

Abstract
Altered metabolite levels are now well-established to drive epigenetic changes in development and disease. However, in many cases the specific protein–metabolite interactions that underlie this process remain enigmatic. In this review, we make the case that this fundamental missing information may be discovered by applying the tools of modern drug target validation to study endogenous metabolite pharmacology. We detail examples in which chemical proteomics has been applied to gain new insights into reversible and covalent metabolite signaling mechanisms, using acetyl-CoA and fumarate as case studies. Finally, we provide a brief survey of nascent chemical biology methods whose application to the study of endogenous metabolite pharmacology may further advance the field.
Introduction
Cancer genome sequencing has specified mutation of chromatin-modifying enzymes as a central mechanism of tumorigenesis [1]. The identification of small molecules that can rebalance aberrant epigenetic signaling thus represents a major focus of oncology research [2]. Related to mutations and molecules, a third mechanism that can regulate epigenetic signaling in cancer is metabolism [3–5]. For example, in 2009 several groups discovered conserved driver mutations in isocitrate dehydrogenase (IDH1/2) genes associated with glioblastoma and acute myeloid leukemia (AML) [6]. Functional studies revealed mutant IDH aberrantly produces an “oncometabolite,” (R)-2-hydroxyglutarate (2-HG), that can competitively inhibit Fe(II)-α-ketoglutarate (KG) dioxygenases involved in epigenetic regulation. Driver mutations in IDH and TET2, an α-KG-dependent epigenetic tumor suppressor, are mutually exclusive in a subset of AML patients [7]. This implies that overproduction of 2-HG can phenotypically mimic mutational inactivation of a chromatin-modifying enzyme, and provides a powerful demonstration of the ability of metabolism to fuel epigenetic mechanisms of disease.
There are now many studies (see [3–5] for reviews) in which changes in the steady-state levels of metabolites have been correlated with altered histone and DNA modifications (Fig. 1). These two processes are linked by endogenous metabolite pharmacology: the ability of metabolites to alter protein activity via reversible and covalent interactions. These pharmacological interactions are analogous in nature to those that small molecule drugs make with their targets, but also have many distinct features that make their unbiased characterization uniquely challenging. Due to this, many of the seminal studies of metabolic regulation of epigenetics have lacked a systematic determination of the protein–metabolite interactions that govern this process, particularly in living cells. In this review, we highlight the importance of this fundamental missing knowledge, and its potential for discovery by studying metabolites as ligands using chemical proteomics.
Figure 1.
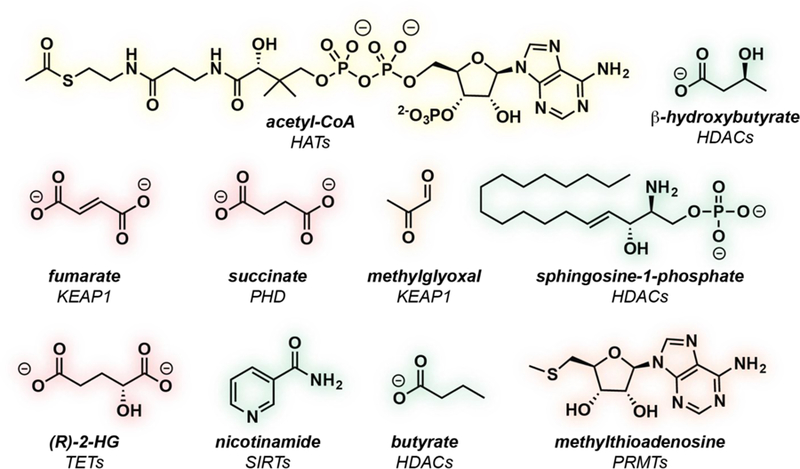
Examples of endogenous metabolites that have been postulated to exert pharmacological effects on epigenetic enzymes in the nucleus, and their validated or proposed biological targets.
Why study endogenous metabolite pharmacology?
The example of IDH1/2 demonstrates the power of endogenous metabolite pharmacology, but also highlights some challenges in the study of this phenomenon. For instance, the mutual exclusivity observed between IDH1/2 and TET2 mutations in AML does not extend to solid tumors driven by mutant IDH, including glioblastoma [6]. This implies that in these tissues, 2-HG may engage a more complex target profile to spur tumorigenesis. Furthermore, while the genetic linkage of IDH and TET2 suggests this chromatin modifier may be a functional target of 2-HG in AML, biochemical studies have found the 2-HG is a poor inhibitor of TET2 relative to other Fe(II)-2-KG dioxygenases [8]. This raises the possibility that 2-HG may target a pathway (or set of pathways) that are redundant with TET2, rather than TET2 itself. One class of plausible alternative targets are Fe(II)-2-KG-utilizing lysine demethylases [9]. Indeed, 2-HG accumulation has been correlated with hypermethylation of histones as well as DNA. However, it cannot be ruled out that this correlation may result from 2-HG’s interaction with unexpected mechanistic effectors, for example transcription factors and RNA modifying enzymes, whose modulation may influence histone modifications indirectly [10,11].
There is little doubt that metabolites pharmacologically interact with chromatin modifiers. However, the correlative studies described above imply rather than demonstrate this interaction, and contrast sharply with how drugs targeting chromatin modifiers are characterized. For these synthetic agents, gold-standard validation includes in vivo assessment of target occupancy [12] as well as rescue of the inhibitor-driven phenotype with a drug-resistant allele [13]. Applying similar approaches to define the specific targets of endogenous metabolite pharmacology represents a vital objective that has the potential to powerfully influence our understanding of the etiology and treatment of human disease. From the perspective of chemoprevention, identifying new links between metabolism, a controllable environmental input, and oncogenic gene expression may offer novel avenues for nutritional intervention. From the perspective of therapy, a knowledge of the functional targets of metabolites may provide new insights into the optimal metabolic contexts for cancer therapy, and facilitate the identification of unexpected collateral vulnerabilities [14]. Finally, it is important to note that unlike protein–drug interactions, which affect a subset of humans during treatment of disease, protein–metabolite interactions impact each and every living organism from fertilization until death. This fact underscores the need for the continued innovation of new approaches to define endogenous metabolite pharmacology, including (but not limited to) chemical proteomics.
Metabolite pharmacology versus drug pharmacology
In considering the application of chemical proteomic methods to study protein–metabolite interactions, it is important to consider several distinct properties of metabolites (Fig. 2):
Figure 2.
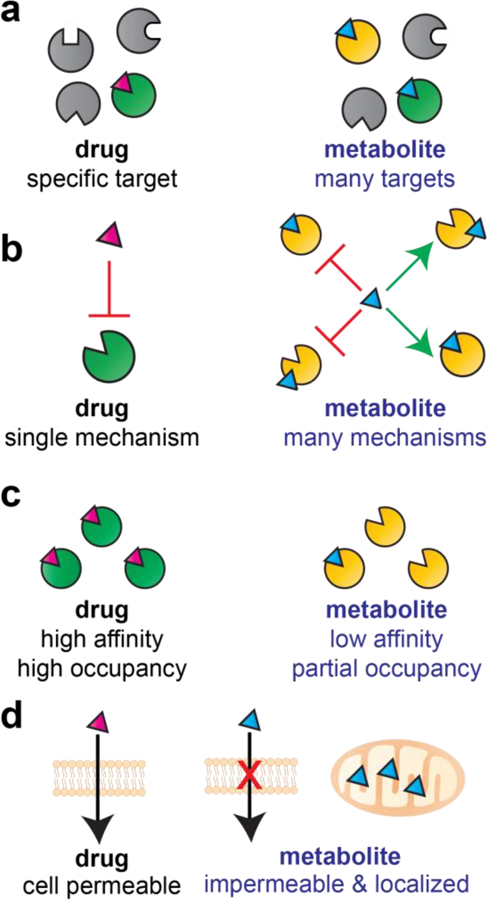
Comparing the pharmacology of drugs and metabolites. (a) Many drugs are designed to target a small number of proteins, while metabolites are polypharmacological and target many proteins. (b) Most drugs act via a single mechanism (e.g. competitive inhibition), while metabolites often facilitate multiple mechanisms functioning as cofactors/substrates, inhibitors, and allosteric modulators of protein function. (c) Drugs are designed to possess high affinity and fully occupy their protein target, while metabolites often make weak interactions and exhibit partial active site occupancy. (d) Drugs are cell-permeable and can be studied via quantitative dose–response profiling, while metabolites are membrane impermeable and often exhibit disparate concentrations in different subcellular compartments.
Metabolites are polypharmacological: Metabolites have co-evolved to interact with multiple cellular enzymes. Therefore, these molecules are expected to be inherently less specific than highly optimized small molecule drugs (Fig. 2a). A less intuitive counterpoint is that metabolites may be more specific than highly promiscuous, synthetic pan-assay interference compounds [15], upon whom no evolutionary pressure for restricted human enzyme interaction has acted.
Metabolites are polymechanistic: In addition to target promiscuity, metabolites also display mechanistic promiscuity. While synthetic drugs exert their activity through a single mechanism (e.g. competitive inhibition), most pharmacologically active metabolites interact with enzymes via multiple mechanisms (Fig. 2b). For example, acetyl-CoA can act as an enzyme cofactor, a competitive inhibitor, an allosteric inhibitor, and a covalent modifier.
Metabolites make weak interactions: Most small molecule drugs have been optimized for potency and exert their effects at nanomolar to micromolar concentrations. In contrast, many metabolites interact with enzymes that utilize them as cofactors with only modest (micromolar) affinity, and may not exert pharmacological effects on physiologically-relevant off targets until they accumulate to millimolar levels (e.g. 2-HG; Fig. 2b).
Metabolites are not drug-like: Most small molecule drugs have been developed keeping in mind Lipinski’s “Rule of Five,” which aims to optimize pharmacological activity by limiting molecular weight (<500 Da), polar atoms, ionizable functional groups, and lipophilicity. These physical properties of drugs contrast with those of endogenous metabolites, whose elevated polarity and lipophilicity help limit their extracellular diffusion and enable subcellular compartmentalization (Fig. 2d). Related to this lack of drug-likeness is the fact that while synthetic molecules are often optimized to limit metabolism, metabolites by their very nature are rapidly turned over and converted into other substances.
These unique features greatly impact both the technical implementation of chemical proteomic studies of metabolites, as well as what may be learned from them (note: in this section we use the technical terms ‘competitive’ and ‘in situ’ chemoproteomics, which are excellently defined elsewhere [12]). For instance, the polar nature of many metabolites impedes their cellular uptake, which can impede assessment of their protein targets by direct or competitive in situ chemoproteomic methods. While steady-state levels of metabolites can be increased by the use of cell-permeable metabolite analogues [16], these agents do not recapitulate the subcellular localization of metabolites, which can drastically affect the concentration at which these ligands are presented to cellular proteins. Furthermore, all chemical proteomic methods report only on a fixed range of ligand-protein binding affinities, the lower limit of which is not well-defined. This raises the possibility that functional protein–metabolite interactions may not be detected due to methodological bias (‘false negative problem’). Most importantly, the polypharmacological nature of metabolite–protein interactions changes the nature of what may be learned from their characterization. Unlike studies of drugs, chemoproteomic profiling of metabolite pharmacology is unlikely to identify a single protein target critical to a cellular phenotype. Instead, these studies benchmark the scope of the ligandable proteome accessible to a given metabolite, and (potentially) highlight new biological processes which it may functionally intersect [17]. The value of endogenous metabolite pharmacology may be considered in some ways analogous to that of target identification efforts for promiscuous ligands (e.g. electrophiles and pan-assay interference compounds) which emerge from phenotypic screening campaigns. For such compounds, evidence of target engagement does not mean the molecule is necessarily a specific or optimizable probe of a protein; instead, flipping the target elucidation paradigm on its head, it suggests the protein target can provide a window into the polypharmacology of the small molecule ligand, as well as new insights into its biological activity [18,19]. To illustrate these tactical considerations, as well as insights that can arise from studies of endogenous metabolite pharmacology, here we review our recent chemoproteomic investigations of two cancer metabolites: acetyl-CoA and fumarate.
Acetyl-CoA: a central metabolite linking metabolism and epigenetics
Acetyl-CoA is a central metabolite that plays a critical role in epigenetic signaling, serving as the requisite cofactor for lysine acetyltransferase (KAT)-catalyzed histone acetylation. Wellen and coworkers first discovered that production of glucose-derived acetyl-CoA by the metabolic enzyme ATP-citrate lyase (ACLY) was required for histone acetylation, as well as acetylation-dependent transcriptional programs necessary for adipocyte differentiation [20]. This “glucose-to-gene link” established one of the first direct connections between metabolism and epigenetic signaling. Subsequent studies expanded on the role of this link in disease, most notably when it was found that Warburg cancer metabolism (driven by Akt signaling) could drive high levels of nucleocytosolic acetyl-CoA, stimulating histone acetylation and pro-proliferative gene expression [21]. By implicating acetyl-CoA as a central node linking cellular bioenergetics and growth signaling, these studies provided strong motivation for analyses of this metabolite’s reversible and covalent pharmacological targets.
Profiling reversible metabolite pharmacology
Chemoproteomic discovery of novel endogenous KAT inhibitors.
An intuitive explanation for the link between acetyl-CoA and histone acetylation is that acetyl-CoA can become rate-limiting for KATs. Supporting this possibility, simultaneous siRNA-based depletion of ACLY and a non-essential KAT (GCN5) was found to result in redundant and non- additive decreases in histone acetylation [20]. Complicating this view are biochemical studies, which have shown that most KATs are kinetically saturated by acetyl-CoA at concentrations (Km~0.5–5 μM) far below its estimated cellular levels (~20–200 μM) [5,22]. Furthermore, mutations in yeast GCN5 that increase the enzyme’s intrinsic acetyl-CoA binding affinity do not alter the responsiveness of histone acetylation to yeast metabolism [23]. These findings have led to the hypothesis that rather than being rate-limiting in itself, acetyl-CoA may promote acetylation by alleviating the inhibition of KATs by endogenous metabolites [5,21]. However, efforts to understand the specificity of this mechanism, i.e. what metabolic pathways communicate with KATs by producing endogenous inhibitors, and what specific effectors of acetylation are conditionally regulated by this process, have been hindered by a lack of methods for the high-throughput biochemical analysis of KAT–acetyl-CoA interactions.
To address this challenge, our group recently developed a chemical proteomic approach to study acetyltransferase enzymes [24]. This method applies active site probes in which the acetyl-CoA cofactor is covalently linked via a thioether functionality to a lysine substrate surrogate. These molecules act as bisubstrate inhibitors of KAT enzymes, and were pioneered by Cole and coworkers as tools for the biochemical and structural analysis of KATs. When immobilized on streptavidin beads, these inhibitors report on the occupancy of the acetyl-CoA-binding active site of KATs and KAT complexes in their endogenous proteomic context. In the first application of these tools to study metabolite pharmacology, they were used to identify novel endogenous inhibitors of KATs [25]. These studies were inspired by the fact that cells contain a diverse repertoire of metabolic acyl-CoAs (i.e. malonyl-, succinyl-, and palmitoyl-CoA), whose concentration reflects the activity of specific metabolic pathways. We hypothesized that if KATs were capable of physically interacting with these molecules, it could provide a novel avenue to integrate changes in metabolism with epigenetic signaling. To explore this phenomenon, we applied a competitive chemoproteomic approach to study the interaction of a small panel of physicochemically diverse metabolic acyl-CoAs with three KATs: GCN5, PCAF, and MOF. As expected, the known cofactor acetyl-CoA and feedback inhibitor CoA efficiently blocked chemoproteomic capture of all three enzymes, consistent with their ability to strongly interact with KAT enzymes. An unexpected finding was that capture was also strongly antagonized by palmitoyl-CoA, a long-chain fatty acyl-CoA [25]. In-depth biochemical studies revealed that a variety of long- and short-chain fatty acyl-CoAs could act as inhibitors of KAT enzymes, and led to the characterization of palmitoyl-CoA as the most potent endogenous KAT inhibitor discovered to date. Structural analysis of GCN5 indicate this likely reflects the ability of these fatty acyl-CoAs to act as endogenous bisubstrate inhibitors, in which anchoring of the CoA moiety in the KAT cofactor binding site competes with acetyl-CoA, while the fatty acyl chain sterically occludes productive substrate binding and acyl transfer [25,26]. This is significant because relatively few KATs have been shown to use elongated acyl-CoAs as cofactors [27], which suggests inhibition as a more widespread mechanism by which acyl-CoA metabolites influence KAT activity. Evidence indicates this pharmacological mechanism of KAT inhibition can also be accessed by non-steroidal anti-inflammatory drugs (NSAIDs) that form acyl-CoA metabolites, including salicylate and ibuprofen [28,29]. Consistent with these kinetic and structural studies, treatment of cells with fatty acid and NSAID-CoA precursors downregulates histone acetylation [25,28]. Furthermore, in mice fed high-fat diet, H3K9 histone acetylation was more well correlated with the ratio of acetyl-CoA:(iso)butyryl-CoA than any other CoA metabolite, consistent with the model that short chain fatty acyl-CoAs can act as functional competitive metabolites in vivo [30]. These studies illustrate how understanding the pharmacological properties of acyl-CoA metabolites may aid in the prediction of epigenetic responses to metabolic stimuli.
Chemoproteomic discovery of novel metabolically-sensitive acetyltransferases.
The above studies expanded our knowledge of the metabolic cues sensed by chromatin modifiers; however, one limitation was their exclusive focus on histone acetyltransferase enzymes. An unexamined question was whether endogenous metabolite pharmacology may act on other effectors of acetylation, and thus serve to link novel signaling pathways to acetyl-CoA and the metabolic state of the cell. Based on previous biochemical studies [22], we hypothesized that metabolically sensitive acetyltransferases may be identifiable based on a unique pharmacological signature: specifically, we would expect them to display relatively high affinity for CoA, a feedback metabolite elevated under conditions of bioenergetic stress, relative to acetyl-CoA, the enzymatic cofactor. To test this hypothesis, we developed a second-generation chemoproteomic probe capable of broad-spectrum profiling of acetyltransferase activity (Fig. 3a) [31]. Using dose-dependent competition and immunoaffinity profiling, we quantified the relative acetyl-CoA and CoA binding affinity of four histone acetyltransferases (GCN5, PCAF, MOF, and HAT1), two N-terminal acetyltransferases (NAA10, NAA50), one non-histone lysine acetyltransferase (ESCO1), and an RNA acetyltransferase (NAT10). A critical technical aspect of this study was its focus on the relative acetyl-CoA versus CoA binding affinity. This provides an internal control which accounts for biases created by differential binding of the affinity matrix to the active sites of individual acetyltransferase enzymes. Applying this strategy, we discovered that cellular acetyltransferases display a broad range of pharmacological interaction with the metabolic feedback inhibitor CoA. Among histone acetyltransferases, GCN5 and PCAF were found to interact most strongly with CoA relative to acetyl-CoA. This served as an important control, as previous biochemical studies have found GCN5 family acetyltransferases to be susceptible to metabolic feedback inhibition [22,23]; chemoproteomic methods now allowed this result to be recapitulated for the first time in an endogenous proteomic context. Also identified in this metabolically sensitive subset was NAT10. NAT10 is a relatively uncharacterized enzyme that has been recently found to acetylate RNA cytidine residues [32]. This posttranscriptional modification forms the minor nucleobase N4-acetylcytidine, which has thus far been characterized in ribosomal RNA, transfer RNA, and messenger RNA [33,34]. Biophysical studies of recombinant NAT10 found the enzyme binds acetyl-CoA and CoA with similar affinity, consistent with its potential for metabolic feedback regulation. Subsequent studies in model organisms have found that nutrient stress reduces N4-acetylcytidine levels in RNA [35]. It is important to note that in addition to interaction with endogenous feedback metabolites, other mechanisms may also contribute to the metabolic regulation of NAT10, including SIRT1-mediated deacetylation [36]. Overall, these studies showcase how chemoproteomic studies of endogenous metabolite pharmacology can identify new metabolically sensitive enzymes within uncharacterized members of large enzyme superfamilies, and suggest NAT10 may constitute a novel enzymatic activity linking acetyl-CoA to the epitranscriptome.
Figure 3.
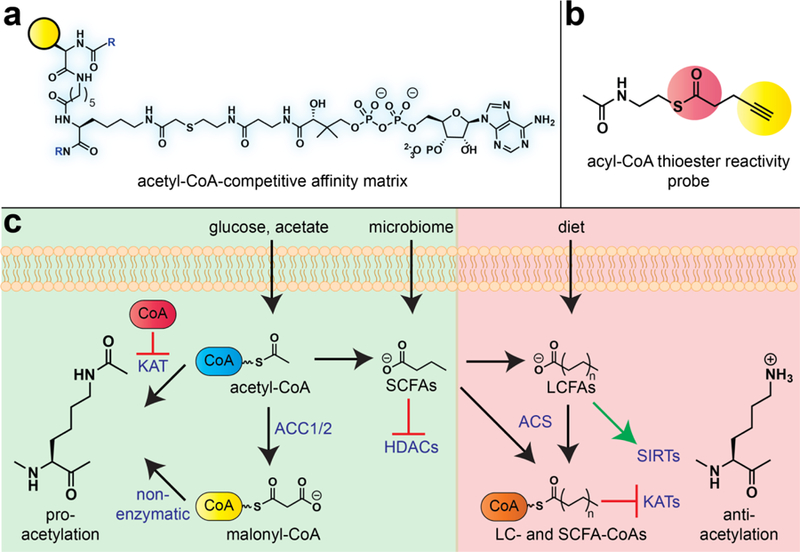
Profiling the pharmacology of the central metabolite acetyl-CoA. (a) Chemoproteomic probe of reversible acetyl-CoA/protein interactions. (b) Chemoproteomic probe of covalent acyl-CoA/protein interactions. (c) Diverse signaling functions of acyl-CoA metabolites. Pro-acetylation signaling functions (green): Acetyl-CoA produced from dietary glucose forms acetyl-CoA, which can free KATs as well as non-lysine acetyltransferases from feedback inhibition by CoA and other acyl-CoA metabolites. Acetyl-CoA carboxylase (ACC) enzymes in the cytosol convert acetyl-CoA to malonyl-CoA, a hyperreactive acyl-CoA which can facilitate non-enzymatic acylation in the cytosol and nucleus. Short chain fatty acids such as butyrate derived from the microbiome can bolster these effects by inhibiting HDACs. Anti-acetylation signaling functions (red): Long-chain and short-chain fatty acids are converted by acyl-CoA synthetases (ACS enzymes) into acyl-CoAs that can act as endogenous inhibitors of KAT enzymes. Long-chain fatty acids can also act as activators of sirtuin (SIRT) deacetylase enzymes. The use of alternative acyl-CoA (e.g. crotonyl-CoA) as substrates by the KAT p300 is not depicted for brevity.
Profiling covalent metabolite pharmacology
Chemoproteomic profiling of non-enzymatic acetylation.
Lysine acetylation is a unique posttranslational modification in that it can arise from both enzymatic and non-enzymatic mechanisms. Specifically, a number of studies have shown the innate electrophilicity of the acetyl-CoA cofactor can cause spontaneous acetylation of nucleophilic protein lysine residues [37]. This pharmacological mechanism is intrinsically metabolically regulated, as it is a bimolecular reaction whose kinetics are controlled by cellular acetyl-CoA concentrations. Non-enzymatic acetylation could constitute an ancient and direct mechanism for proteins to respond to fluctuating acetyl-CoA levels or, alternatively, a covalent history of cell metabolism. However, a significant obstacle to exploring the covalent pharmacology of acetyl-CoA is that enzymatic and non-enzymatic acetylation events are chemically indistinguishable. To address this challenge, our laboratory developed a reactivity-based approach to characterize the proteome-wide targets of non-enzymatic acetylation [38]. The design of this approach was inspired by structural studies of KATs, which have observed that molecular recognition of acetyl-CoA and the reactive thioester moiety are often spatially sequestered. This suggested a straightforward strategy whereby structurally truncated acyl-CoA cofactors would not be recognized by KATs, but would retain acyl-CoA thioester reactivity, allowing them to be used as surrogate reporters of non-enzymatic acetylation (Fig. 3b). Model studies showed a truncated thioester incorporating an alkyne was not utilized as a cofactor by KAT enzymes, and enabled reactivity-dependent proteome labeling that was competed by native thioester metabolites such as acetyl-CoA [38]. Proteomic profiling using a panel of cancer cell lines led to the recurrent identification of several glycolytic proteins as targets of non-enzymatic acetylation, including GAPDH. Considering the diversity of thioester metabolites, we profiled a series of cytosolic acyl-CoAs for their ability to biochemically inhibit GAPDH and found malonyl-CoA to be a potent, time-dependent, and irreversible inhibitor of the enzyme. Mechanistic studies revealed that malonyl-CoA’s potent inhibition stems from its increased reactivity relative to acetyl-CoA, which favors more rapid covalent protein modification. Consistent with this model, we found the mitochondrial metabolite succinyl-CoA exhibits even greater reactivity than malonyl-CoA, which correlates with increased covalent labeling and enzyme inhibition. Concurrently with our studies, Hirschey and coworkers also discovered that reactive acyl-CoA species formed in the mitochondria can mediate rapid non-enzymatic acylation and covalent enzyme inhibition, providing critical cross-validation of this finding [39]. Finally, our group showed that covalent metabolite pharmacology can be directly manipulated using cell-permeable malonylation reagents. Direct electrophilic stimulation allows the effects of malonyllysine on targets such as GAPDH and mTOR to be studied, while avoiding the ancillary metabolic effects of FASN inhibition [38,40]. These studies demonstrate the ability of reactivity-based chemoproteomics to provide new insights into the covalent pharmacology of acyl-CoA metabolites, and provide a set of endogenous posttranslational modifications (lysine malonylation and lysine succinylation) that can be used to monitor non-enzymatic acylation in the cytosol and mitochondria. More broadly, these studies inform new polymechanistic models for how nutrient-derived acyl-CoAs and acyl-CoA precursors modulate the balance of macromolecular acetylation (Fig. 3c).
Chemoproteomic profiling of a covalent oncometabolite: fumarate
Another disease-relevant example of covalent metabolite pharmacology occurs in the genetic cancer susceptibility syndrome hereditary leiomyomatosis and renal cell carcinoma (HLRCC). In this disorder, mutations in the primary metabolic enzyme fumarate hydratase (FH) cause the TCA cycle metabolite fumarate to accumulate to supraphysiological levels (1–10 mM) [41]. Similar to 2-HG, fumarate has been found to reversibly inhibit Fe(II)-KG-dependent dioxygenase enzymes [5]. These reversible interactions were characterized in a recent chemoproteomic study, which found fumarate to interact strongly with the Fe(II)-KG-dependent RNA demethylase METTL3 [42]. Similar to acetyl-CoA, fumarate also harbors the capacity to react with proteins covalently, forming the non-enzymatic posttranslational modification S-succination (Fig. 4a). This characteristic is unique relative to other oncometabolites, and has been suggested to contribute to the unique transcriptional profiles, tissue specificity, and clinical prognosis exhibited by FH-deficient tumors [43]. However, outside of a handful of candidate targets (e.g. KEAP1), few covalent protein targets of fumarate are known.
Figure 4.
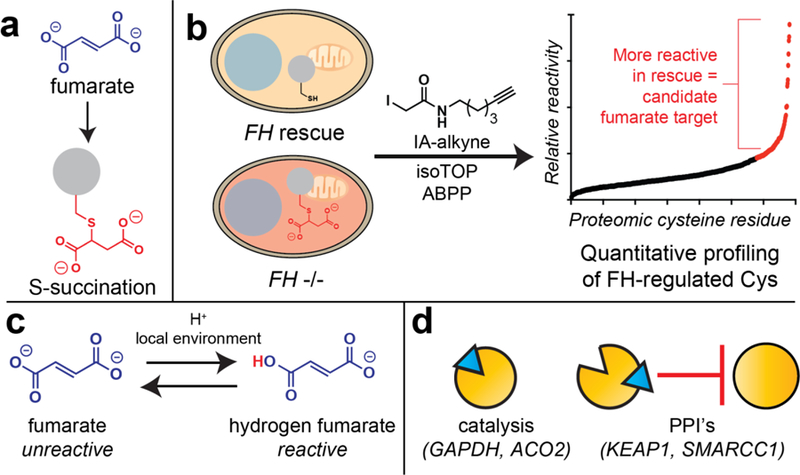
Profiling the pharmacology of the covalent oncometabolite fumarate. (a) Fumarate modifies proteomic cysteine residues to form the non-enzymatic protein modification cysteine S-succination. (b) Comparative chemoproteomic profiling of endogenous oncometabolites. Proteomes are isolated from isogenic cell lines harboring high fumarate (FH−/−) or low fumarate (FH rescue). Changes in cysteine reactivity are analyzed using an iodoacetamide alkyne (IA-alkyne) affinity probe and a quantitative proteomics method derived from isoTOP-ABPP. Proteins found to be equally abundant, but more reactive in rescue (low fumarate cells) constitute candidate covalent targets of fumarate. (c) While fumarate is a relatively unreactive electrophile, high cellular concentrations, and acidic global or microenvironments can form the active electrophile hydrogen fumarate. (d) Fumarate can act via multiple mechanisms including inhibition of protein catalytic activity and interruption of protein-protein interactions (PPIs).
To better understand the pharmacological basis for FH-dependent tumorigenesis, our group recently reported a competitive chemoproteomic strategy to profile covalent targets of fumarate [44]. This approach uses a general cysteine reactive affinity reagent (iodoacetamide alkyne) in combination with isotopically-labeled cleavable affinity tags, to quantitatively assess cysteine occupancy changes across the proteome (Fig. 4b), and has previously been powerfully applied to study the polypharmacology of covalent kinase inhibitors and reactive drug fragments [18]. To sample changes in cysteine occupancy driven by pathophysiological and subcellularly localized fumarate, we performed comparative profiling of proteomes from FH−/− and FH-rescue HLRCC cells that harbor high and low levels of fumarate, respectively. An important aspect of these studies is that they are mechanism-agnostic, and can identify cysteine reactivity changes driven by fumarate as well as other stimuli produced by FH mutation, such as reactive oxygen species [45]. Applying this approach, we identified >100 cysteines whose reactivity was altered >2-fold upon FH mutation, the majority of which had not been previously characterized as fumarate targets. These cysteines were distinct from those identified in a recent chemical proteomic study of the multiple sclerosis drug dimethyl fumarate (Tecfideria) [19], highlighting the oncometabolite’s unique reactivity. A surprising finding arose from flanking motif analysis and comparison to legacy chemoproteomic datasets, which revealed that FH-regulated cysteines were strikingly anti-correlated with overall cysteine nucleophilicity. This unique local sequence environment of FH-regulated cysteines suggested that protonation of fumarate may facilitate covalent reactivity. Kinetic analyses revealed that in contrast to most electrophiles, fumarate displays elevated thiol reactivity at acidic pH. This is consistent with a model in which hydrogen fumarate, rather than fumarate, serves as a covalent oncometabolite in HLRCC (Fig. 4c) [44]. The conditional reactivity of fumarate offers an explanation for why high levels of covalent S-succination are not observed in healthy mitochondria, and suggests that FH inactivation may collaborate with metabolic features such as the Warburg effect in order to remodel the cellular cysteine-ome [41]. In addition to this chemical insight, bioinformatic analyses specified many FH-sensitive cysteines whose modification was expected to alter protein function. In particular, an FH-sensitive cysteine in SMARCC1 was found to lie at a key protein–protein interface in the SWI-SNF complex. Covalent modification of this residue may contribute to the modest SWI-SNF dysfunction and FH-dependent EZH2 inhibitor sensitivity displayed by HLRCC cells. Overall, these studies exemplify how chemoproteomics can provide chemical and biological insights into metabolite function, and suggest that in addition to inhibition of catalytic activity, covalent metabolite pharmacology may play a role in the functional regulation of protein–protein interactions in the nucleus (Fig. 4d).
Conclusion and Future Directions
Chemical proteomics provides one powerful approach to characterize epigenetic regulation by endogenous metabolite pharmacology. While we have focused in this perspective on two metabolites that are known to alter histone acetylation and methylation in the nucleus (acetyl-CoA and fumarate), it is important to note that chemoproteomic methods have been powerfully applied to study the pharmacology of many other endogenous molecules, including signaling lipids, polar metabolites, and enzyme cofactors [17,46,47]. In the future, we envision that the application of these tools to characterize additional models of development and disease will lead to the identification of new intersections between metabolism and epigenetics. In addition to generation of these datasets another important goal will be integration in order to define the relative scope of these metabolite–protein interactions, i.e. does a metabolite have 2000, 200, or 20 targets, and where do different metabolites fall on this scale. Functional analysis of these interactions also represents a daunting task, with recent review articles offering some views on potential routes forward [48]. Finally, it is interesting to project forward and consider what other tools that have been used to effectively characterize drug pharmacology may provide useful insights into the metabolic regulation of epigenetics. Label-free proteomic approaches such as cellular thermal shift assay (CETSA) and limited proteolysis-coupled mass spectrometry (LiP-MS) have already proven complementary to affinity-based chemoproteomic methods for analysis of metabolite–protein interactions [49]. Genetically-encoded biosensors [50], chemical sensors [51,52], and bioluminescent reporters of target engagement [53] may prove useful for studying metabolite occupancy directly in cells. The continued development of improved cell-permeable pro-metabolite analogues [54], approaches to rapidly degrade metabolic enzymes [55], and genetic methods for subcellular metabolite depletion [56] will be necessary tools for hypothesis testing, allowing targeted manipulation of metabolite levels. An awareness that metabolite and drug pharmacology are cut from the same cloth has the potential to provide both methodological inspiration, as well as biological illumination, within this rapidly expanding field.
Acknowledgements
We are grateful to members of the Meier lab for proofreading and helpful comments. We sincerely apologize to the many authors whose primary literature could not be cited due to space limitations, and whose work was instead referenced via reviews. This work was supported by National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIABC011488).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW: Cancer genome landscapes. Science 2013, 339:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfister SX, Ashworth A: Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov 2017, 16:241–263. [DOI] [PubMed] [Google Scholar]
- ••3.Kinnaird A, Zhao S, Wellen KE, Michelakis ED: Metabolic control of epigenetics in cancer. Nat Rev Cancer 2016, 16:694–707. [DOI] [PubMed] [Google Scholar]
- ••4.Kaelin WG Jr., McKnight SL: Influence of metabolism on epigenetics and disease. Cell 2013, 153:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••5.Meier JL: Metabolic mechanisms of epigenetic regulation. ACS Chem Biol 2013, 8:2607–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]; The above three reviews provide an excellent primer on metabolic regulation of epigenetics, including references to the primary literature that could not be cited here due to space constraints.
- •6.Losman JA, Kaelin WG Jr.: What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 2013, 27:836–852. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides a more in-depth discussion of the role of mutant IDH in cancer.
- 7.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. : Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG Jr.: (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339:1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •9.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. : IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483:474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important illustration of a metabolism-epigenetic correlation studied by traditional biological methods.
- 10.Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. : R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018, 172:90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. : Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483:484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••12.Simon GM, Niphakis MJ, Cravatt BF: Determining target engagement in living systems. Nat Chem Biol 2013, 9:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]; Provides an excellent introduction to competitive, in situ, and reactivity-based chemoproteomic approaches that are discussed in this review.
- ••13.Kaelin WG Jr.: Common pitfalls in preclinical cancer target validation. Nat Rev Cancer 2017, 17:425–440. [DOI] [PubMed] [Google Scholar]; Excellent review discussing gold standard practices for validating a small molecule’s activity is due to engagement of a specific protein target. Important for considering the analogies between drug target validation and the pharmacology of metabolites.
- 14.McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, Sarosiek KA, Briggs KJ, Robbins AK, Sewastianik T, et al. : Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175:101–116 e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahlin JL, Nelson KM, Strasser JM, Barsyte-Lovejoy D, Szewczyk MM, Organ S, Cuellar M, Singh G, Shrimp JH, Nguyen N, et al. : Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat Commun 2017, 8:1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zengeya TT, Kulkarni RA, Meier JL: Modular synthesis of cell-permeating 2-ketoglutarate esters. Org Lett 2015, 17:2326–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niphakis MJ, Lum KM, Cognetta AB 3rd, Correia BE, Ichu TA, Olucha J, Brown SJ, Kundu S, Piscitelli F, Rosen H, et al. : A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 2015, 161:1668–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, et al. : Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, Cravatt BF: Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci Signal 2016, 9:rs10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •20.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB: ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important illustration of a seminal metabolism-epigenetic connection whose pharmacological basis has been challenging to ascertain using traditional biological methods.
- 21.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. : Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 2014, 20:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanner KG, Langer MR, Kim Y, Denu JM: Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem 2000, 275:22048–22055. [DOI] [PubMed] [Google Scholar]
- 23.Cai L, Sutter BM, Li B, Tu BP: Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell 2011, 42:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montgomery DC, Sorum AW, Meier JL: Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J Am Chem Soc 2014, 136:8669–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••25.Montgomery DC, Sorum AW, Guasch L, Nicklaus MC, Meier JL: Metabolic Regulation of Histone Acetyltransferases by Endogenous Acyl-CoA Cofactors. Chem Biol 2015, 22:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important example applying chemical proteomics as a high-throughput biochemical profiling method to identify new endogenous inhibitors of histone acetyltransferases.
- 26.Ringel AE, Wolberger C: Structural basis for acyl-group discrimination by human Gcn5L2. Acta Crystallogr D Struct Biol 2016, 72:841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, Dai L, Shimada M, Cross JR, Zhao Y, et al. : Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell 2015, 58:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirakawa K, Wang L, Man N, Maksimoska J, Sorum AW, Lim HW, Lee IS, Shimazu T, Newman JC, Schroder S, et al. : Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shrimp JH, Garlick JM, Tezil T, Sorum AW, Worth AJ, Blair IA, Verdin E, Snyder NW, Meier JL: Defining Metabolic and Nonmetabolic Regulation of Histone Acetylation by NSAID Chemotypes. Mol Pharm 2018, 15:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •30.Carrer A, Parris JL, Trefely S, Henry RA, Montgomery DC, Torres A, Viola JM, Kuo YM, Blair IA, Meier JL, et al. : Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. J Biol Chem 2017, 292:3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides a comprehensive catalogue of metabolic and histone modification changes in response to high-fat diet consistent with the hypothesis that short chain fatty acyl-CoAs are functional inhibitors of KATs in vivo.
- ••31.Montgomery DC, Garlick JM, Kulkarni RA, Kennedy S, Allali-Hassani A, Kuo YM, Andrews AJ, Wu H, Vedadi M, Meier JL: Global Profiling of Acetyltransferase Feedback Regulation. J Am Chem Soc 2016, 138:6388–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]; Important example applying chemical proteomics as a high-throughput biochemical profiling method to identify new acetyltransferases susceptible to reversible pharmacology of feedback inhibitor CoA.
- 32.Montgomery DC, Sorum AW, Meier JL: Defining the orphan functions of lysine acetyltransferases. ACS Chem Biol 2015, 10:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD, et al. : Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175:1872–1886 e1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma S, Langhendries JL, Watzinger P, Kotter P, Entian KD, Lafontaine DL: Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 2015, 43:2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Delft P, Akay A, Huber SM, Bueschl C, Rudolph KLM, Di Domenico T, Schuhmacher R, Miska EA, Balasubramanian S: The Profile and Dynamics of RNA Modifications in Animals. Chembiochem 2017, 18:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Cai S, Zhang C, Liu Z, Luo J, Xing B, Du X: Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res 2018, 46:9601–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagner GR, Payne RM: Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem 2013, 288:29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••38.Kulkarni RA, Worth AJ, Zengeya TT, Shrimp JH, Garlick JM, Roberts AM, Montgomery DC, Sourbier C, Gibbs BK, Mesaros C, et al. : Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem Biol 2017, 24:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]; Details a chemical approach to discriminate targets of enzymatic and non-enzymatic acetylation based on selective molecular recognition, and uses this method to discover novel classes of hyperreactive acyl-CoA cofactors.
- ••39.Wagner GR, Bhatt DP, O’Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD, et al. : A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab 2017, 25:823–837 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]; Contemporaneous study of metabolite reactivity discovering that succinyl-, glutaryl-, and hydroxymethylglutaryl-CoA are hyperreactive thioesters that can modulate enzyme activity.
- 40.Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, Dubois C, Cantelmo AR, Chen R, Loroch S, et al. : Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab 2018, 28:866–880 e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, et al. : Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 2009, 29:4080–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •42.Joberty G, Boesche M, Brown JA, Eberhard D, Garton NS, Humphreys PG, Mathieson T, Muelbaier M, Ramsden NG, Reader V, et al. : Interrogating the Druggability of the 2-Oxoglutarate-Dependent Dioxygenase Target Class by Chemical Proteomics. ACS Chem Biol 2016, 11:2002–2010. [DOI] [PubMed] [Google Scholar]; Only example to date exploring the reversible proteome-wide engagement of oncometabolites with Fe(II)-KG-dependent dioxygenases.
- 43.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, et al. : Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••44.Kulkarni RA, Bak DW, Wei D, Bergholtz SE, Briney CA, Shrimp JH, Alpsoy A, Thorpe AL, Bavari AE, Crooks DR, et al. : A chemoproteomic portrait of the oncometabolite fumarate. Nat Chem Biol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study details one of the first applications of reactivity-based chemoproteomics to study a covalent oncometabolite. In addition to chemical and biological insights it provides a strategic roadmap for future studies.
- 45.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS: The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 2013, 51:236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoegl A, Nodwell MB, Kirsch VC, Bach NC, Pfanzelt M, Stahl M, Schneider S, Sieber SA: Mining the cellular inventory of pyridoxal phosphate-dependent enzymes with functionalized cofactor mimics. Nat Chem 2018, 10:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu M, Chong LS, Perlman DH, Resnick AC, Fiedler D: Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms. Proc Natl Acad Sci U S A 2016, 113:E6757–E6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •48.Diether M, Sauer U: Towards detecting regulatory protein-metabolite interactions. Curr Opin Microbiol 2017, 39:16–23. [DOI] [PubMed] [Google Scholar]; This review highlights approaches to move from cataloguing protein–metabolite interactions to establishing their functional regulatory importance.
- ••49.Piazza I, Kochanowski K, Cappelletti V, Fuhrer T, Noor E, Sauer U, Picotti P: A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172:358–372 e323. [DOI] [PubMed] [Google Scholar]; This is one of the largest studies of protein–metabolite interactions catalogued to date using a label-free proteomic approach.
- 50.Sallin O, Reymond L, Gondrand C, Raith F, Koch B, Johnsson K: Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kulkarni RA, Briney CA, Crooks DR, Bergholtz SE, Mushti C, Lockett SJ, Lane AN, Fan TW, Swenson RE, Marston Linehan W, et al. : Photoinducible Oncometabolite Detection. Chembiochem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zengeya TT, Garlick JM, Kulkarni RA, Miley M, Roberts AM, Yang Y, Crooks DR, Sourbier C, Linehan WM, Meier JL: Co-opting a Bioorthogonal Reaction for Oncometabolite Detection. J Am Chem Soc 2016, 138:15813–15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasta JD, Corona CR, Wilkinson J, Zimprich CA, Hartnett JR, Ingold MR, Zimmerman K, Machleidt T, Kirkland TA, Huwiler KG, et al. : Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem Biol 2018, 25:206–214 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sinclair WR, Shrimp JH, Zengeya TT, Kulkarni RA, Garlick JM, Luecke H, Worth AJ, Blair IA, Snyder NW, Meier JL: Bioorthogonal pro-metabolites for profiling short chain fatty acylation. Chem Sci 2018, 9:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. : The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 2018, 14:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cracan V, Titov DV, Shen H, Grabarek Z, Mootha VK: A genetically encoded tool for manipulation of NADP(+)/NADPH in living cells. Nat Chem Biol 2017, 13:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]