

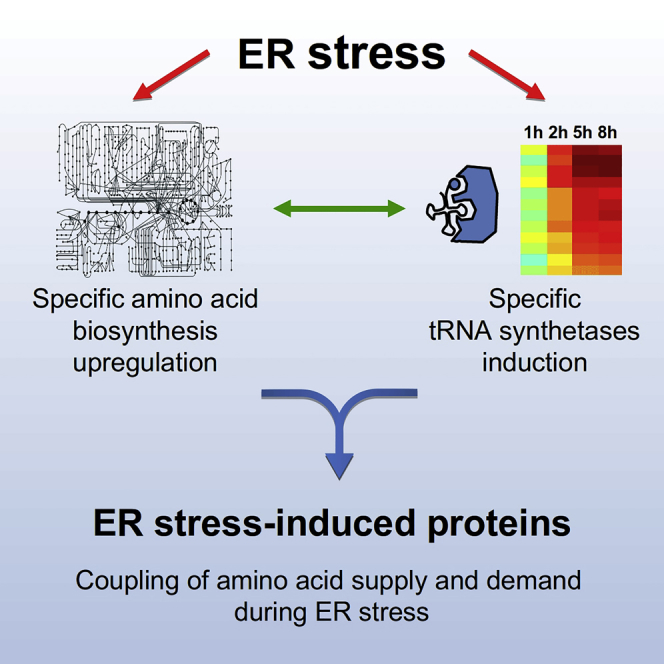
Summary
The endoplasmic reticulum (ER) stress response, also known as the unfolded protein response (UPR), is a complex cellular response to ER protein misfolding that involves transcriptional regulatory branches and a PERK-mediated translational regulatory branch.
Here we revealed that amino acid biosynthesis regulation is coupled to protein synthesis demands during ER stress. Specifically, we demonstrated that the UPR leads to PERK-dependent induction in the biosynthesis of specific amino acids, and to upregulation of their corresponding tRNA synthetases. Furthermore, we found that sequences of UPR-upregulated proteins are significantly enriched with these UPR-induced amino acids. Interestingly, whereas the UPR leads to repression of ER target proteins, we showed that secreted proteins tended to escape this repression and were highly enriched for the UPR-induced amino acids.
Our results unravel coordination between amino acid supply, namely, biosynthesis and tRNA loading, and demand from UPR-induced proteins under ER stress, thus revealing an additional regulatory layer of protein synthesis.
Subject Areas: Cell Biology, Expression Study, Membrane System, Transcriptomics
Graphical Abstract
Highlights
-
•
Coordination of amino acid supply and protein synthesis demand during ER stress
-
•
Specific amino acid biosynthesis and cognate tRNA synthetases induction by the UPR
-
•
UPR-induced amino acids support amino acid demand of UPR-upregulated proteins
-
•
UPR-induced amino acids are highly enriched within secreted proteins
Cell Biology; Expression Study; Membrane System; Transcriptomics
Introduction
Endoplasmic reticulum (ER) stress elicits a complex cellular program, also termed the unfolded protein response (UPR). This response enables cells to cope with dynamic changes in protein folding and processing demands in the ER (Ron and Walter, 2007). The UPR in mammalian cells consists of three major branches: the PERK branch, which leads to global inhibition of translation initiation through eIF2α phosphorylation, and two transcriptional branches, namely, the IRE1-XBP1 and the ATF6 branches (Pavitt and Ron, 2012). Importantly, one of the major secondary effectors downstream of PERK is the transcription factor ATF4, the translation of which is highly induced in an eIF2α phosphorylation-dependent manner during ER stress (Vattem and Wek, 2004) and is known to induce the transcription of genes essential for ER stress adaptation (Rutkowski and Kaufman, 2007).
We and others have characterized another hallmark of the UPR: enhanced repression of ER-targeted proteins (Gonen et al., 2019, Guan et al., 2017, Reid et al., 2014). We found that this repression is mainly at the level of translation and occurs in many cell types (Gonen et al., 2019). Furthermore, using ribosome footprint profiling, we identified three major ER stress gene expression programs in mouse embryonic fibroblasts (MEFs): early induction, late induction, and repression, all of which are PERK dependent (Gonen et al., 2019). Notably, late induction genes are enriched for amino acid biosynthesis pathways (Gonen et al., 2019).
Amino acid biosynthesis genes were previously shown to be induced in response to ER stress. Harding et al. demonstrated that amino acid metabolism is upregulated in response to ER stress, in an ATF4-dependent manner (Harding et al., 2003). Furthermore, ATF4−/− MEFs were found to have an amino acid deficiency, and thus require supplemental non-essential amino acids for their growth (Harding et al., 2003). ATF4−/− cells have also been shown to experience oxidative stress, and supplementation of cysteine, glutathione (GSH), or other reducing agents rescues their growth defects (Harding et al., 2003). In chronic ER stress, ATF4 binds to the promoters of various amino acid biosynthesis genes, and also to amino acid transporters (Han et al., 2013). Moreover, serine metabolism induction was highlighted in response to ER stress (Rendleman et al., 2018). In addition, ATF4 target genes were found to be enriched with tRNA synthetases (Han et al., 2013). In pancreatic β-cells, increased protein synthesis during chronic ER stress involved the transcriptional induction of an amino acid transporter network, and of tRNA synthetases, in an ATF4-dependent manner (Krokowski et al., 2013). The ATF4-induced network of amino acid transporters has also been shown to be involved in alleviating the inhibition of protein synthesis during prolonged ER stress, by promoting mTOR activation (Guan et al., 2014). In turn, mTOR can contribute to ATF4 expression in an eIF2α phosphorylation-independent manner, by mediating ATF4 mRNA stabilization (Ben-Sahra et al., 2016, Park et al., 2017).
Here, we set out to further characterize the late response to ER stress. Using ribosome footprint profiling in ER-stressed wild-type (WT) and PERK−/− MEFs (treated with thapsigargin [Tg]) (Gonen et al., 2019), we found that ER stress, particularly a late-specific ER stress induction gene expression program, is highly enriched in two categories: amino acid biosynthesis and tRNA synthetases, in line with previous studies. Herein, we observed induction in the biosynthesis of a specific subset of amino acids, with highly significant congruent upregulation of their corresponding tRNA synthetases. This observation led us to hypothesize that this coordinated induction during ER stress might be coupled to differential demands of amino acids from the amino acid sequences of the UPR-induced proteins. Indeed, our analyses showed that sequences of induced proteins are significantly enriched with these amino acids. Finally, we showed that this UPR amino acid signature is preferentially enriched within subclasses of ER targets that tended to escape the enhanced repression during ER stress, namely, secreted and extracellular matrix proteins.
Results
Secreted Proteins Escape the Repression of ER Targets during ER Stress
Previously, we and others (Gonen et al., 2019, Guan et al., 2017, Reid et al., 2014) described widespread enhanced repression of ER-targeted proteins during ER stress. Furthermore, we showed that this repression occurs mainly at the level of translation and starts as early as 1 h into the stress, thus affecting the expression of hundreds of mRNAs encoding ER targets (Gonen et al., 2019). These include membrane proteins, glycoproteins, and disulfide bond-containing proteins (Gonen et al., 2019).
We therefore wished to perform a more fine-tuned analysis of expressed ER targets. One of the major mechanisms of ER localization is that of the signal recognition particle, which is responsible for the localization and co-translational import of many ER processed proteins that contain a signal peptide in their N terminus (Keenan et al., 2001). We therefore utilized our ribosome footprint profiling dataset of MEFs that were subjected to short and long ER stress induced by the SERCA inhibitor Tg (Gonen et al., 2019) and aimed to examine all signal peptide-encoding mRNAs (see Methods). We performed hierarchical clustering of the expression levels of the subset of signal peptide-encoding mRNAs that changed at least 2-fold during a time course of short and long ER stress treatments. Our analysis revealed that whereas about 57% of the signal peptide-containing proteins were significantly repressed, the remaining proteins were actually induced (Figure 1A). We then sought to identify the functional differences between the repressed and the enhanced signal peptide-encoding mRNAs. We observed that over 60% of the repressed signal peptide-encoding mRNAs were membrane proteins, consistent with our previous results (Gonen et al., 2019). On the other hand, signal peptide-encoding mRNAs with induced expression during ER stress were selectively enriched for extracellular region and secreted proteins (Figure 1B). Therefore, it seems that different subsets of ER targets are regulated differently, with membrane proteins being selectively repressed, whereas many secreted proteins escaping this repression and being actually enhanced.
Figure 1.

Analysis of Signal Peptide Encoding mRNAs Reveals that Secreted Proteins Tend to Escape Repression upon ER Stress
(A) Hierarchical clustering analysis was performed using 236 signal peptide-encoding mRNAs, with translation change of at least 2-fold compared with control in any of the time points examined following Tg treatment (1, 2, 5, and 8 h) in WT MEFs. The heatmap depicts hierarchal clustering of mRNA translation levels (transcripts per million [TPM] of ribosome footprint profiling in the respective samples, log2), according to Spearman's correlation. Z score normalization was further performed for visualization purposes. The mRNAs were clustered into two distinct groups: 102 induced mRNAs (blue square) and 134 repressed mRNAs (red square) following Tg treatment.
(B) Functional enrichment of induced signal peptide encoding genes was analyzed using DAVID (Huang da et al., 2009). The enriched terms were extracellular region and secreted proteins. False discovery rate (FDR)-corrected p values are indicated.
Late-Specific ER Stress-Induced Genes Include Amino Acid Biosynthesis Genes and tRNA Synthetases
Next, we set out to further characterize the candidate genes important for adaptation to ER stress, and whose regulation is specific to the late stages of the response. To this end, we performed a differential expression analysis to identify genes that vary in their expression between early (1 and 2 h) and late (5 and 8 h) ER stress treatments (see Methods). The analysis resulted in 120 significantly changing mRNAs, of which 60% were induced at the late time points (Figure 2A). Interestingly, both specific late-induced and late-repressed gene sets were PERK dependent, as their induction or repression in PERK−/− cells was highly impaired (Figures 2A and S1A–S1D). Reassuringly, functional enrichment analysis revealed that the induced gene set was enriched for genes involved in the ER stress response (Figure 2B). Furthermore, the late-specific induced gene set was also highly enriched with two additional functional categories: amino acid biosynthesis and tRNA synthetases (Figure 2B). This result is in agreement with our previously identified late ER stress gene induction program, which was enriched for response to ER stress and amino acid biosynthesis (Gonen et al., 2019), as well as with earlier reports of changes in amino acid biosynthesis using mRNA expression profiling (Harding et al., 2003). In addition, specific amino acids, such as cysteine and leucine, were shown to be essential for cell survival under prolonged ER stress (Harding et al., 2000, Harding et al., 2003). Interestingly, we found that 25% of the late-specific induced mRNAs were ATF4 targets (p = 4.4 × 10−49, see Methods). Indeed, ATF4 transcriptional output is known to be a component of the adaptive UPR, which occurs at the late time points.
Figure 2.

Late-Specific ER Stress-Induced mRNAs Include Specific Amino Acid Biosynthesis Pathways and Their Cognate tRNA Synthetases
(A) Hierarchical clustering analysis was performed using 120 mRNAs that were differentially translated (identified using DESeq2, FDR-corrected p value < 0.1) between the group of 1- and 2-h ER-stressed WT MEFs and a second group of 5- and 8-h treated WT MEFs. The heatmap depicts hierarchically clustered log2 fold change in translation levels (transcripts per million [TPM] of ribosome footprint profiling in the respective samples, Tg/control, log2), according to Spearman's correlation. Clustering was performed according to WT MEF samples. As shown, PERK−/− cells display very little change in these mRNAs.
(B) Functional enrichment of the late-specific induced genes was analyzed using DAVID (Huang da et al., 2009). False discovery rate (FDR)-corrected p values are indicated.
(C) An amino acid biosynthesis metabolic map that was generated using the KEGG mapper (Kanehisa and Goto, 2000) of late-specific ER stress-induced genes shows induction of biosynthesis pathways (induced reaction arrows are colored in red) for eight non- or partly essential amino acids: Ser, Cys, Gly, Ala, Asp, Asn, Glu, and Pro.
(D) Hierarchical clustering analysis of translation log2 fold changes (as described in A) for the group of all tRNA synthetases, identified a cluster of ER stress-induced, PERK-dependent, tRNA synthetases (marked by a red square).
Biosynthesis of a Specific Subset of Amino Acids Is Induced in the Late ER Stress Response
After observing that amino acid biosynthesis pathways are enriched within late-specific ER stress-induced genes, we analyzed the expression patterns of all mRNAs related to amino acid biosynthetic pathways (see Methods). Although overall, amino acid biosynthesis pathway mRNAs showed significant, PERK-dependent, induction in ER stress (Figures S2A and S2B), hierarchical clustering analysis showed that only about half of these mRNAs were induced, whereas the rest were largely unchanged (Figure S2E). To distinguish the pathways that were induced, we examined the amino acid biosynthesis metabolic map (see Methods) and overlaid on it the data of the late-specific induced mRNA set that was characterized above (Figure 2A). Using this map, we identified a subset of eight non- or partly essential amino acids that showed induction of their related biosynthetic pathways: Ser, Cys, Gly, Ala, Asn, Asp, Glu, and Pro (Figure 2C).
Significant Overlap between Amino Acids with ER Stress-Induced Biosynthesis and Upregulated tRNA Synthetases Defines a UPR Amino Acid Signature
Next, having observed that aminoacyl-tRNA synthetases were also enriched within late-specific ER stress-induced mRNAs, we examined the expression pattern of all tRNA synthetases. The overall induction of 37 tRNA synthetases was highly significant and PERK dependent, as tRNA synthetases were not induced in PERK−/− MEFs at any of the ER stress time points (Figures S2C, S2D, and 2D). Nevertheless, clustering analysis revealed that here too, not all tRNA synthetases were equally induced in WT MEFs (Figure 2D). Rather, a subset of 12 cytosolic tRNA synthetases was highly upregulated during ER stress (Figure 2D). Notably, these specific tRNA synthetases were not induced in PERK−/− MEFs (Figure 2D). Other tRNA synthetases were either slightly induced (a subset of mitochondrial tRNA synthetases) or remained unchanged. Importantly, none of the tRNA synthetases was selectively repressed during ER stress.
Having identified a specific subset of eight amino acids whose biosynthesis was induced during the late stages of the ER stress response on one hand, and a subset of 12 highly upregulated tRNA synthetases on the other hand, we were prompted to examine the overlap between these two subsets. We found that seven of the eight amino acids whose biosynthesis was induced, showed upregulation of their cognate tRNA synthetase: Ser, Cys, Gly, Ala, Asn, Glu, and Pro (Table S1, p value for the overlap = 0.0012, using the hypergeometric test). We thus termed this subset of seven amino acids with both ER stress-induced biosynthesis and upregulated tRNA synthetases the UPR amino acid signature.
The other induced tRNA synthetases were related to essential amino acids. Indeed, we observed that several amino acid transporters were highly induced during the late stages of ER stress (Figure S3), in line with previous reports (Krokowski et al., 2013). However, as various transporters can import a broad range of amino acids, we decided not to include the essential amino acids in this signature.
The UPR Amino Acid Signature Is Coupled to Differential Amino Acid Demand from ER Stress-Induced Proteins
The identification of a UPR amino acid signature led us to hypothesize that the coordinated induction of biosynthesis of a specific subset of amino acids and their cognate tRNA synthetases might be coupled to changes in the ER stress cellular translatome. More specifically, we hypothesized that induction in the biosynthesis of the identified UPR amino acid signature may correspond with the amino acid composition of proteins that comprise the ER stress induction program. To test this hypothesis, we defined a metric termed amino acid demand, which is simply the fraction of an amino acid, or a group of amino acids, within the sequence of each protein. We calculated the UPR amino acid demand as the demand of the set of the seven amino acids comprising the UPR amino acid signature, in each protein sequence in the mouse genome. We then examined the amino acid demand within the three ER stress gene expression programs that we previously identified (Gonen et al., 2019), namely, early UPR induction, late UPR induction, and UPR repression, and compared these with the background UPR amino acid demand of all expressed proteins (Figure 3A). We observed that whereas proteins belonging to the ER stress repression program were significantly depleted for the UPR amino acid signature (p = 3.8 × 10−5), both early and late induction program proteins were highly enriched for this amino acid set (Figure 3A, p = 3 × 10−4 and p = 1.2 × 10−4 for the early and late induction gene expression programs, respectively). Importantly, examination of the proteins that were induced or repressed by ER stress in the PERK−/− MEFs did not reveal enrichment or depletion of the UPR amino acid signature (Figure 3B).
Figure 3.

The UPR Amino Acid Signature Is Enriched in UPR-Induced Proteins, and in Secreted Proteins
(A) The UPR amino acid demand, namely, the fraction of the seven amino acids comprising the UPR amino acid signature, in protein sequences within the UPR-repressed (blue), early UPR-induced (green), and late UPR-induced (red) gene expression programs from Gonen et al. (2019) compared with the background of all expressed proteins (gray) using a cumulative distribution function (CDF) plot. The plot shows the cumulative distribution of the fraction of proteins within each of the groups (y axis), with different UPR amino acid demand scores (x axis). Proteins belonging to the UPR repression program were significantly depleted for the UPR amino acid signature, as indicated by a shift in distribution to more negative values compared with the background distribution. In contrast, both early and late UPR induction program proteins were highly enriched for this amino acid set, as indicated by a significant shift in distribution to more positive values compared with the background distribution. Kolmogorov-Smirnov (KS) test p values are indicated.
(B) Amino acid demand CDF plot (as in A) for all ER stress-induced (red) and ER stress-repressed (blue) mRNAs in the PERK−/− MEFs (at least 2-fold at any of the time points compared with control). None of the groups showed enrichment or depletion for the UPR amino acid signature; KS test p values are indicated.
(C) The UPR amino acid demand of secreted proteins (red) and extracellular matrix proteins (green) compared with the background of all expressed proteins (gray) is shown using a CDF plot (as in A). Both secreted and extracellular matrix proteins were highly enriched for the UPR amino acid signature, KS test p values are indicated.
To investigate whether this finding can be recapitulated in another dataset, we examined an ER stress dataset in which MEFs were stressed for 16 h using Tg, and translatome was mapped using polysome sequencing analysis (Guan et al., 2017). Here too, we identified a UPR amino acid signature relevant for a 16-h ER stress, which was similar to the signature that we identified for the 5- and 8-h ER stress in our dataset (Table S1). Almost all cytosolic tRNA synthetases were induced by 16 h of ER stress (Figure S4A), whereas the biosynthesis of fewer amino acids was upregulated at this time point (Figure S4B). Thus, the 16-h UPR amino acid signature consisted of the amino acids Ser, Gly, Cys, Asp, and Glu (Table S1), a similar subset to the late (5–8 h) ER stress signature above. Nevertheless, when considering the amino acid demand originating from the induced proteins at the 16-h ER stress time point of the Guan et al. translatome data, we again observed significant enrichment of the 16-h UPR amino acid signature within induced proteins (Figure S4C, p = 1.5 ×10−10).
Taken together, regulation at the level of amino acid biosynthesis and tRNA synthetases during ER stress is coupled to changes in the demand of amino acids originating from induced proteins.
UPR-Mediated Regulation of Amino Acid Metabolism Coupled with Amino Acid Demand of Secreted Proteins
Next, we sought to examine whether this mode of regulation applies to specific pathways or protein classes. Our observation that secreted proteins tended to escape the ER stress-mediated repression of ER-targeted proteins (Figure 1) prompted us to examine the amino acid composition of this group of proteins. Interestingly, secreted protein sequences demonstrated significant enrichment in the UPR amino acid signature (Figure 3C). Notably, although overall, secreted proteins showed elevated amino acid demand for this signature, secreted proteins that were induced were more significantly enriched for the signature than were those that were repressed (Figure S5D). Even greater enrichment of the UPR amino acid signature was observed in the sequences of extracellular matrix proteins (Figure 3C). This result was not due to the particular amino acid composition of the signal peptide, as exclusion of the signal peptide region from the analyzed protein sequences did not affect any of the trends (Figures S5A and S5B). In addition, this trend was not confounded by overlap with the ER stress induction protein expression programs (from Figure 3A), as it remained significant when we removed these proteins (Figure S5C).
The UPR Amino Acid Signature Shows Higher Codon Optimality in Both UPR-Induced and Secreted Proteins
We next decided to analyze an additional level of protein synthesis regulation, namely, the level of codon optimality. To this end, we calculated for each protein, the tRNA adaptation index (tAI, see Methods), a widely used metric for codon optimality that takes into account the corresponding tRNA copy numbers of each codon (dos Reis et al., 2003). We compared the tAI of secreted proteins to those of all expressed proteins, and observed a highly similar distribution (Figure 4A, p = 4.7 × 10−1). Surprisingly, when the tAI calculation was restricted to the UPR amino acid signature, the codon optimality for secreted proteins was significantly increased (p = 8.4 × 10−6, using Kolmogorov-Smirnov [KS] test, Figures 4B and 4C). Importantly, this finding was not due to the signal peptide region, as it remained after exclusion of this region from the analysis (Figures S6A–S6C). Further examination of the set of late ER stress-induced proteins revealed a similar trend, whereby codon optimality (measured using tAI) for late UPR-induced proteins was elevated for the UPR amino acid signature (Figures S6D–S6F). Thus, the UPR amino acid signature is coupled to UPR-induced protein expression demands, as well as to the amino acid demand of secreted proteins and shows increased codon optimality for these specific groups.
Figure 4.

The UPR Amino Acid Signature Shows Higher Codon Optimality for UPR-Induced Proteins and for Secreted Proteins
Codon optimality was calculated for all expressed proteins using the tRNA adaptation index (see Methods), using all amino acids (A), only the amino acids comprising the UPR amino acid signature (B), or only the amino acids that are not part of the UPR amino acid signature (C). tAI distributions were plotted for the group of secreted proteins (red) versus the background distribution of all expressed proteins (gray), using cumulative distribution function (CDF) plots (as in Figure 3). p values for the differences between these two groups, calculated using the Kolmogorov-Smirnov test, showed no difference in the codon optimality of all amino acids (A); however, the codon optimality of the UPR amino acid signature is significantly higher for secreted proteins (B).
In summary, our analyses unraveled an additional mode of protein synthesis regulation as part of the UPR, which involves rewiring of amino acid metabolism and tRNA synthetase induction, and the coupling of these processes to the amino acid demand of proteins whose synthesis is induced during ER stress.
Discussion
Here we analyzed ribosome footprint profiling data of a time course of ER stress treatments and identified an additional mode of protein synthesis regulation that occurs in the late adaptive stage of the response. Specifically, we found coordinated induction in certain amino acid biosynthetic pathways, together with upregulation of the expression of their cognate tRNA synthetases. In agreement with our hypothesis that this coordination may be related to protein synthesis demands, we found indeed that these signature amino acids are enriched within UPR-induced proteins.
Codon bias was shown to vary according to categories of gene expression. Upon stress, yeast cells were shown to induce the expression of tRNAs whose codons are enriched within stress-induced mRNAs (Torrent et al., 2018). Furthermore, codon usage was found to differ between proliferation-related genes, whose expression is altered in cancer, and differentiation-related genes, and this was accompanied by shifts in the expression of respective pools of tRNAs (Gingold et al., 2014). In addition, secreted proteins were shown to have particular codon bias properties in several organisms, from yeast to human (Mahlab and Linial, 2014). Therefore, organisms seem to have evolved means of coupling the expression of their tRNA pools to match demands of varying protein expression programs (Quax et al., 2015). Here we describe a phenomenon in which amino acid biosynthesis and tRNA synthetase expression are coupled to the amino acid content of induced proteins during the course of the ER stress response. In codon bias, shifts in tRNA pools involve synonymous codons, whereas the signs of a metabolic shift reported here were related to the amino acid content. Nonetheless, it is interesting that the UPR amino acid signature we identified also shows higher codon optimality for secreted proteins and for late UPR-induced proteins, thus manifesting a combined-level regulation.
Amino acid biosynthesis induction was previously discussed in the context of ER stress. Some studies suggested that this upregulation is adaptive (Harding et al., 2000, Harding et al., 2003), whereas others claimed that the ATF4-mediated induction of amino acid biosynthesis and tRNA synthetases enhances protein synthesis in a manner that could become harmful upon prolonged stress (Krokowski et al., 2013). Here we suggest that the induced biosynthesis of specific amino acids is coupled to protein synthesis demands, which supports the notion that this is indeed an adaptive mechanism. We propose that amino acid composition and shifts in amino acid metabolism have co-evolved such as to yield an improved regulatory program that fine-tunes supply and demand for proteins that are preferentially synthesized upon ER stress.
An alternative possibility is that feedback regulation senses depletion in some amino acids, and thereby acts to enhance biosynthesis, which then meets the increased demand. However, we note that the cells were grown throughout the experiment in a media that was supplemented with non-essential amino acids (Gonen et al., 2019). Therefore, it is highly unlikely that the cells were depleted for these amino acids, but rather that an intrinsic program led to the shift in amino acid metabolism.
We wondered whether the UPR amino acid signature we identified is the optimal one, given the set of induced proteins. Our analysis found that the UPR amino acid signature is only in the top 37th percentile in terms of optimal compatibility to the set of induced proteins and in the top 30th percentile of optimality for secreted proteins (see Figure S7). In other words, other combinations of seven amino acids would more optimally meet the specific demands of amino acids for induced protein expression programs. Importantly, other roles have been ascribed to various amino acid biosynthesis pathways upon ER stress (Harding et al., 2000, Harding et al., 2003). Thus, it is highly likely that amino acid demand is only partly responsible for the observed UPR amino acid signature.
For example, the serine-glycine biosynthesis pathway contains four enzymes, all of which were highly induced in our data in a PERK-dependent manner (Figures 2C and S8A). This pathway was also found to be elevated, in an ATF4-dependent manner, and to increase glutathione production in non-small-cell lung cancer cells, thus contributing to their aggressiveness (DeNicola et al., 2015). Indeed, glutathione has an important role in protection from oxidative stress in general, and in ATF4−/− cells in particular (Harding et al., 2003). Furthermore, mitochondrial tetrahydrofolate, which is another important derivative of serine-glycine metabolism, has been shown to play important roles in protecting from mitochondrial stress in response to glucose starvation, and in maintaining proper mitochondrial translation upon stress (Minton et al., 2018). Indeed, our data showed that mitochondrial translation is largely maintained during the course of ER stress, as evident by the increased ratio of mitochondrially encoded gene translation (Figure S8C), whereas cytosolic translation was overall repressed (Gonen et al., 2019). Therefore, it is possible that the induction of serine biosynthesis actually leads to increased conversion of serine into glycine and tetrahydrofolate, which is adaptive for cell fate and helps maintain healthy mitochondria during ER stress.
Proline, which is generated by a series of reactions from glutamate, has also been shown to have protective roles in stress. Proline starvation results in unresolved ER stress in cancer cells (Sahu et al., 2016). Furthermore, proline was shown to be limiting in clear cell renal cell carcinoma tumors, in which the PYCR1 enzyme responsible for the final step of proline production from glutamate was induced, whereas proline catabolism enzymes were downregulated (Loayza-Puch et al., 2016). Our data show high induction of PYCR1, along with two additional enzymes in the glutamate to proline biosynthesis pathway, PYCR2 and ALDH18A1, together with a nearly 2-fold reduction in the expression of the proline catabolic enzyme ALDH4A1 (Figure S8B). Therefore, the observed induction in glutamate may be only an intermediate step that promotes the biosynthesis of proline.
Following the above, we investigated whether removal of serine and glutamate would generate a more optimal UPR amino acid signature. Indeed, when we removed one or both of these from the UPR amino acid signature, the signature became more optimal (4.9th and 2nd top percentiles for late-induced proteins and secreted proteins, respectively, Figure S9). Therefore, it is possible that some induced amino acids serve as precursors for certain derivatives, which are themselves involved in protection from various effects of ER stress, or from secondary related oxidative stress. Our observation of a favorable, yet suboptimal set of induced amino acids corroborates with the possibility of other underlying causes for the induction of some of the induced amino acid biosynthesis pathways. This supports the notion that fine-tuned changes in amino acid biosynthesis coincide with amino acid demands that originate particularly from induced protein expression programs. The extent to which these metabolic shifts in the biosynthesis of amino acids or their metabolic derivatives is congruent with specific amino acid-related functions, or with protein synthesis demands, remains to be resolved.
Limitations of the Study
Our study describes an additional regulatory layer that occurs during ER stress, involving coordination between supply and demand of amino acids to fine-tune UPR protein synthesis demands. Furthermore, the same amino acid signature shows improved codon optimality, further supporting the adaptiveness of this mode of regulation. Nevertheless, amino acid biosynthesis generates multiple derivatives, some of which have been shown to play specific roles in protection from prolonged ER stress and other secondary stresses, as discussed above. Future additional experiments, such as tRNA-charging assays and metabolomics, would be required to fully disentangle the effects of amino acid supply-demand regulation from those of secondary derivatives and fully quantify their specific contributions.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Oded Lewinson for useful discussions of the results and critical reading of the manuscript. We thank Ruth Hershberg for critical reading of the manuscript. This project received funding from the European Research Council under the European Union's Horizon 2020 program Grant 677776.
Author Contributions
R.S. conceived and supervised the study. N.G., A.M., and N.S. performed all data analyses. R.S. wrote the manuscript with input from N.S. and A.M.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.07.022.
Data and Code Availability
The datasets analyzed in this study are available on GEO. Gonen et al. (Gonen et al., 2019) data are available as GEO: GSE118660. Guan et al. polysome sequencing data (Guan et al., 2017) are available as GEO: GSE90070.
Supplemental Information
References
- Ben-Sahra I., Hoxhaj G., Ricoult S.J.H., Asara J.M., Manning B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola G.M., Chen P.H., Mullarky E., Sudderth J.A., Hu Z., Wu D., Tang H., Xie Y., Asara J.M., Huffman K.E. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M., Wernisch L., Savva R. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003;31:6976–6985. doi: 10.1093/nar/gkg897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H., Tehler D., Christoffersen N.R., Nielsen M.M., Asmar F., Kooistra S.M., Christophersen N.S., Christensen L.L., Borre M., Sorensen K.D. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Gonen N., Sabath N., Burge C.B., Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci. Rep. 2019;9:4330. doi: 10.1038/s41598-019-38705-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B.J., Krokowski D., Majumder M., Schmotzer C.L., Kimball S.R., Merrick W.C., Koromilas A.E., Hatzoglou M. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2alpha. J. Biol. Chem. 2014;289:12593–12611. doi: 10.1074/jbc.M113.543215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan B.J., van Hoef V., Jobava R., Elroy-Stein O., Valasek L.S., Cargnello M., Gao X.H., Krokowski D., Merrick W.C., Kimball S.R. A unique ISR program determines cellular responses to chronic stress. Mol. Cell. 2017;68:885–900.e6. doi: 10.1016/j.molcel.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Back S.H., Hur J., Lin Y.H., Gildersleeve R., Shan J., Yuan C.L., Krokowski D., Wang S., Hatzoglou M. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan R.J., Freymann D.M., Stroud R.M., Walter P. The signal recognition particle. Annu. Rev. Biochem. 2001;70:755–775. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- Krokowski D., Han J., Saikia M., Majumder M., Yuan C.L., Guan B.J., Bevilacqua E., Bussolati O., Broer S., Arvan P. A self-defeating anabolic program leads to beta-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J. Biol. Chem. 2013;288:17202–17213. doi: 10.1074/jbc.M113.466920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F., Rooijers K., Buil L.C., Zijlstra J., Oude Vrielink J.F., Lopes R., Ugalde A.P., van Breugel P., Hofland I., Wesseling J. Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature. 2016;530:490–494. doi: 10.1038/nature16982. [DOI] [PubMed] [Google Scholar]
- Mahlab S., Linial M. Speed controls in translating secretory proteins in eukaryotes–an evolutionary perspective. PLoS Comput. Biol. 2014;10:e1003294. doi: 10.1371/journal.pcbi.1003294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton D.R., Nam M., McLaughlin D.J., Shin J., Bayraktar E.C., Alvarez S.W., Sviderskiy V.O., Papagiannakopoulos T., Sabatini D.M., Birsoy K. Serine catabolism by SHMT2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol. Cell. 2018;69:610–621.e5. doi: 10.1016/j.molcel.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Reyna-Neyra A., Philippe L., Thoreen C.C. mTORC1 balances cellular amino acid supply with demand for protein synthesis through post-transcriptional control of ATF4. Cell Rep. 2017;19:1083–1090. doi: 10.1016/j.celrep.2017.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavitt G.D., Ron D. New insights into translational regulation in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2012;4:a012278. doi: 10.1101/cshperspect.a012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax T.E., Claassens N.J., Soll D., van der Oost J. Codon bias as a means to fine-tune gene expression. Mol. Cell. 2015;59:149–161. doi: 10.1016/j.molcel.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid D.W., Chen Q., Tay A.S., Shenolikar S., Nicchitta C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell. 2014;158:1362–1374. doi: 10.1016/j.cell.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendleman J., Cheng Z., Maity S., Kastelic N., Munschauer M., Allgoewer K., Teo G., Zhang Y.B.M., Lei A., Parker B. New insights into the cellular temporal response to proteostatic stress. Elife. 2018;7:e39054. doi: 10.7554/eLife.39054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski D.T., Kaufman R.J. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Sahu N., Dela Cruz D., Gao M., Sandoval W., Haverty P.M., Liu J., Stephan J.P., Haley B., Classon M., Hatzivassiliou G. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab. 2016;24:753–761. doi: 10.1016/j.cmet.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Torrent M., Chalancon G., de Groot N.S., Wuster A., Madan Babu M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018;11:eaat6409. doi: 10.1126/scisignal.aat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattem K.M., Wek R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed in this study are available on GEO. Gonen et al. (Gonen et al., 2019) data are available as GEO: GSE118660. Guan et al. polysome sequencing data (Guan et al., 2017) are available as GEO: GSE90070.