


Abstract
Nucleotide excision repair (NER) consists of global genomic NER (GG-NER) and transcription coupled NER (TC-NER) subpathways. In eukaryotic cells, genomic DNA is wrapped around histone octamers (an H3–H4 tetramer and two H2A–H2B dimers) to form nucleosomes, which are well known to profoundly inhibit the access of NER proteins. Through unbiased screening of histone H4 residues in the nucleosomal LRS (loss of ribosomal DNA-silencing) domain, we identified 24 mutations that enhance or decrease UV sensitivity of Saccharomyces cerevisiae cells. The histone H4 H75E mutation, which is largely embedded in the nucleosome and interacts with histone H2B, significantly attenuates GG-NER and Rad26-independent TC-NER but does not affect TC-NER in the presence of Rad26. All the other histone H4 mutations, except for T73F and T73Y that mildly attenuate GG-NER, do not substantially affect GG-NER or TC-NER. The attenuation of GG-NER and Rad26-independent TC-NER by the H4H75E mutation is not due to decreased chromatin accessibility, impaired methylation of histone H3 K79 that is at the center of the LRS domain, or lowered expression of NER proteins. Instead, the attenuation is at least in part due to impaired recruitment of Rad4, the key lesion recognition and verification protein, to chromatin following induction of DNA lesions.
INTRODUCTION
Nucleotide excision repair (NER) is a conserved DNA repair pathway that removes bulky and/or helix-distorting DNA lesions, such as ultraviolet (UV) induced cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (1,2). The serial steps in NER are similar in organisms from bacteria to complex mammals and plants, and involve lesion recognition and verification, incision of the DNA on both sides of the lesion, excision of an oligonucleotide containing the lesion, repair synthesis copying the opposite undamaged strand, and ligation. Global genomic NER (GG-NER) is an NER subpathway that removes lesions throughout the genome, whereas transcription coupled NER (TC-NER) is the other NER subpathway that is dedicated to rapid removal of lesions in the transcribed strand of active genes. The two NER subpathways differ at the damage-recognition step, but share common factors in the later steps of the repair process. In Saccharomyces cerevisiae, Rad26, the yeast homolog of mammalian CSB, is well known to facilitate TC-NER (3). However, a substantial level of TC-NER still occurs in rad26Δ cells, and Rpb9, a nonessential subunit of RNA polymerase II (RNAPII), is largely responsible for Rad26-independent TC-NER (4). Sen1, a 5′ to 3′ RNA and DNA helicase has also been shown to facilitate TC-NER in yeast (5). Rad7, Rad16 (6) and Elc1 (7) are known to be specifically required for GG-NER in yeast. Rad4 is the yeast sequence homolog of mammalian XPC. However, unlike XPC that is specifically required for GG-NER in mammalian cells, Rad4 is essential for both GG-NER and TC-NER in yeast (2). Rad4 has a sequence- and damage-independent DNA binding domain that anchors the protein on the DNA and a damage specific binding domain that senses the single-stranded character induced by the damage without directly interacting with the damage (8). Rad4 interacts with and is stabilized by Rad23, which in addition to functioning in NER, plays a central role in targeting ubiquitylated proteins for proteasomal degradation (9). Among other core NER proteins shared by TC-NER and GG-NER are Rad1, Rad2, Rad10, Rad14 and TFIIH, a 10 subunit complex that is required for both transcription initiation and NER (2).
In eukaryotic cells, genomic DNA is wrapped around histone octamers (an H3–H4 tetramer and two H2A–H2B dimers) to form nucleosomes, which are further folded into higher order chromatin structures (10). The chromatin structure is needed to package the large genome but profoundly inhibits the access of NER proteins. Indeed, NER has been shown to be inhibited within nucleosomes in vitro (11) and in vivo (12–14). A single histone H4 R45H or R45C mutation, which is located in the SIN (Switch-independent) domain of the nucleosomal surface (Figure 1A), increases UV resistance and NER in yeast (15). The H4 R45 side chain protrudes into the minor groove of the nucleosomal DNA, and a mutation at this site may create a more accessible landscape for NER proteins (15).
Figure 1.

Features of a nucleosome. (A) Structure of a yeast nucleosome (1ID3) (45). The approximate SIN and LRS domains are indicated by the yellow and red circles, respectively. (B and C) The LRS domain and surrounding nucleosomal areas. The locations of certain residues are indicated. The dotted blue lines are hydrogen bonds between the side chain of H4H75 and those of H2BE96 and H2BT99.
The LRS (loss of ribosomal DNA-silencing) domain is another nucleosomal surface structure (Figure 1A) that is required for heterochromatin formation and transcriptional repression at specific yeast loci (16,17). Although they map to completely different regions of the nucleosome, the three-dimensional histone-fold structures and the interacting nucleosomal DNA of the SIN and LRS domains can be superimposed following rotation of 180° around a symmetry axis (17). However, mutations in the LRS domain do not alleviate the need for the Swi/Snf chromatin remodeling factors during transcription, nor do they disrupt higher order nucleosomal folding as mutations in the SIN domain (18).
At the center of the LRS domain is the side chain of histone H3 lysine 79 (H3K79) (Figure 1), which can be methylated by the methyltransferase Dot1 in yeast (19). A role for H3K79 methylation in NER was suggested by the observation that dot1 deletion or H3K79E mutation is epistatic to deletion of rad1, the essential gene for NER in yeast (20). Direct analyses of UV induced CPDs in yeast cells showed that the H3K79 methylation is required for efficient NER at least in certain genes (21,22). To date, however, how H3K79 methylation affects NER remains unknown. The side chain of H3K79 is largely surrounded by histone H4 residues in the LRS domain (Figure 1). NER may be facilitated by (i) methylated H3K79 itself, (ii) a specific structure jointly formed by the methylated H3K79 and certain surrounding residues or (iii) a structure formed by the surrounding residues that can be efficiently accessed following H3K79 methylation. Even if the methylated H3K79 itself can facilitate NER, the surrounding residues are likely to play significant roles by affecting either the methylation efficiency or the accessibility of the methylated residue. To clarify the different scenarios, we performed UV sensitivity screening of random histone H4 mutations located in the LRS domain in yeast. We found that histone H4 H75E (H4H75E) mutation dramatically attenuates GG-NER and Rad26-independent TC-NER, but does not affect TC-NER in the presence of Rad26. The attenuation is not due to decreased chromatin accessibility, impaired H3K79 methylation, or lowered expression of NER proteins. Instead, the attenuation appears to be at least in part due to impaired recruitment of Rad4 to chromatin following induction of DNA lesions in yeast cells. Our findings challenge the typical view that nucleosome structure just passively inhibits NER and the NER machinery battles to overcome the inhibition. A nucleosomal feature conferred by H4H75 may actually play an active role in the repair process by enhancing damage recognition and verification in the chromatin.
MATERIALS AND METHODS
Yeast strains
All yeast strains used in this study were derivatives of YBL574 [MATa, leu2Δ1, his3Δ200, ura3–52, trp1Δ63, lys2–128Δ, (hht1-hhf1)Δ::LEU2, (hht2-hhf2)Δ::HIS3 Ty912Δ35-lacZ::his4, (pDM9)], which has its divergent HHT1-HHF1 and HHT2-HHF2 gene pairs (both pairs encode histones H3 and H4) deleted and complemented with a centromeric URA3 plasmid (pDM9) bearing the wild type HHT1-HHF1 gene pair (23). Deletions of additional genes were accomplished by using procedures as described previously (4). Strains with their genomic genes tagged with three consecutive FLAG (3 × FLAG) sequences were created by using PCR products amplified from plasmid p3FLAG-KanMX, as described previously (24).
UV sensitivity screening and confirmation of random histone H4 mutations located in the LRS domain
The screening strategy is outlined in Figure 2. Plasmid pHTF2 was created by inserting the wild type HHT2-HHF2 gene pair (2.1 kb) into the XhoI and SacII sites of the centromeric TRP1 plasmid pRS414 (25). Using pHTF2 as a backbone, 17 sets of plasmids were created, each of which contains random mutations at one of the 17 codons for histone H4 residues 64–80. The random mutations were introduced by solid-phase synthesis of a PCR primer, which was then used for amplification of a HHF2 gene fragment. To maximize the possibility that all 20 possible natural amino acids can be encoded by each of the 17 randomly mutated codons, 4000–5000 independent E. coli transformants were obtained for each of the 17 sets of plasmids. Equal amounts of the 17 sets of plasmids were pooled and transformed into YBL574 cells to generate ∼1 million independent yeast transformants. The pDM9 plasmid bearing the wild type HHT1-HHF1 gene was removed from the cells by selection with 5-fluoorotic acid (5-FOA) (26). One billion of the 5-FOA selected yeast cells were further irradiated with 60 J/m2 of UV (254 nm, from a 15 W UV germicidal bulb, General Electric), which kills ∼90% of wild type yeast cells. The UV irradiated cells were allowed to grow for 10 cell divisions to let the UV sensitive histone H4 mutants ‘dilute’ and the UV resistant ones enrich in the cell population. The TRP1 plasmids encoding the histone H4 mutants were isolated from the unirradiated and UV-irradiated yeast cells. The region containing the random histone H4 mutations in the original pooled plasmid libraries and in the UV-irradiated and unirradiated cells were sequenced. The sequencing reads were translated into amino acid sequences using EMBOSS TRANSEQ (27). The abundances of the randomly mutated histone H4 codons were calculated.
Figure 2.

Screening of UV sensitive or resistant histone H4 mutations in the LRS domain. (A) Shuffling of a TRP1 plasmid library bearing HHT2-HHF2 with randomly mutated histone H4 codons with a URA3 plasmid bearing the wild type HHT1-HHF1. Stripped gray arrows denote the deleted genomic HHT1-HHF1 and HHT2-HHF2 (hht1-hhf1Δ and hht2-hhf2Δ). (B) Assessments of viabilities and UV sensitivities of H4 mutations by next generation DNA sequencing. Black lines indicate the TRP1-plasmid-born HHF2 gene with random mutations. Dots of different colors denote different mutated codons. The region containing the mutations in the original pooled plasmid libraries and in the UV-irradiated and unirradiated cells shuffled with the libraries were sequenced. The abundancies of the amino acid codons in the mutated region of the HHF2 gene in the different samples were calculated. Lethal mutations (shown as red dots) are indicated by their substantial presence in the original plasmid libraries but not in the shuffled (unirradiated) yeast cells. UV sensitive (shown as gray and green dots) or resistant (shown as blue and purple dots) mutations are indicated by their ‘dilution’ or enrichment, respectively, in the UV irradiated yeast cells (compared to the unirradiated ones).
To confirm the UV sensitive or resistant histone H4 mutations, TRP1 plasmids that bear the HHT2-HHF2 genes with the candidate mutations were created and shuffled into YBL574 cells.
UV sensitivity assay
Yeast cells were grown in synthetic dextrose (SD) medium at 30°C to saturation, sequentially 10-fold diluted and spotted onto YPD (1% yeast extract, 2% peptone and 2% dextrose) plates. After different doses of UV irradiation, the plates were incubated in the dark at 30°C for 3–6 days before being photographed.
Repair analysis of UV induced CPDs
Yeast cells were grown in SD medium at 30°C to late log phase (A600 ≈ 1.0), irradiated with 120 J/m2 of UV and incubated in YPD medium in the dark at 30°C. At different times of the repair incubation, aliquots were removed, and the genomic DNA was isolated. Nucleotide-level analyses of repair of CPDs were performed as described previously (28).
Chromatin accessibility assay
Micrococcal nuclease (MNase) digestion was done essentially as described previously (29). Briefly, yeast cells were grown in SD medium at 30°C to late log phase. Half of the culture was irradiated with 120 J/m2 of UV followed by incubation at 30°C for 1 h. Cells from 45 ml of the irradiated and unirradiated samples were treated with 50 units of Zymolyase (Zymo Research) in 5 ml zymolyase buffer (50 mM Tris, pH 7.8, 1 M sorbitol, 5 mM β-ME, 0.5 mM PMSF) at 30°C for 40 min. The resulting spheroplasts from each sample were then suspended in 2 ml of spheroplast digestion buffer (50 mM Tris, pH 7.8, 1 M sorbitol, 50 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 1 mM β-ME, 0.5 mM PMSF and 0.075% v/v NP-40), divided into 300-μl aliquots and digested with varying concentrations of MNase for 10 min at 37°C. The reactions were terminated by mixing with 60 μl of stop solution (6% SDS, 250 mM EDTA) and immediately incubated at 65°C for 3 h. The genomic DNA was isolated from the aliquots and fractionated on 1.2% agarose gels.
Measurements of expression and chromatin-association of NER proteins
Cells were cultured in SD medium at 30°C to late log phase. Half of the cultures were irradiated with 120 J/m2 of UV. At different times of incubation at 30°C, aliquots were removed. To measure the cellular levels of NER proteins, whole cell protein extracts were prepared from the UV irradiated and unirradiated cell aliquots by using the procedure as described previously (30).
To measure the association of NER proteins with chromatin, the UV irradiated and unirradiated cell aliquots were treated with 1% formaldehyde at room temperature for 30 min, quenched with 125 mM glycine, washed and then pelleted. The cells from a 40 ml aliquot were mixed with 0.5 ml ice cold cell lysis buffer [20 mM HEPES (pH 8.0), 60 mM KCl, 15 mM NaCl, 10 mM MgCl2, 1 mM CaCl2, 10 mM N-butyric acid, 0.8% Triton X-100, 0.25 M sucrose, 2.5 mM spermidine, 0.5 mM spermine, 3× concentrated protease inhibitor cocktail (78438, Thermo Fisher Scientific), 1 mM PMSF] and 0.5 ml acid washed beads. Cells were lysed by 8 × 30 s pulses of bead-beating (kept on ice for 1 min between beat-beating). Residual intact cells were removed by centrifugation at 500 g for 5 min. The cell lysate was centrifuged at 2000 g for 20 min. The pellet (chromatin fraction) was washed once with ice-cold cell lysis buffer. The protein pellet was dissolved in 60 μl 2× SDS-PAGE gel loading buffer and boiled for 30 min to reverse formaldehyde crosslinks.
Proteins of interest were detected by western blots. Anti-FLAG antibody (M2) was from Sigma. Antibodies against mono-, di- and tri-methylated H3K79 and total histone H3 were from Abcam.
RESULTS
Identification of UV sensitive or resistant histone H4 mutations in the LRS domain
We performed UV sensitivity screening of random mutations of histone H4 residues 64–80 (Figure 2), which are largely located in the nucleosomal LRS domain (Figure 1). Only one (R67F) (Supplementary Table S1, marked with ‘−’) out of the 340 (17 × 20) possible amino acids at the 17 randomly mutated residues of histone H4 was not adequately covered by our high throughput screening. For an unknown reason, we obtained < 10 sequencing reads for the R67F mutation from the original pool of the plasmid libraries. We obtained at least 50 sequencing reads for all other mutations from the original plasmid library. Among the 339 amino acids covered by our screening, 106 mutations were lost in the unirradiated yeast cells shuffled with the plasmid libraries (Supplementary Table S1, marked with ‘×’). This indicates that ∼ 1/3 of the histone H4 mutations are inviable. Substitutions of amino acids whose side chains are nucleosomal surface-exposed (e.g. R67, V70, T71, E74, K77 and K79) appeared to be more likely to be viable than those whose side chains are embedded. A previous report showed that a large fraction of alanine and certain other amino acid substitutions of histones H3 and H4 are viable, although these proteins are highly conserved (31). Our results agree generally well with the previous report. Among the 233 viable histone H4 mutations we screened, 24 appeared to be ≥3 times more UV sensitive than wild types (Supplementary Table S1, shown in red). Interestingly, 18 mutations appeared to be ≥ 3 times more UV-resistant than wild types (Supplementary Table S1, shown in green). The more UV-resistant mutations tend to cluster within the stretch of residues 64–71 and the more UV-sensitive ones within the stretch of residues 73–80 (Supplementary Table S1).
To confirm the screening results, we created yeast strains specifically expressing the mutant histone H4. Twenty-four of the histone H4 mutations were confirmed to be truly UV sensitive or resistant although their degrees of UV sensitivity or resistance might be somewhat different from those of the screening results (compare the data in Supplementary Tables S1 and S2).
H4H75E mutation attenuates GG-NER and Rad26-independent TC-NER
To determine the effects of the confirmed histone H4 mutations on GG-NER, we measured repair of UV induced CPDs in the non-transcribed strand of the RPB2 gene in cells with these mutations (Supplemental Figures S1 and S2; Figure 3A and B). The repair was significantly slower in the H4H75E mutant than in the wild type cells (Figure 3A–C), indicating that the mutation attenuates GG-NER. The repair in the H4T73F and H4T73Y mutants were also somewhat slower than that in the wild type cells (compare Supplemental Figure S2B and C with Figure 3A). However, all the other histone H4 mutants did not show obvious defects in GG-NER (Supplemental Figures S1 and S2).
Figure 3.

H4H75E mutation attenuates GG-NER and Rad26-independent TC-NER. (A and B) Sequencing gels showing CPDs remaining in the nontranscribed strand of the RBP2 gene in the wild type and H4H75E cells at the indicated repair time (h). ‘U’ indicates samples from unirradiated cells. Approximate nucleotide positions relative to the transcription start site (+1) of the RPB2 gene are indicated on the left. (C) Percent CPDs remaining in the nontranscribed strand of the RPB2 gene. (D and E) Sequencing gels showing CPDs remaining in the transcribed strand of the RBP2 gene in the rad7Δ and rad7Δ H4H75E cells. (F) Percent CPDs remaining in the coding region (between nucleotides +1 and +940) of the transcribed strand of the RPB2 gene. (G and H) Sequencing gels showing CPDs remaining in the transcribed strand of the RBP2 gene in the rad7Δ rad26Δ and rad7Δ rad26Δ H4H75E cells. (I) Percent CPDs remaining in the coding region of the transcribed strand of the RPB2 gene.
To determine the effects of the confirmed histone H4 mutations on TC-NER, we measured repair of UV induced CPDs in the transcribed strand of the RPB2 gene in rad7Δ and rad7Δ rad26Δ cells with these mutations (Supplementary Figures S3 and S4; Figure 3D-I). As expected, the repair was fast, starting at about 40 nucleotides upstream (-40) of the transcription start site of the RPB2 gene in rad7Δ cells (Figure 3D). No apparent repair can be seen in the region over 40 nucleotides upstream (below nucleotide –40 on the gel) of the RPB2 gene, where only GG-NER but not TC-NER has been known to be operative (Figure 3D). The repair in rad7Δ H4H75E cells was similar to that in rad7Δ cells (Figure 3D–F). As expected, rad7Δ rad26Δ cells have significantly decreased repair in the transcribed strand of the RPB2 gene, except for certain sites, especially those in the short region (between nucleotides +1 and +50) immediately downstream of the transcription start site, where TC-NER has been known to be independent of Rad26 (4) (Figure 3, compare G with D). Essentially no repair can be seen in the rad7Δ rad26Δ H4H75E cells (Figure 3H and I). These results indicate that the H4H75E mutation does not significantly affect TC-NER in the presence of Rad26 but dramatically attenuates Rad26-independent TC-NER. All the other confirmed histone H4 mutations, including H4T73F and H4T73Y, did not substantially affect TC-NER in the presence of Rad26 (not shown) or Rad26-independent TC-NER (Supplementary Figures S3 and S4).
The H4H75E mutation increased the UV sensitivity of otherwise wild-type (Figure 4A) and rad7Δ rad26Δ (Figure 4C) cells ∼10-fold. However, the mutation caused little increase of UV sensitivity in rad7Δ (Figure 4B) and rad14Δ (which is completely defective for both GG-NER and TC-NER) (Figure 4D) cells. These results are in line with the above notion that the H4H75E mutation primarily attenuates GG-NER and Rad26-independent TC-NER. We noticed that the H4H75E mutant cells grow much slower than wild type cells (Figure 4). To the best of our knowledge, a slow growth does not seem to correlate with slow NER, presumably due to the fact that the NER speed is limited by the time-consuming lesion-recognition, rather than the subsequent steps (including repair synthesis) (32). In support of this notion, TC-NER (in the presence of Rad26) occurred normally in the H4H75 mutant cells (see above).
Figure 4.

The effects of H4H75E mutation on UV sensitivity. (A–D) Images of spotted yeast cells following irradiation with the indicated doses of UV.
H4H75E mutation enhances chromatin accessibility but does not affect either H3K79 methylation or NER protein expression
To determine if the attenuation of GG-NER and Rad26-independent TC-NER in the H4H75E mutant were caused by decreased accessibility of chromatin, we examined the cleavage of cellular chromatin DNA by MNase. Decreased chromatin accessibility allows less MNase cleavage of nucleosomal linker DNA, generating longer nucleosomal DNA (i.e., associated with more nucleosomal repeats). As can be seen in Figure 5A, at the same amount of MNase, relatively shorter nucleosomal DNA was generated in the H4H75E mutant than in the wild type cells, indicating that chromatin in the H4H75E mutant is more, rather than less, accessible. UV irradiation did not significantly affect the accessibility in either the wild type or mutant cells (Figure 5A).
Figure 5.

H4H75E mutation enhances chromatin accessibility to MNase but does not affect methylation of H3K79. (A) Top, gel images of bulk chromatin DNA following digestion with the indicated amount of MNase. M, D and T denote mono-, di- and tri-nucleosomal DNA, respectively. Bottom, scans of signal intensities of the chromatin DNA shown at the top. (B) Western blots showing mono-, di- and tri-methylation of H3K79 in the indicated cells. Asterisks indicate nonspecific bands. Total histone H3 serves as loading control.
On the nucleosome, H4H75 is in close proximity to H3K79 (Figure 1), whose methylation has been shown to attenuate GG-NER (21,22). We therefore tested if the H4H75E mutation affected H3K79 methylation. As expected, no H3K79 methylation (mono-, di- or tri-methylation) can be detected in dot1Δ cells (Figure 5B). In agreement with previous reports (e.g. (33)), cells lacking Rtf1, one of the 5 subunits of the RNAPII-associated factor 1 complex (Paf1C), showed no tri-methylation, dramatically reduced di-methylation and increased mono-methylation of H3K79 (Figure 5B). By modulating mono-ubiquitination of histone H2B K123, Paf1C is known to be absolutely required for tri-methylation, partially required for di-methylation and dispensable for mono-methylation of H3K79 (34). The H4H75E mutation did not seem to affect the tri-, di- or mono-methylation of H3K79 (Figure 5B).
We also tested if the H4H75E mutation affected the expression of NER proteins, including Rad3, a TFIIH subunit that is well known to be required for NER. The levels of all key NER proteins in the H4H75E mutant were similar to those in the wild type cells (Supplementary Figure S5 and Figure 6A). Also, UV irradiation did not seem to significantly affect the levels of the NER proteins in either the wild type or mutant cells.
Figure 6.
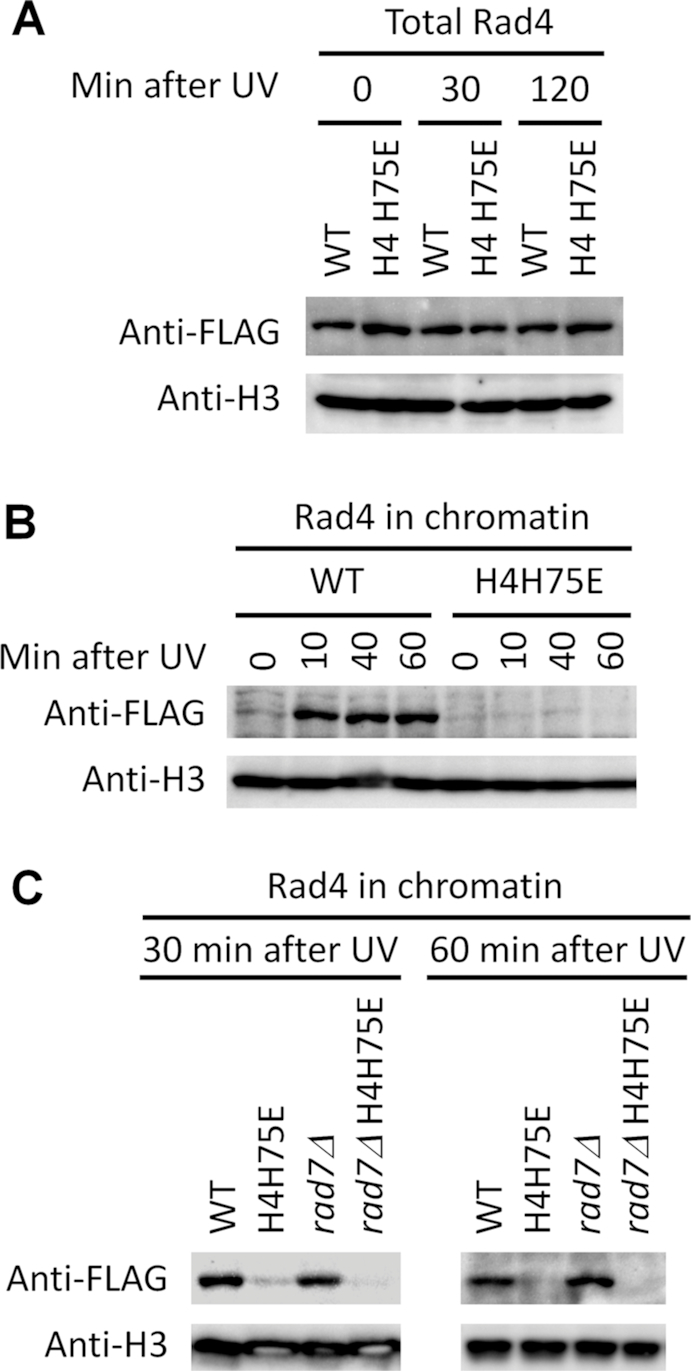
H4H75E mutation impairs recruitment of Rad4 to chromatin following induction of UV damage. (A) Western blots showing the levels of total 3 × FLAG-tagged Rad4 in the indicated cells at the indicated times after UV irradiation. (B and C) Western blots showing chromatin-associated 3 × FLAG-tagged Rad4 in the indicated cells at the indicated times after UV irradiation. Histone H3 serves as loading control.
H4H75E mutation impairs recruitment of Rad4 to chromatin following UV irradiation
To gain insights into how H4H75E mutation might attenuate GG-NER and Rad26-independent TC-NER, we measured recruitment of NER proteins to chromatin. In agreement with a previous report showing that Rad4 is recruited to chromatin upon induction of UV DNA lesions (35), the association of Rad4 with chromatin was dramatically increased in the wild type cells shortly after UV irradiation (Figure 6B). However, no such an increase was observed in the H4H75E mutant (Figure 6B). The increased association could also be seen in rad7Δ but not rad7Δ H4H75E cells (Figure 6C), indicating that the recruitment of Rad4 to chromatin after UV irradiation is independent of the GG-NER factor Rad7 but is impaired by the H4H75E mutation.
Following UV irradiation, the associations of other NER proteins with chromatin were not nearly as dramatically changed (if any) as Rad4 in either the wild type or H4H75E mutant cells (Supplementary Figure S6).
DISCUSSION
Through unbiased screening of histone H4 residues in the nucleosomal LRS domain, we identified novel UV sensitive and resistant mutations. Most of these mutations, except for H4H75E (and to a lesser extent, H4T73F and H4T73Y), do not significantly affect NER but may be implicated in other as-yet to be characterized DNA repair or damage tolerance pathways. The H4H75E mutation appears to attenuate GG-NER and Rad26-independent TC-NER at least in part by impairing the recruitment of Rad4 to chromatin upon induction of UV DNA lesions.
Rad4 is well-known to be involved in lesion recognition and verification during NER (36). Rad4 forms a complex with Rad23, which in addition to functioning with Rad4 in NER, plays a central role in targeting ubiquitylated proteins for proteasomal degradation (9). An in vitro study demonstrated that Rad4–Rad23 randomly diffuses along naked undamaged DNA and forms stable complexes with DNA containing a fluorescein-modified deoxythymidine, a model NER substrate (37). However, Rad4–Rad23 does not form stable complex with DNA containing a less helical distorting CPD, although its motion along the DNA becomes constrained around the CPD (37). It was therefore proposed that recognition and verification of lesions with minimal helical distortions (e.g., CPDs) might be accomplished by co-operative actions of Rad4, Rad14 (the yeast homolog of human XPA) and TFIIH (37). The increased association of Rad4 with chromatin in wild type cells following UV irradiation observed by us here and by others previously (35) may reflect the formation of lesion recognition and verification complex following remodeling of the nucleosome structure. Rad4 and Rad23 have been shown to become more tightly associated with the SWI/SNF (38) and INO80 (35) chromatin remodeling complexes and target them to chromatin to facilitate NER in yeast cells. The H4H75 residue, especially its side chain, is largely embedded in the nucleosome and interacts with histone H2B (Figure 1). It is therefore quite unlikely that the residue is directly involved in interactions with factors other than histone H2B. However, the H4H75E mutation will likely cause a conformational change to the nucleosome, as evidenced by enhanced accessibility of chromatin DNA to MNase in the mutant cells. The conformational change may impair the recruitment of chromatin remodeling complexes and/or inhibit the remodeling of nucleosome structure, thereby preventing the formation of lesion recognition and verification complex containing Rad4.
The recruitment of Rad4 to chromatin in wild type cells after UV irradiation should in theory lead to increased recruitment of downstream NER proteins. However, we did not detect dramatic changes in chromatin associations of the other NER proteins, including the Rad4 interacting partner Rad23. Rad23 has Rad4-independent functions and is much more abundant than Rad4 (9), which may be why we did not detect significantly increased recruitment of Rad23 to chromatin. The formation of the lesion recognition and verification complex containing Rad4 may be the rate-limiting step during NER and relatively long-lived. In contrast, the downstream reactions may be fast and involve only transient recruitment of the downstream NER factors (32), which might be one reason that we missed the detection of chromatin-recruitment of the downstream factors. Also, certain NER factors may be inherently associated with chromatin, and upon induction of DNA lesions, the factors may simply redistribute to the lesion sites within chromatin. Our chromatin fractionation analyses here would not detect intra-chromatin redistributions of the NER factors. Indeed, certain NER factors have been shown to be recruited to a gene promoter along with TFIIH and RNAPII in human cells in the absence of exogenously induced DNA damage (39). Rad7 and Rad16 have been shown to inherently bind to intergenic regions of the genome, and upon UV irradiation, redistribute to gene coding regions (40).
TC-NER is believed to be triggered by stalling of RNAPII at DNA lesions in the transcribed strand of actively transcribed genes (3,41). To the best of our knowledge, the requirement of a nucleosomal feature for TC-NER has not been documented. Here, we found that the H4H75E mutation impairs Rad26-independent TC-NER but does not affect TC-NER in the presence of Rad26. We recently found that Rad26 moderately evicts Spt5, a key transcription elongation factor and TC-NER repressor, from the RNAPII complex and promotes transcriptional bypass of UV induced DNA lesions (42). Unlike its mammalian homolog XPC, which is only required for GG-NER but completely dispensable for TC-NER, Rad4 is required for both GG-NER and TC-NER in yeast. In the presence of Rad26, the moderate eviction of Spt5 may allow a lesion-stalled RNAPII to efficiently recruit NER factors (including Rad4), making the recruitment of Rad4 to chromatin unnecessary for efficient TC-NER. However, in the absence of Rad26, the tight association of Spt5 and other TC-NER repressors with RNAPII (41) may impair the recruitment of NER factors, making the recruitment of Rad4 to chromatin necessary for efficient TC-NER.
In mammalian cells, the UV-DDB complex consisting of DDB1 and DDB2 plays an important role in GG-NER of minimally helix-distorting lesions such as CPDs by recognizing and handing over the lesions to XPC (36). It has been proposed that the yeast GG-NER complex consisting of Rad7 and Rad16 may be the functional counterpart of UV-DDB although no sequence homology exists between the two complexes (43). However, we found that the recruitment of Rad4 to chromatin after UV irradiation is independent of Rad7 (Figure 6C), which argues against the possibility that the recruitment of Rad4 to chromatin results from handing over of lesions by the yeast GG-NER complex. One possibility is that the yeast GG-NER complex function after the recruitment of Rad4 to chromatin. Indeed, a previous report showed that Rad7 and Rad16 are required for GG-NER at a step after incision of the damaged DNA (44).
The accessibility of cellular chromatin to MNase was not significantly changed upon UV irradiation even in the wild type cells (Figure 5), indicating that our assay missed the detection of structural changes of nucleosomes caused by CPDs and the remodeling of nucleosomes during NER. This may be due to the fact that only a small fraction (≤1/5) of the cellular nucleosomes would contain a minimally helix-distorting CPD (the UV dose of 120 J/m2 we used would induce ∼1 CPD/kb of double-stranded DNA), and the chromatin remodeling events during NER were transient. Also, the enhancement of chromatin accessibility to MNase in H4H75E cells compared to wild type cells was modest (Figure 5). Clearly, elucidating exactly how the H4-H75E mutation impacts nucleosome structure and interferes with recruitment of Rad4 will require further study and a more sensitive technique for mapping of nucleosome structure will be needed.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Scott Herke at LSU Genomics Facility for help with next-generation sequencing.
Notes
Present address: Sheikh Arafatur Rahman, Department of Pathobiology, Faculty of Veterinary Medicine and Animal Science, Bangabandhu Sheikh Mujibur Rahman Agricultural University, Gazipur-1706, Bangladesh.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1615550]. Funding for open access charge: National Science Foundation [MCB-1615550].
Conflict of interest statement. None declared.
REFERENCES
- 1. Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst.). 2015; 36:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tatum D., Li S.. Storici F. DNA Repair - On the Pathways to Fixing DNA Damage and Errors. 2011; Rijeka, Croatia: Tech Open Access Publisher; 97–122. [Google Scholar]
- 3. Li S. Transcription coupled nucleotide excision repair in the yeast Saccharomyces cerevisiae: the ambiguous role of Rad26. DNA Repair (Amst.). 2015; 36:43–48. [DOI] [PubMed] [Google Scholar]
- 4. Li S., Smerdon M.J.. Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J. 2002; 21:5921–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li W., Selvam K., Rahman S.A., Li S.. Sen1, the yeast homolog of human senataxin, plays a more direct role than Rad26 in transcription coupled DNA repair. Nucleic Acids Res. 2016; 44:6794–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verhage R., Zeeman A.M., de Groot N., Gleig F., Bang D.D., van de Putte P., Brouwer J.. The RAD7 and RAD16 genes, which are essential for pyrimidine dimer removal from the silent mating type loci, are also required for repair of the nontranscribed strand of an active gene in Saccharomyces cerevisiae. Mol. Cell Biol. 1994; 14:6135–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lejeune D., Chen X., Ruggiero C., Berryhill S., Ding B., Li S.. Yeast Elc1 plays an important role in global genomic repair but not in transcription coupled repair. DNA Repair (Amst.). 2009; 8:40–50. [DOI] [PubMed] [Google Scholar]
- 8. Min J.H., Pavletich N.P.. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007; 449:U570–U577. [DOI] [PubMed] [Google Scholar]
- 9. Dantuma N.P., Heinen C., Hoogstraten D.. The ubiquitin receptor Rad23: at the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst.). 2009; 8:449–460. [DOI] [PubMed] [Google Scholar]
- 10. Luger K., Dechassa M.L., Tremethick D.J.. New insights into nucleosome and chromatin structure: an ordered state or a disordered affair. Nat. Rev. Mol. Cell Biol. 2012; 13:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hara R., Mo J.Y., Sancar A.. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 2000; 20:9173–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S., Smerdon M.J.. Dissecting transcription-coupled and global genomic repair in the chromatin of yeast GAL1-10 genes. J. Biol. Chem. 2004; 279:14418–14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mao P., Smerdon M.J., Roberts S.A., Wyrick J.J.. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wellinger R.E., Thoma F.. Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J. 1997; 16:5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nag R., Gong F., Fahy D., Smerdon M.J.. A single amino acid change in histone H4 enhances UV survival and DNA repair in yeast. Nucleic Acids Res. 2008; 36:3857–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kruger W., Peterson C.L., Sil A., Coburn C., Arents G., Moudrianakis E.N., Herskowitz I.. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995; 9:2770–2779. [DOI] [PubMed] [Google Scholar]
- 17. Park J.H., Cosgrove M.S., Youngman E., Wolberger C., Boeke J.D.. A core nucleosome surface crucial for transcriptional silencing. Nat. Genet. 2002; 32:273–279. [DOI] [PubMed] [Google Scholar]
- 18. Fry C.J., Norris A., Cosgrove M., Boeke J.D., Peterson C.L.. The LRS and SIN domains: two structurally equivalent but functionally distinct nucleosomal surfaces required for transcriptional silencing. Mol. Cell Biol. 2006; 26:9045–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Leeuwen F., Gafken P.R., Gottschling D.E.. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002; 109:745–756. [DOI] [PubMed] [Google Scholar]
- 20. Bostelman L.J., Keller A.M., Albrecht A.M., Arat A., Thompson J.S.. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair (Amst.). 2007; 6:383–395. [DOI] [PubMed] [Google Scholar]
- 21. Chaudhuri S., Wyrick J.J., Smerdon M.J.. Histone H3 Lys79 methylation is required for efficient nucleotide excision repair in a silenced locus of Saccharomyces cerevisiae. Nucleic Acids Res. 2009; 37:1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tatum D., Li S.. Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J. Biol. Chem. 2011; 286:17530–17535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakanishi S., Sanderson B.W., Delventhal K.M., Bradford W.D., Staehling-Hampton K., Shilatifard A.. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 2008; 15:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gelbart M.E., Rechsteiner T., Richmond T.J., Tsukiyama T.. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol. Cell Biol. 2001; 21:2098–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sikorski R.S., Hieter P.. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989; 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sikorski R.S., Boeke J.D.. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991; 194:302–318. [DOI] [PubMed] [Google Scholar]
- 27. Olson S.A. EMBOSS opens up sequence analysis. European Molecular Biology Open Software Suite. Brief Bioinform. 2002; 3:87–91. [DOI] [PubMed] [Google Scholar]
- 28. Li W., Selvam K., Ko T., Li S.. Transcription bypass of DNA lesions enhances cell survival but attenuates transcription coupled DNA repair. Nucleic Acids Res. 2014; 42:13242–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kent N.A., Mellor J.. Chromatin structure snap-shots: rapid nuclease digestion of chromatin in yeast. Nucleic Acids Res. 1995; 23:3786–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kushnirov V.V. Rapid and reliable protein extraction from yeast. Yeast. 2000; 16:857–860. [DOI] [PubMed] [Google Scholar]
- 31. Dai J., Hyland E.M., Yuan D.S., Huang H., Bader J.S., Boeke J.D.. Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell. 2008; 134:1066–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luijsterburg M.S., von Bornstaedt G., Gourdin A.M., Politi A.Z., Mone M.J., Warmerdam D.O., Goedhart J., Vermeulen W., van Driel R., Hofer T.. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell Biol. 2010; 189:445–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tatum D., Li W., Placer M., Li S.. Diverse roles of RNA polymerase II-associated factor 1 complex in different subpathways of nucleotide excision repair. J. Biol. Chem. 2011; 286:30304–30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shahbazian M.D., Zhang K., Grunstein M.. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol. Cell. 2005; 19:271–277. [DOI] [PubMed] [Google Scholar]
- 35. Sarkar S., Kiely R., McHugh P.J.. The Ino80 chromatin-remodeling complex restores chromatin structure during UV DNA damage repair. J. Cell Biol. 2010; 191:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mu H., Geacintov N.E., Broyde S., Yeo J.E., Scharer O.D.. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair (Amst.). 2018; 71:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kong M.W., Liu L.L., Chen X.J., Driscoll K.I., Mao P., Bohm S., Kad N.M., Watkins S.C., Bernstein K.A., Wyrick J.J. et al.. Single-molecule imaging reveals that Rad4 employs a dynamic DNA damage recognition process. Mol. Cell. 2016; 64:376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gong F., Fahy D., Smerdon M.J.. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol. 2006; 13:902–907. [DOI] [PubMed] [Google Scholar]
- 39. Le May N., Mota-Fernandes D., Velez-Cruz R., Iltis I., Biard D., Egly J.M.. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell. 2010; 38:54–66. [DOI] [PubMed] [Google Scholar]
- 40. Yu S.R., Evans K., van Eijk P., Bennett M., Webster R.M., Leadbitter M., Teng Y.M., Waters R., Jackson S.P., Reed S.H.. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res. 2016; 26:1376–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li W., Li S.. Facilitators and repressors of transcription-coupled DNA repair in saccharomyces cerevisiae. Photochem. Photobiol. 2017; 93:259–267. [DOI] [PubMed] [Google Scholar]
- 42. Selvam K., Ding B., Sharma R., Li S.. Evidence that moderate eviction of Spt5 and promotion of error-free transcriptional bypass by Rad26 facilitates transcription coupled nucleotide excision repair. J. Mol. Biol. 2019; 431:1322–1338. [DOI] [PubMed] [Google Scholar]
- 43. Reed S.H., Gillette T.G.. Nucleotide excision repair and the ubiquitin proteasome pathway - Do all roads lead to Rome. DNA Repair (Amst.). 2007; 6:149–156. [DOI] [PubMed] [Google Scholar]
- 44. Reed S.H., You Z., Friedberg E.C.. The yeast RAD7 and RAD16 genes are required for postincision events during nucleotide excision repair. In vitro and in vivo studies with rad7 and rad16 mutants and purification of a Rad7/Rad16-containing protein complex. J. Biol. Chem. 1998; 273:29481–29488. [DOI] [PubMed] [Google Scholar]
- 45. White C.L., Suto R.K., Luger K.. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001; 20:5207–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.