


Abstract
LeuO is a pleiotropic LysR-type transcriptional regulator (LTTR) and co-regulator of the abundant nucleoid-associated repressor protein H-NS in Gammaproteobacteria. As other LTTRs, LeuO is a tetramer that is formed by dimerization of the N-terminal DNA-binding domain (DBD) and C-terminal effector-binding domain (EBD). To characterize the Escherichia coli LeuO protein, we screened for LeuO mutants that activate the cas (CRISPR-associated/Cascade) promoter more effectively than wild-type LeuO. This yielded nine mutants carrying amino acid substitutions in the dimerization interface of the regulatory EBD, as shown by solving the EBD’s crystal structure. Superimposing of the crystal structures of LeuO-EBD and LeuO-S120D-EBD suggests that the Ser120 to Asp substitution triggers a structural change that is related to effector-induced structural changes of LTTRs. Corresponding functional analyses demonstrated that LeuO-S120D has a higher DNA-binding affinity than wild-type LeuO. Further, a palindromic DNA-binding core-site and a consensus sequence were identified by DNase I footprinting with LeuO-S120D as well as with the dimeric DBD. The data suggest that LeuO-S120D mimics an effector-induced form of LeuO regulating a distinct set of target loci. In general, constitutive mutants and determining the DNA-binding specificity of the DBD-dimer are feasible approaches to characterize LTTRs of unknown function.
INTRODUCTION
LysR-type transcription regulators (LTTRs) constitute the largest family of transcription regulators in bacteria (1,2). LTTRs are involved in the control of a broad spectrum of cellular processes including regulation of metabolic pathways, stress-responses, virulence and biofilm formation, and others (2,3). LeuO is a pleiotropic LTTR that is conserved in the class of Gammaproteobacteria and has been best studied in the species Escherichia coli, Salmonella enterica (Enterobacteriaceae) and Vibrio cholerae (Vibrionaceae) (4).
LeuO functions both as activator and as repressor, and it targets more than 100 loci in E. coli and in S. enterica, as identified by ChIP-on-chip, genome scale SELEX and microarray analyses (5–8). LeuO regulates multi-drug efflux systems and outer membrane porins, as well as the H-NS (nucleoid-structuring protein) repressed cas operon encoding the CRISPR-associated Cascade complex among other loci in E. coli and in S. enterica (9–13). Many LeuO-activated loci are H-NS-repressed, and genomics data revealed a significant overlap of co-regulation of LeuO targets by H-NS (78% in E. coli and 40% in S. enterica) (5–8,11). The abundant H-NS represses transcription by forming extended nucleoprotein complexes on AT-rich DNA including DNA acquired by horizontal transfer (14–16). Therefore, LeuO is considered an H-NS antagonist (11,12). However, LeuO also functions as a repressor and presumptive ‘back-up’ of H-NS. For example, in case of the S. enterica pathogenicity island 1 (SPI-1), LeuO indirectly causes repression under conditions that impair SPI-1 repression by H-NS (17). Yet, in E. coli and S. enterica the leuO gene itself is repressed by H-NS and StpA, a H-NS paralog (16), while signals that induce leuO transcription remain elusive (7,18). The E. coli and S. enterica LeuO proteins are 90% identical to each other and 50% identical to the V. cholerae LeuO. In V. cholerae, LeuO has been associated with regulatory pathways of various virulence-related features such as acid-stress sensitivity, susceptibility to cationic antimicrobial peptides (CAMPs), as well as inhibition of expression of the crucial pathogenicity determinants cholera toxin (CT) and the toxin co-regulated pilus (TCP). In several of these pathways, LeuO has been identified as additional repressor of H-NS-repressed gene loci and thus seems to functionally cooperate with H-NS. Further, leuO expression is activated by the membrane bound transcription regulator ToxR, which links leuO expression to various stimuli that activate the ToxR-dependent virulence regulon of V. cholerae (19–22).
LeuO presumably binds DNA as a tetramer (23), similar to other LTTRs. LTTRs are comprised of a conserved and characteristic N-terminal winged helix-turn-helix (wHTH) DNA-binding domain (DBD) that connects via a helical linker and flexible hinge to a C-terminal effector-binding domain (EBD, also called regulatory domain, RD) (see Figure 1) (1–3,24). The EBD is less well conserved at the amino acid sequence level; however, EBDs share a conserved domain structure with two α/β subdomains (also named RD-1 and RD-2 domains) that are connected by two extended, antiparallel β-strands. A cleft in between the two EBD subdomains, RD-1 and RD-2, is crucial for control of the LTTR’s activity (see below). Both domains, the EBD and DBD, dimerize. Dimerization of the DBD in dyadic symmetry is mediated by the helical linker that connects the DBD to the EBD. Dimerization of the EBD in antiparallel orientation involves a conserved dimerization interface. Accordingly, most of the characterized LTTRs are tetramers containing two DBD-dimers and two EBD-dimers (2,24). Tetrameric LTTRs, with their two dimeric DBDs, bind two adjacent imperfect palindromic DNA-binding sites (often referred to as regulatory DNA-binding site, RBS and activation DNA-binding site, ABS), as shown initially for OxyR and IlvY (25–27). The DNA-binding affinity and pattern, or the topology of the LTTR-DNA complex can be affected by an effector-induced structural change that transmits from the EBD to the DBD (2,3,28). As effector, either a metabolite binds to the LTTR’s EBD or an amino acid residue is modified in response to a signal or specific stress. For example, in case of the oxidative stress regulator, OxyR, oxidation of a cysteine in the cleft of the EBD changes the relative distance and angle of the two dimeric DBDs to each other (25,29,30). This change triggers a ‘sliding’ of one DBD dimer along the DNA to the activation of DNA-binding site, ABS; while binding of the LTTR’s second DBD dimer to the higher affinity regulatory DNA-binding site, RBS, is maintained (29). Consequently, the OxyR-regulated oxyS gene is induced (25,29). Similarly, oxidation of Corynebacterium glutamicum OxyR and octopine binding to Agrobacterium tumefaciens OccR, respectively, can lead to an altered DNA-binding topology (30,31). Effector-binding to the LTTR IlvY changes IlvY-induced DNA-bending resulting in induction of the IlvY-regulated ilvC gene (encoding a ketol-acid reductoisomerase for branched-chain amino acid synthesis) (26,27). In case of the E. coli LTTR ArgP, binding of one effector, lysine, inhibits DNA-binding at 3 of 4 characterized target loci (lysP, dapB and gdhA); while it triggers trapping of RNA polymerase at the fourth ArgP-regulated locus, argO, encoding an arginine exporter. Intriguingly, binding of arginine, a positive second effector of ArgP induces DBD-dimer sliding and activation of argO by ArgP (32–34). Further, for several LTTRs constitutive mutants have been identified, all of which carry mutations that map to the EBD, suggesting that these mutations prompt a structural change similar to effector-induced allosteric changes. These examples of mutants include the LTTRs AmpR, AphB, BenM, CysB, DarR, NodD, NahR, NdhR, OxyR, TsaR and XapR, among others (3,30,35–47). Taken together, these examples highlight the flexibility and plasticity of transcriptional regulation by LTTRs including LeuO.
Figure 1.

Domain architecture and secondary structure elements of LeuO and LeuO-S120D. Schematic presentation of the LeuO domain and secondary structure, with the N-terminal DNA-binding domain (DBD) carrying a wHTH motif and linker helix as well as the C-terminal effector-binding domain (EBD), connected by a flexible hinge. Substitutions of residues that render LeuO hyperactive are shown atop and additional replaced amino acids are depicted below the scheme. The secondary structure of the EBD that is derived of the solved crystal structures is depicted in a schematic representation showing α helices as round filled cylinders, η helices as round unfilled cylinders, β sheets as arrows and non-solved areas as white boxes. The secondary structure of the DBD (shown in light gray) is based on a structural model (https://swissmodel.expasy.org/repository/uniprot/P10151).
For LeuO up to date only a rather degenerate consensus DNA-binding sequence has been identified (5), and no effector or condition has been identified that modulates the activity of the LeuO protein. The lack of a strong DNA-binding consensus sequence may be based on a weak DNA-binding specificity of LeuO and/or a preference for structural features that are related to AT-rich sequences, which has recently been discussed as ‘indirect read-out’ (48). This would be in agreement with LeuO’s presumptive general function as H-NS co-regulator. Another possibility is that LeuO responds to a signal that triggers an effector-induced structural change typical for LTTRs.
In this study, we screened for and isolated LeuO mutants that are hyperactive in activation of the cas promoter and in positive autoregulation of leuO in E. coli. The mutants were functionally characterized and the amino acid substitutions were mapped onto the crystal structure of the EBD, which was solved both for wild-type LeuO and for hyperactive LeuO-S120D. Our data suggest that LeuO-S120D (and putatively the other hyperactive mutants) mimics an effector-induced LeuO.
MATERIALS AND METHODS
Bacterial strains, plasmids and media
Escherichia coli K12 strains (7,49,50) and strain constructions are described in Table 1. Strains were constructed by integration of reporter constructs at the phage λ attachment site attB, as described (51,52). The plasmids (53–56) and plasmid constructions are described in Supplementary Table S1, and oligonucleotides are given in Supplementary Table S2. Bacterial cultures were grown in LB medium (5 g/l Bacto Yeast extract, 10 g/l Bacto Tryptone, 5 g/l NaCl), and for plates 15 g/l Bacto Agar was added. Antibiotics were added in final concentrations of 50 μg/ml ampicillin, 15 μg/ml chloramphenicol and 25 μg/ml kanamycin, if required. IPTG (isopropyl-β,D-thiogalactopyranoside) was added to liquid cultures at a concentration of 1 mM and to plates at a concentration of 200 μM. X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) was added at a final concentration of 40 μg/ml to tryptone plates for screening of leuO mutants.
Table 1.
Escherichia coli K12 strains
| Strain | Genotypea | Reference/Constructionb |
|---|---|---|
| BW30270 | MG1655 rph+ (motile with P-flhDC::IS1) (lab storage number S3839) | CGSC#7925 |
| C41(DE3) | BL21(DE3) derivate selected for expression of toxic proteins | (49) |
| XL1-Blue | F’::Tn10 (TetR) proA+B+ lacIq Δ(lacZ)M15 / recA1 endA1 gyrA96 (NalR) thi-1 hsdR17 (rk–mk–) glnV44 relA1 lac | Stratagene ® |
| S4197 | BW30270 ilvG+ΔlacZ (non-motile) | (7) |
| T314 | S4197 ΔleuOFRT Δ(yjjP-yjjQ-bglJ)FRT | (7) |
| T308 | S4197 attB::(PleuO lacZ aadA) ΔleuOFRT Δ(yjjP-yjjQ-bglJ)FRT | (7) |
| T352 | S4197 attB::(PleuO lacZ aadA) ΔleuOFRT Δ(yjjP-yjjQ-bglJ)FRT ΔhnsFRTstpA::tet | (7) |
| T862 | S4197 attB::(PleuO lacZ aadA) ΔleuOFRT bglJC | (7) |
| T568 | T314 attB::(Pbgl t1RAT bglG lacZ aadA) | (7) |
| T1281 | T314 attB::(Pcas lacZ aadA) | T314/pLDR8 x pKESL34 |
| T1610 | T1281/ F’ Tn10 (TetR) proA+B+ lacIq Δ(lacZ)M15 | T1281 x F’ (XL1-Blue) conjugation |
aThe following abbreviations and genetic designations are used: FRT for Flp recombinase target site, aadA coding for spectinomycin resistance, tet coding for tetracycline resistance. Allele bglJC refers to allele yjjQ/bglJ-Y6::mTn10-cmR, directing constitutive expression of bglJ (50).
bConstruction of strains was performed by integration promoter lacZ reporter fusions into the chromosomal attB site (indicated as strain/pLDR8 x plasmid designation) as described (51). In brief, integration of promoter lacZ constructs into the phage λ attB site was performed using replication origin-less re-ligated BamHI fragments of the indicated plasmids carrying a promoter lacZ fusion, the attP-site and the aadA gene for selection by spectinomycin. All listed alleles of the strains constructed in this work were confirmed by PCR.
Expression analysis
For expression analyses of promoter-lacZ fusions, β-galactosidase assays were performed, as described (57). Briefly, exponential cultures were inoculated from a fresh overnight culture to an OD600 0.1 in LB medium that was supplemented with the specific antibiotic in case of transformants. Where indicated, IPTG was added for induction both to the overnight and the exponential culture to a final concentration of 1 mM. Cultures were grown at 37°C to OD600 0.5 and then harvested on ice. The β-galactosidase assays were repeated at least three times from independent biological replicates.
Protein purification
For purification of C-terminally histidine-tagged LeuOHis6, LeuO-S120DHis6 and LeuO-DBDHis6, expression strain BL21(DE3)C41 was transformed with plasmids carrying the respective genes under the control of a T7 promoter (Supplementary Table S1). About 2 L cultures of LB medium with ampicillin were inoculated to an OD600 of 0.1 and grown at 37°C to OD600 of 0.6. At this point, protein expression was induced by adding IPTG to a final concentration of 200 μM, and the cultures were grown for further 5 h at 28°C. The cultures were harvested and the bacteria were pelleted by centrifugation. The cells were resuspended in resuspension buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl), and again pelleted for storage at −80°C. Lysate preparation was performed at 4°C; the cell pellets were resuspended in lysis buffer [4 ml/g of cells; 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10 mM Imidazole, 20 μg/ml DNase I (5000 U/ml; New England Biolabs, USA)] and lysed by sonication (40% amplitude, 4 min with a 2-s pulse/ 2-s pause; Sonics Vibracell VCX750 High-Volume Ultrasonic Cell Disrupter). The lysate was cleared by centrifugation (35 000 rpm, 45 min, Ti70 rotor, Beckman Coulter XL70) and the supernatant was loaded onto 1 ml HisTrap HP column (GE Healthcare, Germany) using an ÄKTA fast protein liquid chromatography (FPLC) system (GE Healthcare, Germany). The column was washed with the same buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl) with increasing imidazole concentrations (10, 20, 40 and 60 mM), with at least 10 column volumes at each step. The proteins were eluted with 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 250 mM imidazole and 1 ml fractions were collected and analyzed by SDS-PAGE. The protein containing fractions were pooled, and the buffer was exchanged with a PD-10 desalting column (GE Healthcare, Germany) to storage buffer (for LeuO-DBDHis6: 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 50 mM NDSB-256; for LeuOHis6 and LeuO-S120DHis6: 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM DTT, 150 mM NDSB-256). Proteins that were purified for SPR were stored in KCl-storage buffer (proteins were stored in 20 mM Tris, pH 8.0, 100 mM KCl, 150 mM NDSB-256). Protein concentrations were measured by Qubit Fluorometric Quantitation system (Invitrogen, Germany). Aliquots of the proteins were stored at −80°C.
Crystallization of LeuO-EBD and LeuO-S120D-EBD
For crystallization, C-terminally histidine-tagged LeuO effector-binding domains LeuO-EBDHis6 and LeuO-S120D-EBDHis6 were purified as described above for the full-length LeuO protein. Immediately after NiNTA affinity chromatography, the protein containing fractions were pooled and concentrated to 2 mg/ml protein using Vivaspin 20 (10 000 MWCO; Sartorius, Germany). Then, the proteins were subjected to size exclusion chromatography (Superdex 200 16/600; GE Healthcare, Germany) at 4°C in 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM DTT, 150 mM NDSB-256 using an ÄKTA purifier FPLC (GE Healthcare, Germany). The eluted fractions were analyzed by SDS-PAGE, and fractions containing pure protein were pooled and concentrated to 12.5 mg/ml.
Both proteins were crystallized using the sitting-drop vapor diffusion method. LeuO-S120D-EBDHis6 protein (12.5 mg/ml) was mixed with precipitant in a ratio 2:1 to form 300 nl drops and incubated at 20°C. Orthorombic crystals (space group I222) grew in a condition containing 1.4 M sodium malonate, pH 7.0, 0.1 M Bis-Tris propane, pH 7.0, and monoclinic crystals (space group C2) grew in a condition containing 0.1 M Tris, 1.2 M sodium/potassium-tartrate, pH 8.0. Crystals with dimensions of about 80 × 50 × 50 μm3 were cryoprotected in the same buffers supplemented with 30% sucrose and flash-cooled in liquid nitrogen. LeuO-EBDHis6 protein (12.5 mg/ml) was mixed with precipitant in a ratio 1:2 to form 300 nl drops and incubated at 20°C. Crystals grew in a condition containing 0.2 M lithium sulfate, 0.1 M Bis-Tris, pH 5.5, 25% polyethylene glycol 3350. Crystals with dimensions of 90 × 90 × 50 μm3 were cryoprotected in 0.2 M lithium sulfate, 0.1 M Bis-Tris, pH 5.5, 32% polyethylene glycol 3350 and flash-cooled in liquid nitrogen.
X-ray data collection, structure determination and refinement
First, X-ray diffraction data from LeuO-S120D-EBD crystals were collected at 100 K in house using the MicroMax 007-HF diffractometer (Rigaku) and a MAR345 imaging plate (marXperts). High-resolution X-ray diffraction data were collected at 100 K at the Swiss Light Source, Paul-Scherrer-Institute, Villigen, Switzerland on beamline X06DA using the Pilatus 2M detector (Dectris). Diffraction data were processed and scaled using XDS (58) and DIALS (59).
The first LeuO-S120D-EBD structure was solved in the orthorhombic space group I222 to 2.61 Å resolution. Initial phases and model were obtained by molecular replacement with the program Phaser (60) using a truncated and cleaned model based on the structure of a putative transcriptional regulator from Vibrio parahaemolyticus (PDB ID: 3OXN, http://www.rcsb.org/structure/3OXN) showing 21% sequence identity to LeuO. Due to a low TFZ score of 4.9 and LLG of 16 of the molecular replacement solution, we used the initial phases and model in iterative runs of phenix.autobuild (61), phenix.refine (62) and extensive manual model building in Coot (63) to obtain a final model of LeuO-S120D-EBD ‘in house’ with Rwork/Rfree of 20.7%/24.0%.
The high-resolution structure of LeuO-S120D-EBD was solved in space group I222 to 2.1 Å resolution by molecular replacement with the program Phaser using the refined LeuO-S120D-EBD ‘in house’ model. The structure of LeuO-EBD was solved in the monoclinic space group C2 to 1.6 Å resolution by molecular replacement with LeuO-S120D-EBD as search model also using Phaser. Refinement of both structures was performed by iterative cycles of phenix.refine and manual model building in Coot. All data collection and refinement statistics 64–67) are given in Table 2 and the figures were prepared using Chimera (68).
Table 2.
Data collection and refinement statistics
| Wild-type | S120D-C2 monoclinic | S120D-I222 orthorhombic | |
|---|---|---|---|
| Wavelength [Å] | 0.9794 | 1.0000 | 1.0000 |
| Resolution rangea | 54.28–1.52 (1.575–1.52) | 21.86–1.74 (1.802–1.74) | 52.44–1.94 (2.009–1.94) |
| Space group | C 1 2 1 | C 1 2 1 | I 2 2 2 |
| Unit cell [Å, deg.] | 63.62 61.22 56.54 90 106.24 90 | 71.96 53.30 55.80 90 113.08 90 | 64.99 88.77 97.20 90 90 90 |
| Total reflections | 126063 (12263) | 64101 (5614) | 270041 (24603) |
| Unique reflections | 31127 (3000) | 19969 (5614) | 21190 (2070) |
| Multiplicity | 4.0 (4.1) | 3.2 (2.9) | 12.7 (11.9) |
| Completeness (%) | 96.95 (93.46) | 99.54 (97.99) | 99.89 (99.76) |
| Mean I/sigma(I) | 10.34 (1.01) | 9.89 (1.12) | 19.75 (1.52) |
| Wilson B-factor /Å2 | 20.12 | 21.57 | 36.93 |
| R-mergeb | 0.051 (0.4834) | 0.062 (0.4267) | 0.082 (1.597) |
| R-meas | 0.059 (0.5548) | 0.074 (0.5225) | 0.086 (1.669) |
| R-pim | 0.029 (0.2689) | 0.040 (0.2974) | 0.024 (0.48) |
| CC1/2c | 0.998 (0.825) | 0.998 (0.766) | 1 (0.757) |
| CC* | 1 (0.951) | 0.999 (0.932) | 1 (0.928) |
| Reflections used in refinement | 31124 (2999) | 19969 (1954) | 21173 (2068) |
| Reflections used for R-freed | 1871 (180) | 1997 (195) | 1090 (110) |
| R-work | 0.1666 (0.2431) | 0.1768 (0.2557) | 0.2094 (0.3574) |
| R-free | 0.1932 (0.2855) | 0.2072 (0.3108) | 0.2340 (0.3819) |
| Number of non-hydrogen atoms | 1793 | 1699 | 1685 |
| macromolecules | 1607 | 1577 | 1649 |
| ligands | 6 | 5 | 7 |
| solvent | 180 | 117 | 29 |
| Protein residues | 192 | 193 | 201 |
| RMS(bonds) [Å] | 0.010 | 0.005 | 0.006 |
| RMS(angles) [deg.] | 1.05 | 1.06 | 0.97 |
| Ramachandran favored (%)e | 96.24 | 97.33 | 97.95 |
| Ramachandran allowed (%) | 3.76 | 2.67 | 2.05 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.00 | 0.00 | 0.54 |
| Clashscore | 3.45 | 4.18 | 1.84 |
| Average B-factor [Å] | 30.20 | 37.36 | 59.71 |
| macromolecules | 29.31 | 37.38 | 59.91 |
| ligands | 36.25 | 54.91 | 72.89 |
| solvent | 37.91 | 36.27 | 45.23 |
| Number of TLS groups | 4 | 6 | 5 |
aStatistics for the highest-resolution shell are shown in parentheses.
b R-merge = ΣΣ|I(hkl;j) - <I(hkl)>|/ ΣΣ<I(hkl)> with I(hkl;j) being the jth measurement of the intensity of the unique reflection (hkl) and <I(hkl)> the mean over all symmetry-related measurements; R-meas = ΣΣ [n(hkl)/(n(hkl-1)]1/2 |I(hkl;j) - <I(hkl)>|/ ΣΣ<I(hkl)> (64);
cCC(1/2) (65);
drandom 5% of working set of reflections (66);
eMolProbity (67)
DNase I footprinting
DNase I footprinting was carried out as described (69), with minor modifications. Briefly, 75 pmol of oligonucleotides were labeled with 75 μCi [γ-32P]ATP (3000 Ci/mmol, Hartmann Analytics, Germany) in the presence of 20 units of T4 polynucleotide kinase (Thermo Fisher Scientific, Germany) in 50 μl T4 polynucleotide kinase buffer for 30 min at 37°C. Unincorporated nucleotides were removed from the labeled oligonucleotides using illustra ProbeQuant G50 Micro columns (GE Healthcare, Germany) that were pre-buffered with PCR buffer. Then, DNA fragments were generated by standard PCR in 50 μl with GoTaq polymerase (Promega, Germany) using 20 pmol of the [32P]-labeled oligonucleotide and a non-labeled oligonucleotide as reverse primer. The DNA fragments were purified by agarose gel electrophoresis and eluted with a gel purification kit (Machery Nagel, Germany). The concentration of the eluted fragments was measured using a Qubit Fluorometric Quantitation system (Invitrogen, Germany), and the counts per minute (cpm) were determined by Cerenkov counting. For each footprint reaction 120 000 cpm (∼40 ng DNA) were used. Binding reactions were carried out for 10 min at 30°C in 40 μl of binding buffer (25 mM HEPES, pH 8.0, 100 mM potassium glutamate, pH 8.0, 0.5 mg/ml BSA) with increasing concentration of the wild-type LeuOHis6, LeuO-DBD (LeuO-DBDHis6) and LeuO-S120DHis6 proteins. Then, 0.02 units (in 4 μl) of DNase I were added (Thermo Fisher Scientific, Germany; diluted 200-fold in 10 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 10 mM CaCl2, 125 mM KCl, 0.1 mM DTT). Samples were incubated for 60 s at 30°C and DNase I digestion was stopped by adding 100 μl Tris-HCl, pH 8.0, buffered phenol and 200 μl stop buffer [0.5 M Na-acetate pH 5.0, 10 μg/ml herring sperm DNA (Ultra-Pure, Thermo Fisher Scientific, Germany), 2.5 mM EDTA] followed by a phenol extraction and ethanol precipitation. The DNA was dried and resuspended in 5 μl H2O, followed by addition of 6 μl loading dye (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol FF, 0.025% ethidium bromide, 0.5 mM EDTA). The samples were heated for 2 min at 90°C and loaded onto a 6% denaturing sequencing gel [6% long ranger (Lonza by Biozym Scientific, Germany), 7 M urea, 72 mM Tris-boric acid, 1.6 mM EDTA] next to a Sanger sequencing ladder, which was generated using the labeled oligonucleotide and the T7 polymerase sequencing kit (USB corporation, USA). The gel was washed with 10% ethanol, 10% acetic acid, transferred to Whatman 3 MM paper and dried in a gel dryer. Imaging was carried out by exposure to phosphorimaging plates and scanning by a Typhoon 7000 imaging system (GE Healthcare, Germany).
Identification of a consensus LeuO DNA-binding site
The DNase I footprint of LeuO DNA-binding site II at the cas promoter region was screened by eye and a 19 bp palindromic sequence of two 7 bp half-sites separated by 5 bp was detected. A similar palindromic sequence was found within DNA-binding site LeuO-I of the leuO regulatory region (7). Then, two 33 bp sequences covering the palindromic motifs of LeuO site II at the cas locus and LeuO site I at the leuO locus (Supplementary Table S3) were submitted to MEME Suite (70) to generate a motif, which was subsequently submitted to FIMO (71) to search the E. coli K 12 genome (substr MG1655 uid225) for putative LeuO DNA-binding sites. MEME suite is an open source application (http://meme-suite.org/). The 13 sequences with top scores (33.3 to 20.6; see 1.score in Supplementary Table S4) were filtered regarding their intergenic position as well as their identification in a microarray (7) and a genomic SELEX screening (6). Six of the 13 putative LeuO-binding sequences fulfilled all criteria (see Supplementary Table S4). In a second motif analysis, 33 bp sequences covering the six putative LeuO-binding sites were submitted to MEME Suite to generate a less stringent motif. When this motif was submitted to FIMO to search the E. coli K12 MG1655 genome for putative LeuO sites, 37 LeuO DNA-binding sequences with the best motif scores (cut off value 14.0; see 2. score in Supplementary Table S4) were identified. These were compared with previous microarray and SELEX data (6,7) and the gene function was taken from the Ecocyc Database (72) (see Supplementary Table S4). A similar motif was obtained with less stringent conditions of filtering the initial 13 sequences.
DNA-binding analysis by surface plasmon resonance (SPR)
SPR assays were performed in a Biacore T200 device (GE Healthcare, Germany) using carboxy-methyl dextran sensor chips pre-coated with streptavidin (XanTec SAD500L, XanTec Bioanalytics GmbH, Germany). All experiments were carried out at a constant temperature of 25°C using HBS-EP+ (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% (v/v) detergent P20) as running buffer, as described (73). 5′-biotinylated DNA fragments A, B and C were generated for SPR by annealing of oligonucleotides (Supplementary Table S2). Before immobilizing the DNA fragments, the chips were equilibrated by three injections of 1 M NaCl/50 mM NaOH applied at a flow rate of 10 μl per minute. Then, the respective double-stranded biotinylated DNA fragment (10 nM) was injected at a flow rate of 10 μl per minute for a total contact time of 420 s. The chips were then washed by injecting 1 M NaCl/50 mM NaOH/50% (v/v) isopropanol. Approximately 300 RU (response units) of the relevant DNA fragment was bound per flow cell. Analyses of the kinetics of interaction of wild-type LeuO and LeuO-S120D and LeuO-DBD, respectively, with the three DNA fragments (60 bp full-length DNA-binding site, 25 bp core-site and 60 bp scrambled sequence as control) were performed at a flow rate of 30 μl per minute in HBS-EP+ buffer at 25°C. Various concentrations of the proteins (10-200 nM for wild-type LeuO, 10-100 nM for LeuO-S120D and LeuO-DBD), dissolved in HBS-EP+ buffer, were passed over the flow cells for 180 s, and the complexes formed were allowed to dissociate for 300 s before the next cycle started. After each cycle, the surface was regenerated by injection of 2.5 M NaCl for 30 s, followed by 0.5% (w/v) SDS for 60 s, at a flow rate of 30 μl per minute. All experiments were performed at 25°C. Sensorgrams were recorded using Biacore T200 Control Software 2.0 and analyzed with Biacore T200 Evaluation Software 2.0 as well as TraceDrawer software 1.5 (Ridgeview Instruments, Sweden). The surface of flow cell 1 was not coated and used to obtain blank sensorgrams for subtraction of the bulk refractive index background. The referenced sensorgrams were normalized to a baseline of 0. Spikes in the sensorgrams at the beginning and the end of the injection are due to the run-time difference between the flow cells for each chip.
Gel filtration chromatography of LeuO-DNA complexes
Gel filtration chromatography was performed with using a Superdex 200 10/300 GL column and ÄKTA fast protein liquid chromatography (FPLC) system (GE Healthcare, Germany) in 20 mM Tris-HCl pH 8.0, 100 mM KCl at a flow rate of 0.5 ml/min. The calibration curve was obtained with a gel filtration marker kit (MWGF1000, Sigma Aldrich). LeuOHis6 (0.18 mg/ml) and LeuO-S120DHis6 (0.27 mg/ml) were purified by Ni-NTA affinity chromatography as described above and stored at −80°C in storage buffer (20 mM Tris-HCl, pH 8.0, 100 mM KCl, 1 mM DTT, 150 mM NDSB-256). A 60 bp DNA fragment encompassing the LeuO DNA-binding site II at the cas promoter region was generated by annealing of oligonucleotides OA745/OA746 at a concentration of 10 pMol/μl in 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0, 100 mM NaCl. To generate DNA-bound complexes of LeuOHis6 and LeuO-S120DHis6, respectively, 1000 pMol of the proteins (concentration of the monomer) were incubated with 200 pMol of the 60 bp DNA fragment for 1 h at room temperature in a total volume of ∼220 μl. Then, the LeuO protein–DNA complexes were subjected to gel filtration chromatography.
RESULTS
Screen for hyperactive LeuO mutants
For the LysR-type regulator LeuO no effector is known, while for other LysR-type regulators effectors have been described and amino acid mutations have been characterized that mimic the effector-bound form and render these regulators constitutively active (3,35–41,43–46). To characterize LeuO, we set up a screen for LeuO mutants that are constitutively active. As a reporter the promoter of the cas operon (encoding the CRISPR-associated Cascade complex) that is activated by LeuO (9,74) was fused to lacZ, and this Pcas-lacZ fusion was integrated into the genome (at the phage λ attachment site attB) of a ΔlacZ ΔleuO background (yielding strains T1281 and T1610). As expected, expression of the Pcas-lacZ reporter was strongly activated by LeuO (from 6 units to 568 units, +IPTG), when LeuO was provided at high levels using a low-to-medium copy plasmid (p15A origin of replication) carrying leuO under the control of the IPTG (isopropyl-β,D-thiogalactopyranoside) induced tac promoter (Figure 2), as shown before (74). At basal expression levels of leuO, without induction of Ptac, the Pcas-lacZ reporter was moderately activated by LeuO (compare 51 units to 6 units of control, Figure 2). Thus, the basal expression level of leuO directed by the leaky Ptac was a suitable condition to screen for constitutively active LeuO mutants. For mutagenesis, the leuO gene fragment was amplified by PCR in parallel reactions using the non-proofreading Taq polymerase, and the PCR fragments were ligated into the Ptac expression plasmid (pKESK22). Transformants of the Pcas-lacZ reporter strain with these ligations were screened for a Lac-positive phenotype on tryptone X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) indicator plates without IPTG. Nine different mutants with substitutions of single amino acids were isolated from 25 independent mutagenesis PCRs. These mutants include 3 independent isolates encoding LeuO-S128P as well as 2 independent isolates each of LeuO-H142R, LeuO-Q210R, LeuO-A237V, and LeuO-H254R, respectively. LeuO-T127I and LeuO-R218C, respectively, were isolated once. LeuO-M244T was isolated as a single mutant and as an independent double mutant with the second amino acid substitution V230I (Supplementary Table S1). In addition, LeuO-S120D was isolated as a triple mutant with additional amino acid substitution at residues E111D and D205N, using a Pbgl-lacZ reporter strain (Supplementary Table S1). Note that initially, a lacZ fusion of the LeuO-activated bgl promoter was used as reporter; however, the Pcas-lacZ fusion turned out to be more sensitive. To analyze the impact of the Ser120 to Asp (S120D) substitution, a single mutant was constructed and used for further analysis. Multiple isolations of the same mutants suggest that the screen for hyperactive LeuO mutants was saturated.
Figure 2.
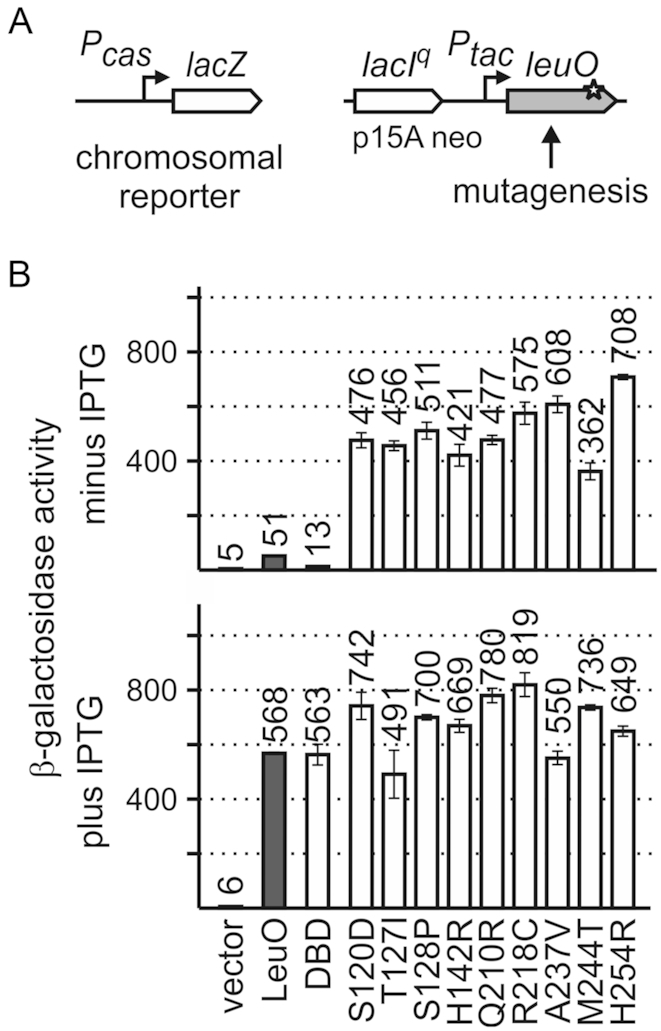
Screen for active LeuO mutants. (A) To screen for active LeuO mutants, a Pcas-lacZ fusion integrated into the chromosomal attB-site was used as reporter for LeuO activity (left). For mutagenesis, leuO gene fragments were amplified in parallel reactions by non-proofreading PCR and cloned in plasmid pKESK22 (p15A replication origin, kanamycin resistance) to place leuO under control of the tac promoter (Ptac) (right). Transformants were selected on tryptone X-Gal, kanamycin indicator plates without IPTG and screened for a Lac-positive phenotype. Of a total of 17 Lac+ clones, the leuO fragment was sequenced, which revealed nine different mutations in leuO (Supplementary Table S1). (B) Activation of Pcas-lacZ expression by the LeuO mutants was determined cultures grown without induction of leuO expression (top panel) and with 1 mM IPTG for induction (bottom panel). Expression was determined in absence (vector control, pKESK22) and in presence of plasmid-provided LeuO (pKETS5), LeuO-DBD (pKESMS63), and mutants S120D (pKESL104), T127I (pKESL74), S128P (pKESL75), H142R (pKESL76), Q210R (pKESL77), R218C (pKESL78), A237V (pKESL80), M244T (pKESL73) and H254R (pKESL101). Cultures for β-galactosidase assays were inoculated from overnight cultures and grown in LB medium with kanamycin to an OD600 of 0.5. Where indicated 1 mM IPTG was added both to the overnight and exponential culture. Mean values of at least three independent biological replicates are shown as bars, and error bars indicate standard deviations.
The hyperactive LeuO mutants were used in a quantitative assay of Pcas-lacZ activation (Figure 2). To this end, transformants of the Pcas-lacZ reporter strain with plasmids carrying the leuO mutants were grown without (no IPTG) and with induction (1 mM IPTG) of leuO expression directed by the tac promoter, and the β-galactosidase activities were determined. When leuO expression was induced, all mutants caused full activation of Pcas-lacZ (Figure 2B, bottom panel). At low leuO expression levels directed by the basal activity of the tac promoter (no IPTG), all LeuO mutants isolated in the screen fully activated the Pcas-lacZ reporter (Figure 2B, top panel). The expression levels ranged from 362 to 708 units of β-galactosidase activity referring to a 7- to 14-fold stronger activation of Pcas-lacZ than by wild-type LeuO (51 units, Figure 2B, top panel). Thus, each of the nine substitutions of single amino acids renders LeuO hyperactive. All the respective residues locate in the effector-binding domain (EBD, amino acids 108–314), and none in the N-terminal DNA-binding domain (DBD, amino acids 1–101; Figure 2).
Crystal structure of the LeuO effector-binding domain (EBD)
The nine mutations causing hyperactivity of LeuO mutants map in the effector-binding domain (EBD) that extends from residue Ala108 to Arg314, according the structure of E. coli LeuO (Figure 3A) and an alignment of S. enterica LeuO with other LysR-type regulators (23) (see Figure 1). To characterize the functional relevance of these residues, the crystal structure of the LeuO-EBD (PDB ID: 6GZ0) was determined (Figure 3B and Table 2). In addition, the structure of the EBD of hyperactive mutant LeuO-S120D was solved in an orthorhombic (space group I222, PDB ID: 6GZ2) and a monoclinic (space group C2, PDB ID: 6GZ1) crystal form (Figure 3A and Table 2). Note that no suitable crystals of the full-length proteins were obtained. In the EBD crystal structures, most residues are well resolved in the electron density maps with the exception of the loop connecting strands β4 and β5 (residues 151–158 including helix α6; secondary structure elements are numbered according to the prediction for the full-length protein), which has weak electron density in the wild-type and in the monoclinic crystal form of the S120D structure, but it could be traced in the orthorhombic S120D crystal form. Furthermore, the loop between β5 and β6 (residues Q151 to R158 and R173 to E175) is not resolved in the wild-type and monoclinic mutant structures and possesses only weak density in the orthorhombic S120D structure.
Figure 3.

Crystal structures of LeuO-EBD and LeuO-S120D-EBD. (A) Cartoon representations of LeuO–S120D–EBD dimer with residues causing hyperactivity when mutated shown in red. (B) LeuO-EBD monomer shown in gradient coloring (blue, N-terminus; red, C-terminus), with secondary structure elements labeled. A bound chloride ion is shown in green and residues causing hyperactivity when mutated are indicated as sticks. (C) Close-up view of dimer LeuO-EBD with bound chloride ion and residues Ser120, Met244 and Arg218 shown as sticks. (D) Superposition of LeuO-EBD in green and LeuO-S120D-EBD crystallized in different space groups in cyan (I222) and purple (C2) in ribbon representation. The Cα atoms of residue Arg112 have a distance of about 4 Å between wild-type and S120D mutant. Numbering of residues is according to the full-length protein sequence of LeuO. The figure was prepared using Chimera (68).
The EBD exhibits the typical fold with two α/β subdomains (also denoted as RD-1 and RD-2 domains) connected by two extended, antiparallel cross-over strands β6 and β11. DALI and PDB FOLD searches revealed the effector-binding domain of Burckholderia sp. DntR as the structurally most similar protein with known structure (PDB ID: 5AE5) (75). In all three crystal structures, the EBDs form a dimer (Figure 3A) that is predicted to be stable in solution under standard conditions as judged by the PISA server (76). This dimer is similar to those observed in structures of other isolated EBDs as well as in full-length structures of LysR-type regulators, e.g. in the structures of AphB (43). As anticipated, there is no tetramer in all three crystal structures presented here, because tetramerization requires the α4 linker helix of the DNA-binding domain that is not present in our constructs.
The 3D structures of wild-type and S120D mutant are similar, however with some important differences (Figure 3D). The root-mean-square (RMS) deviation of all 192 equivalent Cα pairs is ∼1.7 Å for the superposition of wild-type and each of the two S120D structures, while superposing the latter results in a significantly lower RMS deviation of 1.2 Å. Flexible alignment by the RAPIDO web server (77) reduced the RMS deviation to ∼0.8 Å for 175 Cα pairs, revealing two rigid bodies. The first rigid body essentially consists of strands β3 and β4 and helices α5 and α10. This becomes more apparent superposing only domains RD-2 (residues 185–285), which results in an RMS deviation of the 101 Cα pairs of 0.4 Å between wild-type and each mutant structure. Superposing in this way the entire structures leads to a good alignment of both S120D structures, while the wild-type differs especially in the position of α5 and α10 of domain RD-1 (Figure 3D) as well as of strands β3 and β4. Remarkably, the Cα atoms of the first N-terminally resolved residue (Arg112) have a distance of ∼4 Å between wild-type and mutant (Figure 3D). Similar differences between inactive and active EBDs have been observed previously, e.g. in the structures of DntR (PDB IDs: 2y7w, 2y7r) (78) and HypT (PDB ID: 5YDW, 5YDO, 5YDV, 5YER, and 5YEZ) (79). Interestingly, the largest deviations of the three structures in the RD-2 domain occur at the ‘arginine elbow’ at residue 218, of which an exchange to glutamic acid, cysteine or alanine results in hyperactivation (see below).
All amino acids whose mutation causes hyperactivity are surface exposed and all map in the putative dimerization interface (Figure 3A). The residues Ser120, Arg218 and Met244 are located in the vicinity of the cleft, which is located in between the two α/β subdomains and which represents a putative effector-binding pocket. Met244 is close to its counterpart of the second monomer (Figure 3C). Arg218 is reasonably well defined in the wild-type structure and points toward the interior of the putative effector-binding pocket, while mutation of residue Ser120 to Asp induces a reorientation of the side-chain of Arg218 (Figure 3D) with much weaker electron density.
Interestingly, in the wild-type protein structure there appears to be a chloride ion bound close to the position in which the carboxylate group of Asp120 is located in the S120D mutant (Figure 3C). The assignment of the electron density peak as chloride is based on the coordination of the ion, the peak height and a weak anomalous signal. The putative chloride ion is coordinated by the backbone amide of Met243, the hydroxyl group of Ser120 and two water molecules. The vicinity is otherwise rather hydrophobic, with the side-chains of Pro121, Ala242, Met243 and Val214 lining the pocket (Supplementary Figure S1). The guanidinium group of Arg218 is ∼4.5 Å apart from the putative chloride ion (Figure 3C).
Characterization of additional LeuO mutants
Residues Ser120, Arg218 and Met244 map close to the putative effector-binding pocket and are involved in intramolecular and intermolecular contacts. To further characterize these residues, they were mutated to alanine, and Arg218 was in addition mutated to glutamic acid. Further, a LeuO-S120D/M244T double mutant was constructed. The activity of these LeuO mutants was analyzed using the Pcas-lacZ reporter. LeuO-S120A and M244A showed a reduced activation of Pcas compared to wild-type LeuO at basal expression levels (no IPTG, Supplementary Figure S2, top panel), while upon induction of their expression (1 mM IPTG) the mutants fully activated Pcas (Supplementary Figure S2). These results show that the hyperactivity is specific for the Ser120 to Asp and Met244 to Thr amino acid substitutions, but not caused by the substitution to alanine (S120A and M244A, Supplementary Figure S2). The double mutant LeuO-S120D/M244T remained hyperactive, causing full activation of Pcas (Supplementary Figure S2). Interestingly, substitution of Arg218 by alanine or glutamic acid rendered the protein similarly hyperactive as the Arg218 to cysteine substitution, which was isolated in the screen (Supplementary Figure S2).
LeuO harbors two cysteine residues at position 117 and 119, located in the cleft of the EBD (Figures 1 and 3B). To test the relevance of Cys117 and Cys119 for LeuO activity, these residues were replaced by a serine and aspartic acid, respectively. The mutant C119D showed increased activation of Pcas compared to wild-type LeuO (increase from 51 units to 148 units at basal expression levels, Supplementary Figure S2). In contrast the other three mutants C117S, C117D and C119S had a slightly reduced activity even when expressed at high levels (Supplementary Figure S2). Thus, substitution of Cys119 by a negatively charged aspartic acid enhances LeuO activity, which may have a similar effect as the substitution of residue Ser120 to the negatively charged aspartic acid in hyperactive LeuO–S120D.
In order to further characterize residues in the cleft of the EBD, several additional residues were replaced (Figure 1 and Supplementary Figures S1 and S2). Pro121 was replaced by the negatively charged aspartic acid and Leu122, Ile125, Tyr168, Phe219, Met243 and Leu263 by negatively charged glutamic acid. The activity of these mutants was analyzed using a low copy number (pSC origin) expression plasmid carrying leuO under control of the moderate lacUV5 promoter (Supplementary Figure S2). Upon induction of leuO expression mutants, F219E and M243E activated the Pcas-lacZ reporter as wild-type LeuO (Supplementary Figure S2, plus IPTG). In contrast, activation of Pcas was strongly decreased for LeuO mutants P121D, L122E, I125E, Y168E and L263E (Supplementary Figure S2, plus IPTG). Thus, these mutants, which include residues Leu122 and Tyr168 lining the cleft and putative effector-binding site, are inactive. In S. enterica a LeuO-P139A mutant is likewise inactive (23), which supports the role of the RD-1 subdomain in control of LeuO activity.
Taken together, the analysis of additional LeuO mutants revealed that hyperactivity is specific for the particular amino acid substitution S120D and M244T, while the activity of mutant C119D is moderately increased. Arg218 may be inhibitory since substitution of this residue by alanine, cysteine and glutamic acid, respectively, causes hyperactivity.
The LeuO DNA-binding domain (DBD) is functional
E. coli LeuO is presumably a tetramer, like S. enterica LeuO and other LTTRs (23). LTTR tetramers contain two pairs of dimeric DNA-binding domains (DBD) and two pairs of dimeric effector-binding domains (EBD). Dimerization of the N-terminal DBD is mediated by the α4 helical linker (Figure 1), while the EBDs provide an additional dimerization interface (Figure 3). The two DBD dimers within one tetramer presumably bind to two adjacent sites on the DNA. Therefore, a single dimeric DBD may be capable to specifically bind DNA as well. To address this, we analyzed whether the LeuO DBD-dimer is sufficient to activate the Pcas-lacZ reporter. The DBD (aa1 to aa101) was provided using the Ptac expression plasmid pKESMS63. Upon induction of Ptac-directed leuODBD expression with IPTG, full activation of Pcas-lacZ was observed (Figure 2). At basal expression levels of leuODBD (no IPTG), Pcas activation by the DBD was 4-fold lower than by wild-type LeuO, but still detectable (Figure 2, compare 13 and 51 units). These data show that the DBD alone is sufficient to activate Pcas, suggesting specific DNA-binding by the LeuO-DBD dimer.
DNase I footprinting by LeuO, LeuO-S120D and the DBD domain
Two extended AT-rich DNA-binding sites for LeuO at the cas promoter region (LeuO sites I and II) have been identified by DNase I footprinting (74). In these footprints a rather high LeuO concentration was used, which corresponds to high levels of wild-type LeuO that is required for activation of Pcas in vivo. In contrast, only low levels of the hyperactive LeuO-S120D (and the other mutants) are required for Pcas activation, indicating that the DNA-binding affinity of S120D may be higher. To address this, DNA-binding of LeuO and the S120D mutant was analyzed by DNase I footprinting using a broad range of protein concentrations. In addition, to narrow down on the DNA-binding sites, the LeuO-DBD was included in the footprinting analysis. The rationale of this was that the full-length protein carrying two DBD dimers presumably contacts DNA at two sites and therefore yields an extended footprint. The DBD presumably occupies only half of the full-length LeuO DNA-binding site, which may allow defining a consensus sequence.
For footprinting, LeuOHis6, LeuO-S120D His6 and LeuO-DBD His6 were purified. DNA fragments covering the full-length LeuO DNA-binding sites I and II of the cas promoter region, respectively, were [32P]-labeled at the 5′-end of the top and bottom strand, respectively, and used for footprinting analysis (Figure 4A). Footprints of DNA fragments ‘a’ and ‘b’ covering site II are shown in Figure 4B and C, while the footprints of site I (fragments ‘c’ and ‘d’) are shown in Supplementary Figure S3. Corresponding to the previously described LeuO footprint at site II, DNase I protection by all three proteins was detected (Figure 4B and C and Supplementary Figure S3). In the middle of this footprint, a non-protected site is apparent (black triangle) and therefore two half-sites ‘IIa’ and ‘IIb’ are labeled in the footprint (Figure 4B and C). Strikingly, DNase I protection by LeuO mutant S120D occurred already at very low protein concentrations (31 nM) suggesting that the Ser120 to Asp substitution causes higher DNA-binding affinity. Further, additional protection sites were detected for S120D that are indicative of a higher order complex (Figure 4B; Supplementary Figure S3). Intriguingly, the LeuO-DBD caused a DNase I protection pattern at low concentrations (31 nM) as well, but only at DNA-binding site IIa, while at higher protein concentrations (125 nM) both DNA-binding half-sites were protected (Figure 4B and C). For wild-type LeuO, a footprint was apparent as of a concentration of 125 nM, but this footprint appears more diffuse, at least at DNA-binding site IIb (Figure 4B and C) indicating that the steric arrangement of the DBDs in wild-type LeuO hinders DNA-binding. For DNA-binding site I (footprints of fragments ‘c’ and ‘d’), all three proteins protected the DNA from DNase I cleavage as of a concentration of 125 nM, confirming previous data (74) (Supplementary Figure S4). Taken together, the DNase I footprints by LeuO-S120D and in particular by LeuO-DBD indicate that DNA-binding half-site IIa is a high affinity DNA-binding site, which is called ‘core-site’ in the following. This core-site is palindromic (indicated by inverted arrows and bold letters, Figure 4B and C). However, no palindromic core-site is apparent in DNA-binding site I (Supplementary Figure S4). Palindromic sequences are typical for DNA-binding of transcription regulators with dimeric DBDs (80). Thus, the palindromic DNA-binding site IIa may represent a close to ideal DNA-binding half-site of LeuO.
Figure 4.

DNase I footprinting of the cas promoter region with LeuOHis6, LeuO-DBDHis6 and LeuO-S120DHis6. (A) Schematic overview of the cas promoter region and fragments for DNase I footprinting. Indicated are LeuO DNA-binding sites I and II (black boxes) (74) and the Pcas transcription start site (bent arrow) (9). The sequence of LeuO DNA-binding site II with its two half-sites: IIa and IIb, and the core-site is shown at the bottom. Fragments (‘a’ to ‘d’) used for DNase I footprinting cover either one or both LeuO DNA-binding sites, as indicated. The fragments were generated by PCR using primer pairs of which one primer was 5′ [32P]-labeled, indicated by an asterisk (fragment ‘a’: oligonucleotides [32P]-OA477/OA474, fragment ‘b’: OA477/[32P]-OA475, fragment ‘c’: [32P]-OA476/OA473, fragment ‘d’: OA477/[32P]-OA473). Thus, fragments ‘a’ and ‘c’ were labeled at the top strand, and fragments ‘b’ and ‘d’ were labeled at the bottom strand. The [32P]-labeled DNA fragments were incubated with increasing protein concentrations of LeuOHis6, LeuO-DBDHis6 and LeuO-S120DHis6, as indicated. Autoradiograms of DNase I footprints of fragment ‘a’ with sequencing ladder (A and T lanes) (B) and fragment ‘b’ (C). LeuO-protected sites are marked by lines labeled I, IIa and IIb. IIa and IIb represent the two half-sites of DNA-binding site II. Additional protections are marked by dotted lines. Samples were separated on 6% denaturing polyacrylamide gels. Filled arrowheads indicate non-protected sites, open arrowheads indicate hypersensitive sites. The sequence of the protected DNA region is given to the right with the palindromic core sequence shown in bold and marked with inverted arrows. Autoradiograms of fragments ‘c’ and ‘d’ covering LeuO site I are shown in Supplementary Figure S3.
DNA-binding consensus sequence of LeuO
For LeuO, a 28 bp large and rather degenerate consensus sequence has been described (5). The DNase I footprint at the cas promoter region revealed a high affinity DNA-binding core-site, IIa, harboring a 19 bp palindromic sequence (shown in bold letters and indicated by arrows in Figure 4B and Supplementary Figure S4). An almost identical palindromic sequence is present within DNA-binding site I at the autoregulated leuO promoter (7) (Supplementary Figure S5). To define a DNA-binding motif, 33 bp sequences covering the palindromic sequences of the LeuO DNA-binding core-sites at the cas and leuO loci (Figure 5 and Supplementary Table S3) were submitted to MEME Suite (70). The resulting motif (Figure 5, top panel motif) was used to search the E. coli K12 MG1655 genome for putative LeuO DNA-binding sites, using FIMO (71). The obtained 13 top scores (Supplementary Table S4) were filtered in respect to their intergenic position as well as their previous identification as presumptive LeuO targets by microarray transcriptome analysis and genomic SELEX screening (6,7). Then, the sequences of the six top score loci (Supplementary Tables S3 and S4) were used to generate a less stringent motif using MEME Suite (Figure 5, bottom panel motif). To validate this motif, it was again submitted to FIMO to search the E. coli K12 MG1655 genome for putative LeuO DNA-binding sites. By this approach we identified 37 sequences with top scores, which include LeuO targets identified by microarray and genome scale SELEX (6,7), but also included sequences mapping at loci, which have not been characterized as LeuO targets before (Supplementary Table S4). Taken together, the LeuO DNA-binding motif (Figure 5) presumably represents the high-affinity DNA-binding half site that is bound by one DBD-dimer of the LeuO tetramer.
Figure 5.
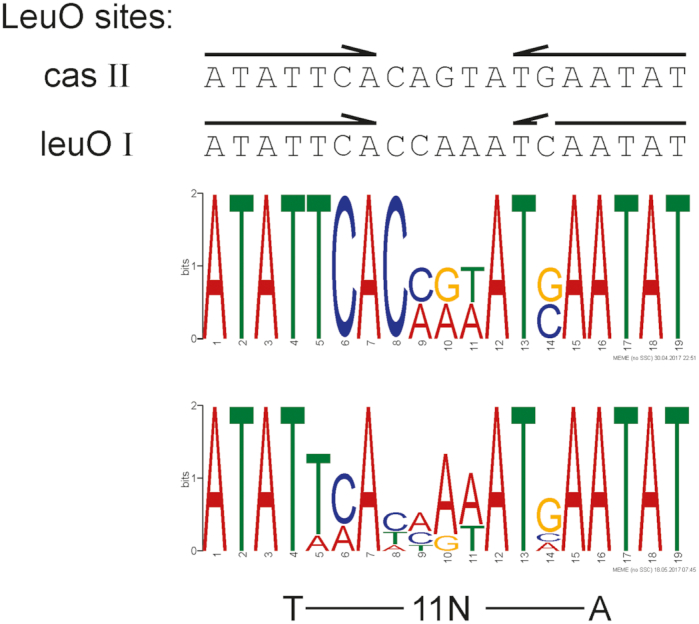
LeuO DNA-binding motif. Sequences of the palindromic core DNA-binding sites of LeuO site II at cas and of LeuO site I at leuO (shown at the top) were used in an iterative approach to determine a DNA-binding consensus sequence. Arrows indicate the palindromic sequence. The top sequence logo represents the motif that is based on the LeuO DNA-binding sites at cas and leuO and was generated with MEME Suite (70) using the sequences listed in Supplementary Table S3. The bottom motif is based on 6 top score hits in the E. coli K12 genome identified by FIMO (see Supplementary Tables S3 and S4). The T-11N-A motif typical for DNA-binding sites of LysR-type regulators (2) is indicated underneath the logo.
DNA-binding affinity of the LeuO, LeuO-S120D and LeuO-DBD
The DNase I footprinting revealed a DNA-binding core-site and an enhanced DNA-binding specificity of LeuO-S120D. To quantitatively determine the DNA-binding affinity of LeuO, LeuO-S120D and the LeuO-DBD, we used plasmon resonance (SPR). For SPR, biotin-labeled DNA fragments covering the full DNA-binding site II of LeuO at the cas locus (60 bp) (Figure 6A) or just the DNA-binding core-site (25 bp) (Figure 6B) as well as a control fragment with a 60 bp scrambled DNA-sequence (Figure 6C) were immobilized onto a sensor chip. Then increasing concentrations of the C-terminally His-tagged wild-type LeuOHis6, LeuO-S120DHis6 and the LeuO-DBDHis6 protein were injected. Wild-type LeuO specifically interacted with the 60 bp full-length binding site (Figure 6A) as well as with the 25 bp core-site DNA fragment (Figure 6B). This interactions exhibit high association (ka) and low dissociation rates (kd) resulting in an high affinity (KD) (Figure 6A and B). However, the highest affinities were observed for LeuO-S120D to both DNA-fragments (0.2 nM for the 60 bp full-site and 0.6 nM for the 25 bp half-site) due to higher association rates compared to wild-type LeuO (0.7 nM for the 60 bp full-site and 0.9 nM for the 25 bp half-site) (Figure 6A and B). Nonetheless, wild-type LeuO binds with high affinity in contrast to other apo-forms of LTTRs as for example HexA (73). Interaction of LeuO-DBD with the fragments encompassing the 60 bp full-length DNA-binding site and the 25 bp core-site, respectively, is characterized by comparable association and dissociation rates. However, the DNA-binding affinity (KD) of the DNA-binding domain (LeuO-DBD) was lower than for wild-type LeuO due to the ∼10-fold higher dissociation rates. No interaction of the LeuO protein variants with the 60 bp scrambled DNA fragment was detected, underlining the sequence specificity of LeuO DNA-binding (Figure 6C). The data suggest that the hyperactivity of the LeuO-S120D in regulation of the cas promoter is based on its enhanced DNA-binding affinity caused by an enhanced association rate compared to wild-type LeuO, while dissociation of LeuO-S120D and wild-type LeuO is comparable.
Figure 6.

DNA-binding affinity of LeuOHis6, LeuO-120DHis6 and LeuO-DBDHis6 to the DNA-binding site II of LeuO at cas (A), the DNA-binding core-site (B) and control fragment (C) determined by SPR. The biotinylated DNA-fragments were captured onto a streptavidin-coated sensor-chip. Different concentrations of LeuOHis6, LeuO-S120DHis6 and LeuO-DBDHis6 (10 nM: purple lines; 15 nM: dark blue lines; 2 × 20 nM: light blue lines; 30 nM: green lines; 50 nM: yellow lines; 100 nM: orange lines; and 200 nM: red lines, the latter only for LeuOHis6) were passed over the chip using a contact (association) time of 180 s, followed by a 300-s dissociation phase. The increase in response units (RU) correlates with increasing LeuO concentrations.
The measured maximal response units (Rmax) for wild-type LeuOHis6 (MW of monomer 36.8 kDa; RU = 260) and LeuO-S120DHis6 (MW of monomer 36.8 kDa; RU = 170) to the 60 bp full-length fragment differs, which may indicate that their binding stoichiometry is different. To address this, we performed gel filtration chromatography of LeuOHis6 and LeuO-S120DHis6 pre-bound to a 60 bp DNA fragment encompassing the LeuO DNA-binding site II (Supplementary Figure S6). The gel filtration data show that the size of the LeuOHis6-DNA and LeuOHis6-S120D DNA complexes are similar; the complexes elute at 12.43 and 12.48 ml, respectively, which corresponds to a molecular weight of ∼170 kDa (LeuOHis6 monomer 36.8 kDa, 60 bp DNA 39.6 kDa). The second peak eluting at 13.64 ml presumably corresponds to the unbound DNA fragment (Supplementary Figure S6). These data suggest that both proteins, LeuOHis6 and LeuO-S120DHis6, bind DNA as tetramers.
Autoregulation of PleuO by hyperactive LeuO variants
To analyze whether the LeuO amino acid substitutions causing hyperactivity in Pcas activation also affect other loci, we analyzed their activity in autoregulation of the leuO promoter. In wild-type cells, LeuO acts as a weak positive autoregulator, when provided at high levels from a plasmid (18). In addition, transcription of leuO is repressed by H-NS and StpA under standard laboratory conditions, and the transcription regulator BglJ-RcsB activates leuO expression (7). However, activation of leuO by BglJ-RcsB and de-repression of leuO in an hns stpA mutant is inhibited by LeuO, when provided at high levels from a plasmid, suggesting that LeuO is also a negative autoregulator (7).
To address autoregulation of leuO expression by the hyperactive LeuO variants and by the LeuO-DBD, the expression of a chromosomal PleuO-lacZ reporter was analyzed in several strain backgrounds. These include a ΔleuO ΔbglJ strain (T308) to assess positive autoregulation of PleuO (Figure 7). Wild-type LeuO, the hyperactive LeuO mutants, and LeuO-DBD were provided using plasmids carrying the leuO alleles under control of Ptac that directs basal expression without IPTG and high expression upon induction by IPTG. The data show that positive autoregulation of the leuO promoter by all hyperactive LeuO mutants is significantly enhanced as compared to wild-type LeuO, when provided at basal levels (Figure 7, minus IPTG). Mutant LeuO-S120D is particularly effective in positive autoregulation, while positive autoregulation by LeuO-M244T is weaker (Figure 7). Upon induction of high levels of LeuO protein synthesis only wild-type LeuO and the DBD acted as positive autoregulators, while in the presence of high levels of the hyperactive LeuO variants, the activity of PleuO remained very low (Figure 7, plus IPTG). These data suggest that the LeuO variants that are hyperactive in the activation of Pcas are also hyperactive in positive autoregulation of PleuO, when they are present in low amounts. The result that high levels of wild-type LeuO and of the hyperactive mutants (Figure 7, plus IPTG) cause autorepression indicates that LeuO variants occupy additional or alternative DNA-sites when present at high levels.
Figure 7.
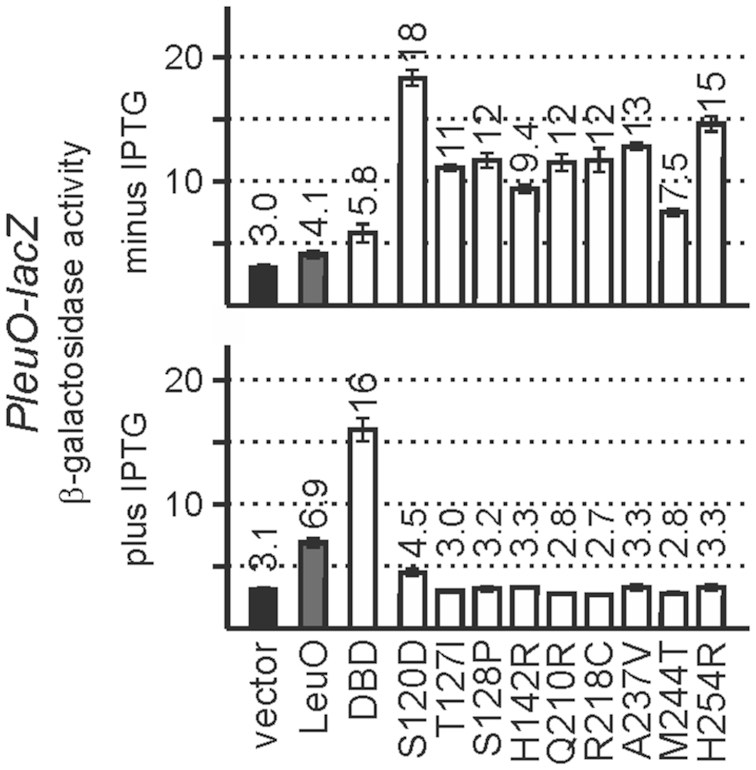
Autoregulation of the leuO promoter. Autoregulation was analyzed using a chromosomal PleuO-lacZ reporter strain T308 (ΔleuO) (Table 1) that was transformed with the vector control (pKESK22), and plasmids providing LeuO (pKETS5), LeuO-DBD (pKESMS63) and LeuO mutants (Supplementary Table S1). Transformants were grown in LB kanamycin medium to an OD600 of 0.5 without induction (top) or with induction by the addition of 1 mM IPTG (bottom). Mean values of at least three independent biological replicates are shown as bars, and error bars indicate standard deviations.
In addition, negative autoregulation of leuO was tested in a BglJ-RcsB positive strain carrying constitutively expressed allele bglJc (T862) and an hns stpA mutant derivative (T352); in both of these strain background PleuO is active. In respect to negative autoregulation, only mutant S120D was significantly different from wild-type LeuO, as basal levels of LeuO-S120D enhanced activation of PleuO by BglJ-RcsB and repressed PleuO in absence of H-NS and StpA (Supplementary Figure S7). High levels of all LeuO variants including the LeuO-DBD function as auto-repressor in the BglJ-RcsB-positive and hns stpA mutant backgrounds. These data suggest that LeuO-S120D is the hyperactive variant that is most effective in autoregulation.
DISCUSSION
Here we characterized E. coli LeuO, a LTTR of pleiotropic function. We identified LeuO mutants that are more effective than wild-type LeuO in activation of the cas promoter and in positive autoregulation of the leuO promoter. All of these hyperactive mutants carry single amino acid substitutions located at the dimerization interface of the C-terminal regulatory effector-binding domain (EBD). In particular a Ser120 to Asp substitution, located at the cleft of the EBD, confers increased DNA-binding affinity as shown by SPR and DNase I footprinting. Further, comparison of the crystal structures of the EBDs of LeuO and of LeuO-S120D revealed a structural change reminiscent of effector-induced structural changes in other LTTRs. This structural change presumably transmits to the steric arrangement of the two dimeric DNA-binding domains (DBDs) present within the tetramer, and thus causes the enhanced DNA-binding affinity. Intriguingly, the dimeric DBD per se was found to bind DNA specifically and to yield a distinct footprint similar to LeuO-S120D. The distinct footprints generated by LeuO-S120D and LeuO-DBD disclosed a 19 bp palindromic DNA-binding core-site, of which a DNA-binding consensus sequence was derived. For other poorly characterized LTTRs, the approach to identify active variants and to use the DBD for characterization of their DNA-binding specificity may be valid as well.
The enhanced DNA-binding affinity of LeuO-S120D in comparison to wild-type LeuO explains why low levels of the hyperactive mutants are sufficient for activation of the H-NS-repressed cas promoter and for positive autoregulation of the H-NS and StpA repressed leuO promoter. The distinct DNase I footprints generated by LeuO-S120D and also by LeuO-DBD are in agreement with binding of a LeuO-S120D tetramer, carrying two dimeric DBDs, to a high-affinity palindromic DNA-binding site (core-site) and secondary less conserved DNA-binding site. Additional protections generated by LeuO-S120D in between the two LeuO DNA-binding sites in the cas regulatory region (sites I and II) indicate that a higher order complex of two or more LeuO-S120D tetramers might be formed (Figure 4B). Thus, the structural change observed in the EBD of LeuO-S120D in comparison to the wild-type LeuO presumably transmits to the DBDs triggering binding of DNA with highest affinity (as shown by SPR and footprinting). This structural change is reflected by a shift in the arrangement of the two subdomains, RD-1 and RD-2, and a re-orientation of the side-chain of Arg218, which is located at the EBD’s cleft between RD1 and RD2. Similar effector-induced or mutation-based rearrangements have been described for other structures of LTTRs including AphB, BenM, DntR, HypT, OccR and OxyR, (29,31,41,43,75,78,79,81). Since the structural change that is induced by mutation S120D is related to effector-induced structural changes of other LTTRs, mutant S120D (and possibly the other hyperactive mutants) may mimic the effector-induced form of LeuO.
Up to date, the biological role of LeuO including the identification of LeuO target genes has been analyzed with native LeuO only. However, the leuO gene is repressed by H-NS (and StpA) in E. coli and S. enterica. Therefore, for these analyses either ectopic leuO expression or late stationary phase bacteria, in which LeuO levels are increased, as well as in vitro SELEX were used (5,6,8). These studies led to the identification of a broad spectrum of target genes and a rather weak consensus sequence for the LeuO DNA-binding site (5,6,8). The palindromic DNA-binding consensus motif defined here for LeuO is not only very specific, but also in agreement with the consensus motif characterized before (5). Top score hits of the DNA-binding consensus motif of LeuO within the E. coli genome sequence includes targets that have been identified for not only native LeuO, but also additional loci. While the overlap of the presumptive target loci validates the consensus motif, top score hits at additional loci suggest that hyperactive LeuO-S120D (and putative mimic of effector-induced LeuO) regulates other genes as well. Thus, the spectrum of LeuO target genes might change upon effector-binding or effector-induced modification of a specific residue, with some targets being more specifically bound, while binding to others may be down-regulated and a third group of targets may not be affected at all. Such a differential regulation of target genes is typical for LTTRs and for example has been shown for the control of E. coli ArgP activity by its effectors lysine (acting inhibitory) and arginine (acting stimulatory) (32–34). A recent comprehensive study on the systematic characterization of transcription factors of unknown function in E. coli included the characterization of two LTTRs (YdcI and YeiI) for which target genes and rather degenerate DNA-binding motifs were identified (82). For LTTRs, combining such a comprehensive approach on the apo-LTTR with constitutive mutants of the LTTR and the DBD dimer may unravel differentially controlled target genes and less degenerate DNA-binding consensus sequences.
LeuO activates the H-NS-repressed promoter of the cas operon encoding the CRISPR–Cascade complex. However, the leuO gene is likewise H-NS and StpA repressed that poses a scientific conundrum for activation of cas. A specific signal that triggers activation of cas by LeuO remains elusive. Effector-induction of LeuO may be sufficient to up-regulate leuO expression and to then activate cas transcription and synthesis of the Cascade-CRISPR complex. Further, uptake of xenogeneic DNA may lead to sequestration of H-NS resulting in de-repression of leuO. Presently, no effector of LeuO is known. However, the structural data revealed that the EBD-cleft and presumptive effector-binding site represent a hydrophobic pocket and that Arg218 is re-oriented in the Ser120 to Asp mutant. Interestingly, mutation of Arg218 to glutamine, alanine and cysteine also causes hyperactivity, indicating that the positively charged guanidinium group of Arginine may be crucial for an inactive LeuO conformation. However, this remains speculative; some LTTRs are bound by several effectors, and for some, several effector-binding sites have been identified, as for example for ArgP and NdhR (34,42).
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers 6GZ0 (wild-type LeuO-EBD), 6GZ1 (S120D-EBD monoclinic) and 6GZ2 (S120D-EBD orthorhombic).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the staff of beamline X06DA at the Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland for their support during data collection and Prof. Kirsten Jung for the use of the Bioanalytics core facility of the LMU Biocenter for SPR analyses.
Notes
Present address: Magdalena Schacherl, Charité - Universitätsmedizin Berlin, Institute of Medical Physics and Biophysics, Charitéplatz 1, 10117 Berlin, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
DFG [SCHN371/10-2, SCHN371/11-1 to K.S.; INST 216/682-1 FUGG to U.B.; SPP1617 He 5247/5-2 to R.H.]. Funding for open access charge: DFG [SCHN371/11-1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Henikoff S., Haughn G.W., Calvo J.M., Wallace J.C.. A large family of bacterial activator proteins. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:6602–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maddocks S.E., Oyston P.C.F.. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008; 154:3609–3623. [DOI] [PubMed] [Google Scholar]
- 3. Schell M.A. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 1993; 47:597–626. [DOI] [PubMed] [Google Scholar]
- 4. The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017; 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dillon S.C., Espinosa E., Hokamp K., Ussery D.W., Casadesús J., Dorman C.J.. LeuO is a global regulator of gene expression in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2012; 85:1072–1089. [DOI] [PubMed] [Google Scholar]
- 6. Shimada T., Bridier A., Briandet R., Ishihama A.. Novel roles of LeuO in transcription regulation of E. coli genome: antagonistic interplay with the universal silencer H-NS. Mol. Microbiol. 2011; 82:378–397. [DOI] [PubMed] [Google Scholar]
- 7. Stratmann T., Pul Ü., Wurm R., Wagner R., Schnetz K.. RcsB-BglJ activates the Escherichia coli leuO gene, encoding an H-NS antagonist and pleiotropic regulator of virulence determinants. Mol. Microbiol. 2012; 83:1109–1123. [DOI] [PubMed] [Google Scholar]
- 8. Ishihama A., Shimada T., Yamazaki Y.. Transcription profile of Escherichia coli: genomic SELEX search for regulatory targets of transcription factors. Nucleic Acids Res. 2016; 44:2058–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pul Ü., Wurm R., Arslan Z., Geißen R., Hofmann N., Wagner R.. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010; 75:1495–1512. [DOI] [PubMed] [Google Scholar]
- 10. Medina-Aparicio L., Rebollar-Flores J.E., Gallego-Hernández A.L., Vázquez A., Olvera L., Gutiérrez-Ríos R.M., Calva E., Hernández-Lucas I.. The CRISPR/Cas immune system is an operon regulated by LeuO, H-NS, and leucine-responsive regulatory protein in Salmonella enterica Serovar Typhi. J. Bacteriol. 2011; 193:2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stoebel D.M., Free A., Dorman C.J.. Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology. 2008; 154:2533–2545. [DOI] [PubMed] [Google Scholar]
- 12. Hernández-Lucas I., Calva E.. The coming of age of the LeuO regulator. Mol. Microbiol. 2012; 85:1026–1028. [DOI] [PubMed] [Google Scholar]
- 13. Shimada T., Yamamoto K., Ishihama A.. Involvement of the leucine response transcription factor LeuO in regulation of the genes for sulfa drug efflux. J. Bacteriol. 2009; 191:4562–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winardhi Ricksen S., Yan J., Kenney Linda J.. H-NS regulates gene expression and compacts the nucleoid: Insights from single-molecule experiments. Biophys. J. 2015; 109:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Landick R., Wade J.T., Grainger D.C.. H-NS and RNA polymerase: a love–hate relationship. Curr. Opin. Microbiol. 2015; 24:53–59. [DOI] [PubMed] [Google Scholar]
- 16. Dillon S.C., Dorman C.J.. Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 2010; 8:185–195. [DOI] [PubMed] [Google Scholar]
- 17. Espinosa E., Casadesús J.. Regulation of Salmonella enterica pathogenicity island 1 (SPI-1) by the LysR-type regulator LeuO. Mol. Microbiol. 2014; 91:1057–1069. [DOI] [PubMed] [Google Scholar]
- 18. Chen C.-C., Wu H.-Y.. LeuO protein delimits the transcriptionally active and repressive domains on the bacterial chromosome. J. Biol. Chem. 2005; 280:15111–15121. [DOI] [PubMed] [Google Scholar]
- 19. Ayala J.C., Wang H., Benitez J.A., Silva A.J.. Molecular basis for the differential expression of the global regulator VieA in Vibrio cholerae biotypes directed by H-NS, LeuO and quorum sensing. Mol. Microbiol. 2018; 107:330–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bina X.R., Taylor D.L., Vikram A., Ante V.M., Bina J.E.. Vibrio cholerae ToxR downregulates virulence factor production in response to Cyclo(Phe-Pro). mBio. 2013; 4:e00366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ante V.M., Bina X.R., Bina J.E.. The LysR-type regulator LeuO regulates the acid tolerance response in Vibrio cholerae. Microbiol. 2015; 161:2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ante V.M., Bina X.R., Howard M.F., Sayeed S., Taylor D.L., Bina J.E.. Vibrio cholerae leuO transcription is positively regulated by ToxR and contributes to bile resistance. J. Bacteriol. 2015; 197:3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guadarrama C., Medrano-López A., Oropeza R., Hernández-Lucas I., Calva E.. The Salmonella enterica serovar Typhi LeuO global regulator forms tetramers: Residues involved in oligomerization, DNA binding, and transcriptional regulation. J. Bacteriol. 2014; 196:2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Momany C., Neidle E.L.. Defying stereotypes: the elusive search for a universal model of LysR-type regulation. Mol. Microbiol. 2012; 83:453–456. [DOI] [PubMed] [Google Scholar]
- 25. Toledano M.B., Kullik I., Trinh F., Baird P.T., Schneider T.D., Storz G.. Redox-dependent shift of OxyR-DNA contacts along an extended DNA-binding site: A mechanism for differential promoter selection. Cell. 1994; 78:897–909. [DOI] [PubMed] [Google Scholar]
- 26. Wek R.C., Hatfield G.W.. Transcriptional activation at adjacent operators in the divergent-overlapping ilvY and ilvC promoters of Escherichia coli. J. Mol. Biol. 1988; 203:643–663. [DOI] [PubMed] [Google Scholar]
- 27. Rhee K.Y., Senear D.F., Hatfield G.W.. Activation of gene expression by a ligand-induced conformational change of a protein-DNA complex. J. Biol. Chem. 1998; 273:11257–11266. [DOI] [PubMed] [Google Scholar]
- 28. Oliver P., Peralta-Gil M., Tabche M.-L., Merino E.. Molecular and structural considerations of TF-DNA binding for the generation of biologically meaningful and accurate phylogenetic footprinting analysis: the LysR-type transcriptional regulator family as a study model. BMC Genomics. 2016; 17:686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jo I., Chung I.-Y., Bae H.-W., Kim J.-S., Song S., Cho Y.-H., Ha N.-C.. Structural details of the OxyR peroxide-sensing mechanism. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:6443–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pedre B., Young D., Charlier D., Mourenza Á., Rosado L.A., Marcos-Pascual L., Wahni K., Martens E., G.de la Rubia A., Belousov V.V. et al.. Structural snapshots of OxyR reveal the peroxidatic mechanism of H2O2 sensing. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E11623–E11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim Y., Chhor G., Tsai C.S., Winans J.B., Jedrzejczak R., Joachimiak A., Winans S.C.. Crystal structure of the ligand-binding domain of a LysR-type transcriptional regulator: transcriptional activation via a rotary switch. Mol. Microbiol. 2018; 110:550–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laishram R.S., Gowrishankar J.. Environmental regulation operating at the promoter clearance step of bacterial transcription. Genes Dev. 2007; 21:1258–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marbaniang C.N., Gowrishankar J.. Role of ArgP (IciA) in lysine-mediated repression in Escherichia coli. J. Bacteriol. 2011; 193:5985–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nguyen Le Minh P., Velázquez Ruiz C., Vandermeeren S., Abwoyo P., Bervoets I., Charlier D.. Differential protein-DNA contacts for activation and repression by ArgP, a LysR-type (LTTR) transcriptional regulator in Escherichia coli. Microbiol. Res. 2018; 206:141–158. [DOI] [PubMed] [Google Scholar]
- 35. Burn J.E., Hamilton W.D., Wootton J.C., Johnston A.W.B.. Single and multiple mutations affecting properties of the regulatory gene nodD of Rhizobium. Mol. Microbiol. 1989; 3:1567–1577. [DOI] [PubMed] [Google Scholar]
- 36. Schell M.A., Brown P.H., Raju S.. Use of saturation mutagenesis to localize probable functional domains in the NahR protein, a LysR-type transcription activator. J. Biol. Chem. 1990; 265:3844–3850. [PubMed] [Google Scholar]
- 37. Bartowsky E., Normark S.. Interactions of wild-type and mutant AmpR of Citrobacter freundii with target DNA. Mol. Microbiol. 1993; 10:555–565. [DOI] [PubMed] [Google Scholar]
- 38. Kullik I., Stevens J., Toledano M.B., Storz G.. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for DNA binding and multimerization. J. Bacteriol. 1995; 177:1285–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Celis R.T.F. Repression and activation of arginine transport genes in Escherichia coli K 12 by the ArgP protein. J. Mol. Biol. 1999; 294:1087–1095. [DOI] [PubMed] [Google Scholar]
- 40. Craven S.H., Ezezika O.C., Haddad S., Hall R.A., Momany C., Neidle E.L.. Inducer responses of BenM, a LysR-type transcriptional regulator from Acinetobacter baylyi ADP1. Mol. Microbiol. 2009; 72:881–894. [DOI] [PubMed] [Google Scholar]
- 41. Ruangprasert A., Craven S.H., Neidle E.L., Momany C.. Full-length structures of BenM and two variants reveal different oligomerization schemes for LysR-type transcriptional regulators. J. Mol. Biol. 2010; 404:568–586. [DOI] [PubMed] [Google Scholar]
- 42. Jiang Y.-L., Wang X.-P., Sun H., Han S.-J., Li W.-F., Cui N., Lin G.-M., Zhang J.-Y., Cheng W., Cao D.-D. et al.. Coordinating carbon and nitrogen metabolic signaling through the cyanobacterial global repressor NdhR. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taylor J.L., De Silva R.S., Kovacikova G., Lin W., Taylor R.K., Skorupski K., Kull F.J.. The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol. Microbiol. 2012; 83:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones R.M., Popham D.L., Schmidt A.L., Neidle E.L., Stabb E.V.. Vibrio fischeri DarR directs responses to D-aspartate and represents a group of similar LysR-type transcriptional regulators. J. Bacteriol. 2018; 200:doi:10.1128/JB.00773-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jørgensen C., Dandanell G.. Isolation and characterization of mutations in the Escherichia coli regulatory protein XapR. J. Bacteriol. 1999; 181:4397–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monferrer D., Tralau T., Kertesz M.A., Dix I., Sola M., Uson I.. Structural studies on the full-length LysR-type regulator TsaR from Comamonas testosteroni T-2 reveal a novel open conformation of the tetrameric LTTR fold. Mol. Microbiol. 2010; 75:1199–1214. [DOI] [PubMed] [Google Scholar]
- 47. Colyer T.E., Kredich N.M. 1996. In vitro characterization of constitutive CysB proteins from Salmonella typhimurium. Mol. Microbiol. 1996; 21:247–256. [DOI] [PubMed] [Google Scholar]
- 48. Dorman C.J., Dorman M.J.. Control of virulence gene transcription by indirect readout in Vibrio cholerae and Salmonella enterica serovar Typhimurium. Environ. Microbiol. 2017; 19:3834–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miroux B., Walker J.E.. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996; 260:289–298. [DOI] [PubMed] [Google Scholar]
- 50. Madhusudan S., Paukner A., Klingen Y., Schnetz K.. Independent regulation of H-NS-mediated silencing of the bgl operon at two levels: upstream by BglJ and LeuO and downstream by DnaKJ. Microbiology. 2005; 151:3349–3359. [DOI] [PubMed] [Google Scholar]
- 51. Diederich L., Rasmussen L.J., Messer W.. New cloning vectors for integration in the lambda attachment site attB of the Escherichia coli chromosome. Plasmid. 1992; 28:14–24. [DOI] [PubMed] [Google Scholar]
- 52. Dole S., Kühn S., Schnetz K.. Post-transcriptional enhancement of Escherichia coli bgl operon silencing by limitation of BglG-mediated antitermination at low transcription rates. Mol. Microbiol. 2002; 43:217–226. [DOI] [PubMed] [Google Scholar]
- 53. Breddermann H., Schnetz K.. Activation of leuO by LrhA in Escherichia coli. Mol. Microbiol. 2017; 104:664–676. [DOI] [PubMed] [Google Scholar]
- 54. Salscheider S.L., Jahn A., Schnetz K.. Transcriptional regulation by BglJ-RcsB, a pleiotropic heteromeric activator in Escherichia coli. Nucleic Acids Res. 2014; 42:2999–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stratmann T., Madhusudan S., Schnetz K.. Regulation of the yjjQ-bglJ operon, encoding LuxR-type transcription factors, and the divergent yjjP gene by H-NS and LeuO. J. Bacteriol. 2008; 190:926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Breddermann H., Schnetz K.. Correlation of antagonistic regulation of leuO transcription with the cellular levels of BglJ-RcsB and LeuO in Escherichia coli. Front. Cell Infect. Microbiol. 2016; 6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miller J.H. A short course in bacterial genetics. A laboratory manual and handbook for Escherichia coli and related bacteria. 1992; NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 58. Kabsch W. XDS. Acta Crystallogr. Sect. D. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Winter G., Waterman D.G., Parkhurst J.M., Brewster A.S., Gildea R.J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I.D. et al.. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D. 2018; 74:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Terwilliger T.C., Grosse-Kunstleve R.W., Afonine P.V., Moriarty N.W., Zwart P.H., Hung L.W., Read R.J., Adams P.D.. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D. 2008; 64:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D.. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D. 2012; 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. Sect. D. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Diederichs K., Karplus P.A.. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 1997; 4:269–275. [DOI] [PubMed] [Google Scholar]
- 65. Diederichs K., Karplus P.A.. Better models by discarding data. Acta Crystallogr. Sect. D. 2013; 69:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brunger A.T. Free R value: cross-validation in crystallography. Methods Enzymol. 1997; 277:366–396. [DOI] [PubMed] [Google Scholar]
- 67. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 69. Gaugué I., Bréchemier-Baey D., Plumbridge J.. DNase I footprinting to identify protein binding sites. Bio-protocol. 2013; 3:e824. [Google Scholar]
- 70. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 71. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Keseler I.M., Mackie A., Santos-Zavaleta A., Billington R., Bonavides-Martínez C., Caspi R., Fulcher C., Gama-Castro S., Kothari A., Krummenacker M. et al.. The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 2017; 45:D543–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Langer A., Moldovan A., Harmath C., Joyce S.A., Clarke D.J., Heermann R.. HexA is a versatile regulator involved in the control of phenotypic heterogeneity of Photorhabdus luminescens. PLoS One. 2017; 12:e0176535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Westra E.R., Pul Ü., Heidrich N., Jore M.M., Lundgren M., Stratmann T., Wurm R., Raine A., Mescher M., Van Heereveld L. et al.. H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol. Microbiol. 2010; 77:1380–1393. [DOI] [PubMed] [Google Scholar]
- 75. Lerche M., Dian C., Round A., Lönneborg R., Brzezinski P., Leonard G.A.. The solution configurations of inactive and activated DntR have implications for the sliding dimer mechanism of LysR transcription factors. Sci. Rep. 2016; 6:19988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 77. Mosca R., Schneider T.R.. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res. 2008; 36:W42–W46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Devesse L., Smirnova I., Lonneborg R., Kapp U., Brzezinski P., Leonard G.A., Dian C.. Crystal structures of DntR inducer binding domains in complex with salicylate offer insights into the activation of LysR-type transcriptional regulators. Mol. Microbiol. 2011; 81:354–367. [DOI] [PubMed] [Google Scholar]
- 79. Jo I., Kim D., No T., Hong S., Ahn J., Ryu S., Ha N.-C.. Structural basis for HOCl recognition and regulation mechanisms of HypT, a hypochlorite-specific transcriptional regulator. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Browning D.F., Busby S.J.W.. Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 2016; 14:638–650. [DOI] [PubMed] [Google Scholar]
- 81. Ezezika O.C., Haddad S., Clark T.J., Neidle E.L., Momany C.. Distinct effector-binding sites enable synergistic transcriptional activation by BenM, a LysR-type regulator. J. Mol. Biol. 2007; 367:616–629. [DOI] [PubMed] [Google Scholar]
- 82. Gao Y., Yurkovich J.T., Seo S.W., Kabimoldayev I., Dräger A., Chen K., Sastry A.V., Fang X., Mih N., Yang L. et al.. Systematic discovery of uncharacterized transcription factors in Escherichia coli K-12 MG1655. Nucleic Acids Res. 2018; 46:10682–10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession numbers 6GZ0 (wild-type LeuO-EBD), 6GZ1 (S120D-EBD monoclinic) and 6GZ2 (S120D-EBD orthorhombic).