- 1.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S and Frisen J. Evidence for cardiomyocyte renewal in humans. Science (New York, NY). 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J and Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nature medicine. 2007;13:970–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soonpaa MH and Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. The American journal of physiology. 1997;272:H220–6. [DOI] [PubMed] [Google Scholar]
- 4.Walsh S, Ponten A, Fleischmann BK and Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovascular research. 2010;86:365–73. [DOI] [PubMed] [Google Scholar]
- 5.Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A and Spiegelman BM. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waring CD, Vicinanza C, Papalamprou A, Smith AJ, Purushothaman S, Goldspink DF, Nadal-Ginard B, Torella D and Ellison GM. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. European heart journal. 2014;35:2722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angert D, Berretta RM, Kubo H, Zhang H, Chen X, Wang W, Ogorek B, Barbe M and Houser SR. Repair of the injured adult heart involves new myocytes potentially derived from resident cardiac stem cells. Circulation research. 2011;108:1226–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S and Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nature medicine. 2007;13:962–9. [DOI] [PubMed] [Google Scholar]
- 9.Bauer M, Cheng S, Unno K, Lin FC and Liao R. Regional cardiac dysfunction and dyssynchrony in a murine model of afterload stress. PloS one. 2013;8:e59915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarnavski O, McMullen JR, Schinke M, Nie Q, Kong S and Izumo S. Mouse cardiac surgery: comprehensive techniques for the generation of mouse models of human diseases and their application for genomic studies. Physiological genomics. 2004;16:349–60. [DOI] [PubMed] [Google Scholar]
- 11.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders S, Pyl PT and Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love MI, Huber W and Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandler H and Dodge HT. LEFT VENTRICULAR TENSION AND STRESS IN MAN. Circulation research. 1963;13:91–104. [DOI] [PubMed] [Google Scholar]
- 16.Hood WP Jr., Rackley CE and Rolett EL. Wall stress in the normal and hypertrophied human left ventricle. The American journal of cardiology. 1968;22:550–8. [DOI] [PubMed] [Google Scholar]
- 17.Grossman W, Jones D and McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. The Journal of clinical investigation. 1975;56:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien CL, Makita T, Lusis AJ, Kumar SR and Sucov HM. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet. 2017;49:1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, Abderrahman Y, Chen R, Garcia JA, Shelton JM, Richardson JA, Ashour AM, Asaithamby A, Liang H, Xing C, Lu Z, Zhang CC and Sadek HA. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature. 2015;523:226–30. [DOI] [PubMed] [Google Scholar]
- 20.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP and Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr., Gojon G Jr., Wang R, Karusula N, Nicholson CK, Calvert JW and Lefer DJ. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W and Sadek HA. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. [DOI] [PubMed] [Google Scholar]
- 23.Carvalho AB and de Carvalho AC. Heart regeneration: Past, present and future. World journal of cardiology. 2010;2:107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hesse M, Doengi M, Becker A, Kimura K, Voeltz N, Stein V and Fleischmann BK. Midbody Positioning and Distance Between Daughter Nuclei Enable Unequivocal Identification of Cardiomyocyte Cell Division in Mice. Circ Res. 2018;123:1039–1052. [DOI] [PubMed] [Google Scholar]
- 25.Bersell K, Arab S, Haring B and Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–70. [DOI] [PubMed] [Google Scholar]
- 26.Wohlschlaeger J, Levkau B, Brockhoff G, Schmitz KJ, von Winterfeld M, Takeda A, Takeda N, Stypmann J, Vahlhaus C, Schmid C, Pomjanski N, Bocking A and Baba HA. Hemodynamic support by left ventricular assist devices reduces cardiomyocyte DNA content in the failing human heart. Circulation. 2010;121:989–96. [DOI] [PubMed] [Google Scholar]
- 27.Canseco DC, Kimura W, Garg S, Mukherjee S, Bhattacharya S, Abdisalaam S, Das S, Asaithamby A, Mammen PP and Sadek HA. Human ventricular unloading induces cardiomyocyte proliferation. Journal of the American College of Cardiology. 2015;65:892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dandel M, Weng Y, Siniawski H, Potapov E, Lehmkuhl HB and Hetzer R. Long-term results in patients with idiopathic dilated cardiomyopathy after weaning from left ventricular assist devices. Circulation. 2005;112:I37–45. [DOI] [PubMed] [Google Scholar]
- 29.Simon MA, Kormos RL, Murali S, Nair P, Heffernan M, Gorcsan J, Winowich S and McNamara DM. Myocardial recovery using ventricular assist devices: prevalence, clinical characteristics, and outcomes. Circulation. 2005;112:I32–6. [DOI] [PubMed] [Google Scholar]
- 30.Mancini DM, Beniaminovitz A, Levin H, Catanese K, Flannery M, DiTullio M, Savin S, Cordisco ME, Rose E and Oz M. Low incidence of myocardial recovery after left ventricular assist device implantation in patients with chronic heart failure. Circulation. 1998;98:2383–9. [DOI] [PubMed] [Google Scholar]
- 31.Oberpriller JO and Oberpriller JC. Response of the adult newt ventricle to injury. The Journal of experimental zoology. 1974;187:249–53. [DOI] [PubMed] [Google Scholar]
- 32.Jopling C, Sleep E, Raya M, Marti M, Raya A and Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.