

Abstract
Aims
Recent genome-wide association studies (GWAS) have identified that the JCAD locus is associated with risk of coronary artery disease (CAD) and myocardial infarction (MI). However, the mechanisms whereby candidate gene JCAD confers disease risk remain unclear. We addressed whether and how JCAD affects the development of atherosclerosis, the common cause of CAD.
Methods and results
By mining data in the Genotype-Tissue Expression (GTEx) database, we found that CAD-associated risk variants at the JCAD locus are linked to increased JCAD gene expression in human arteries, implicating JCAD as a candidate causal CAD gene. We therefore generated global and endothelial cell (EC) specific-JCAD knockout mice, and observed that JCAD deficiency attenuated high fat diet-induced atherosclerosis in ApoE-deficient mice. JCAD-deficiency in mice also improved endothelium-dependent relaxation. Genome-wide transcriptional profiling of JCAD-depleted human coronary artery ECs showed that JCAD depletion inhibited the activation of YAP/TAZ pathway, and the expression of downstream pro-atherogenic genes, including CTGF and Cyr61. As a result, JCAD-deficient ECs attracted fewer monocytes in response to lipopolysaccharide (LPS) stimulation. Moreover, JCAD expression in ECs was decreased under unidirectional laminar flow in vitro and in vivo. Proteomics studies suggest that JCAD regulates YAP/TAZ activation by interacting with actin-binding protein TRIOBP, thereby stabilizing stress fiber formation. Finally, we observed that endothelial JCAD expression was increased in mouse and human atherosclerotic plaques.
Conclusion
The present study demonstrates that the GWAS-identified CAD risk gene JCAD promotes endothelial dysfunction and atherosclerosis, thus highlighting the possibility of new therapeutic strategies for CAD by targeting JCAD.
Keywords: Coronary artery disease, GWAS, JCAD/KIAA1462, Atherosclerosis, Endothelial function , Therapeutic target
See page 2409 for the editorial comment on this article (doi: 10.1093/eurheartj/ehz370)
Translational perspective
Large-scale GWAS have identified genetic variants of JCAD gene as a risk allele for coronary artery disease (CAD). However, the role and function of JCAD in the pathophysiology of CAD remains elusive. Here, we show that CAD-associated risk variants near JCAD are linked to increased JCAD gene expression in human arteries. Furthermore, both global and endothelial cell-specific knockout of JCAD attenuates the development of atherosclerosis in mice. Mechanistically, JCAD regulates the Hippo/YAP/TAZ pathway, and promotes endothelial dysfunction. Our findings suggest that genetic or pharmacological inhibition of JCAD could be a promising therapeutic strategy for CAD.
Introduction
Coronary artery disease (CAD), the leading cause of death, imposes immense health and economic burdens worldwide.1–3 CAD is a complex disease with the involvement of multiple mechanisms, cell types, and is influenced by multiple risk factors such as diet, physical inactivity, smoking, environment, and genetics.4,5 Genetics plays a central role in CAD development, accounting for approximately 50% heritability of CAD.6,7 Recently, several large-scale genome-wide association studies (GWAS) have identified dozens of candidate genetic loci associated with risk for CAD and myocardial infarction (MI).8–10 However, the genetic mechanisms and causative genes for CAD loci are not fully resolved.11 To translate GWAS findings into novel vascular discoveries, more studies are warranted to link associated locus to CAD pathogenesis. Recently, functional analysis in cultured cells and genetically engineered mice indicated that these CAD prioritized genes are important core drivers of CAD risk by modulating vascular remodelling,12,13 macrophage function,14 endothelial function,15,16 and vascular tone.17,18
JCAD (Junctional cadherin 5 associated, also known as KIAA1462, encoding junctional protein associated with CAD) is one of more than 160 GWAS-identified genes. Genetic variants near JCAD were significantly associated with CAD and MI.9,19–21 Previous studies showed that JCAD is a cell–cell junctional protein in endothelial cells (ECs),22 which regulates pathological angiogenesis, rather than developmental angiogenesis.23 JCAD depletion also increased EC apoptosis and reduced EC proliferation, migration, and angiogenesis.24 However, direct evidence showing the involvement of JCAD in regulating CAD is lacking. Further elucidation of the function of JCAD is expected to shed light on the pathophysiology of CAD and exemplify the translation of novel CAD-associated genetic variants into new therapeutic strategies for CAD.
Since atherosclerosis is a common cause of CAD, mouse models that mimic human diseases have been critical to understand the biology and mechanisms of CAD risk genes in human atherosclerosis.25 Here, we hypothesized that deficiency of JCAD would reduce the atherosclerotic burden in athero-susceptible apolipoprotein E deficient (ApoE−/−) mice. We now provide the first report that JCAD deficiency suppresses vascular inflammation, improves vascular activity, and inhibits atherogenesis in ApoE−/− mice, suggesting a new avenue to treat atherosclerosis via inhibiting JCAD expression.
Methods
Mice
JCAD floxed mice were generated by the Mouse Genome Editing Resource at the University of Rochester Medical Center. JCAD global and endothelium specific knockout mice were generated by breeding JCAD floxed mice with CMV-Cre mice (Jackson Laboratory, Stock No. 006054) and Cdh5-Cre mice (Jackson Laboratory, Stock No. 006137), respectively, to achieve targeted deletion of exon 2 of JCAD gene. The mouse strains were back-crossed with C57BL/6J and ApoE−/− mice for studying atherosclerosis. All experimental protocols were approved by the University Committee of Animal Resources at the University of Rochester.
Statistical analysis
Data was analyzed using GraphPad Prism 5 software (GraphPad Software, Inc) and expressed as mean ± SEM. Differences between two groups were analyzed by two-tailed Student’s t test. Differences between multiple group comparisons for single and two variables were analyzed by one-way and two-way ANOVA, respectively. P < 0.05 was considered statistically significant.
For a detailed description of all methods, please see Supplementary material online.
Results
CAD-associated risk variants at JCAD locus are associated with increased JCAD gene expression in human aortic artery tissues
Emerging GWAS findings have identified several genetic variants near JCAD that are associated with cardiovascular diseases including CAD/MI.9,19–21 The latest meta-analysis of CAD GWAS from the Coronary ARtery DIsease Genome wide Replication and Meta-analysis (CARDIoGRAM) plus The Coronary Artery Disease (C4D) Genetics and UK Biobank studies26 identified a synonymous variant rs9337951 as another lead single nucleotide polymorphism (SNP) at the JCAD locus (Figure 1A and B). By mining genotype and transcriptomic data in the Genotype-Tissue Expression (GTEx) database, we mapped the direction of lead GWAS risk variants (e.g. rs9337951), to be associated with increased JCAD expression in human aortic artery tissues. This is consistent with summary-based Mendelian randomization (SMR) colocalization results identifying JCAD as the target gene at this locus using Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) and GTEx datasets (PSMR = 2.05E-07) (Supplementary material online, Table S1). Moreover, many of these GWAS variants were identified as tissue-specific eQTLs for JCAD in aortic artery tissues (Figure 1C, and Supplementary material online, Figure S1 and S2). Given the important potential role of JCAD in human diseases, we then examined the distribution of JCAD in a variety of tissues from C57BL/6J mice and humans. We observed that JCAD was highly expressed in EC-enriched organs/tissues (such as aorta, lung, and brain) from mice (Supplementary material online, Figure S3A). Consistently, we also observed that JCAD expression was highly expressed in endothelial cell-enriched human tissues (aorta, lung, and coronary artery) in GTEx (Supplementary material online, Figure S3B). These data strongly implicate JCAD as a candidate causal gene at this CAD locus and suggest that reduced JCAD expression could confer protective effects against CAD.
Figure 1.

CAD-associated risk variants at JCAD locus are associated with increased JCAD gene expression in human aortic artery tissues. (A) Identification of lead single nucleotide polymorphism (SNP) rs9337951 in the CARDIoGRAMplusC4D and UK Biobank meta-analysis8. (B) Box plots showing expression quantitative trait loci data from GTEx (v7) datasets for the lead genome-wide association studies SNP (rs9337951) associated with normalized JCAD gene expression in aortic artery tissue. Both genome-wide significant expression quantitative trait loci P-values and normalized effect size are shown for indicated SNPs. (C) Single-tissue expression quantitative trait loci normalized effect sizes for rs9337951 are shown for 48 different tissues in GTEx (v7), depicted as a color-coded forest plot with 95% confidence intervals. METASOFT based m-values representing the posterior probability of tissue-specific expression quantitative trait loci’s are also shown with their respective single-tissue expression quantitative trait loci P-values.
JCAD deficiency in mice improves endothelium-dependent vasorelaxation under high fat-diet feeding conditions
JCAD was recently reported to be an EC junctional protein that regulates pathological angiogenesis, EC proliferation and migration.23,24 It remains unknown whether JCAD regulate endothelial function in vivo. To better understand the role of JCAD in regulating endothelial function in vivo, we generated JCAD knockout mice by targeted deletion of exon 2 of mouse JCAD using a CMV-Cre mediated germline deletion strategy. We first confirmed the deletion of JCAD in mouse heart, lung, and brain tissues (Supplementary material online, Figure S4). Considering that JCAD is an endothelial cell junctional protein, it is possible that JCAD may regulate vascular permeability. However, we did not observe any significant effect of JCAD deletion on vascular permeability induced by lipopolysaccharide (LPS) or vascular endothelial growth factor (VEGF) (Supplementary material online, Figure S5), which agrees with a recent report using a cellular model of mustard-elicited vessel permeability.23 Human JCAD depletion by siRNA treatment in human umbilical vein endothelial cells (HUVECs) or genetic deletion in primary mouse lung ECs did not affect the expression of vascular endothelial junctional protein VE-cadherin (also known as CDH5, Supplementary material online, Figure S6). We next evaluated the role of JCAD in endothelium-dependent relaxation. Under normal chow diet feeding, JCAD deletion had no significant impact on acetylcholine-mediated endothelium-dependent relaxation or nitric oxide (NO) donor sodium nitroprusside-mediated relaxation (Supplementary material online, Figure S7). However, under high fat diet feeding (HFD) condition, JCAD deficiency partially attenuated HFD-induced impairment of endothelium-dependent relaxation, while sodium nitroprusside-mediated relaxation remained unaffected (Figure 2A and B).
Figure 2.

JCAD deficiency in mice improves endothelium-dependent vasorelaxation under high fat-diet feeding conditions. (A) Endothelium-dependent vasodilation was determined by relaxation of aortic rings pre-constricted with phenylephrine (10−6 mol/L). The dose-response curves of aortic rings to the vasodilator acetylcholine (10−9–10−5 mol/L) in JCAD−/− mice (n = 6) and JCAD+/+ (n = 9) mice under high fat diet conditions for 12 weeks. (B) The dose-response curves of aortic rings to NO donor sodium nitroprusside (10−9–10−5 mol/L) in JCAD−/− mice (n = 6) and JCAD−+/+ (n = 9) mice fed a high fat diet for 12 weeks.
JCAD deficiency reduces atherosclerosis in ApoE−/− mice fed a western diet
Since impaired endothelium-dependent relaxation is a crucial initiating event in the development of atherosclerosis, we next explored whether or not JCAD affects atherosclerosis development in ApoE−/− mice under HFD. By performing lesion analysis in en face aorta and aortic sinus tissues, we observed that both male and female JCAD−/−; ApoE−/− mice showed significantly reduced atherosclerotic burden compared with littermate controls (JCAD+/+; ApoE−/− mice) (Figure 3A and B). Next, we assessed whether JCAD deficiency would modulate plaque composition by analyzing the contents of macrophages, smooth muscle cells (SMCs), and collagen in aortic sinus with immunofluorescent staining and Masson’s trichrome staining, respectively. Our data suggest that mouse aortic sinus with JCAD deletion had no significant difference in SMC content (alpha-smooth muscle actin–positive signal), but had reduced macrophage (CD68-positive staining) and increased collagen content (blue signal by Masson’s trichrome staining) (Figure 3C–F). JCAD deletion does not affect the number of TER-119+ erythroid cells within the plaques (Supplementary material online, Figure S8), and resulted in a trend decrease (but not statistically significant) in necrotic core size (Supplementary material online, Figure S9). We also observed that endothelial marker CD31-positive staining was localized at the endothelial lining of the intima, and the level of CD31-positive staining was similar between the two study groups (Supplementary material online, Figure S10). Analysis of serum lipid profiles indicated that JCAD global knockout did not alter serum levels of total cholesterol and triglycerides (Supplementary material online, Tables S2 and S3). We also observed that JCAD-deficient macrophages expressed comparable levels of genes involved in inflammation, lipid uptake, cholesterol efflux, and M1/M2 polarization, and exhibited similar capacity in monocyte transendothelial migration and oxidized low-density lipoprotein (oxLDL)-induced foam cell formation (Supplementary material online, Figure S11–S16).
Figure 3.

JCAD deficiency reduces atherosclerosis in ApoE−/− mice fed a western-type diet. (A-B) En face Oil Red O–staining of whole aorta from male (A, n = 15–16) and female (B, n = 7–12) JCAD+/+; ApoE−/− and JCAD−/−; ApoE−/− mice after 12 weeks of western-type diet feeding. Quantification of en face lesion area. Data were represented as percent Oil Red O positive area of the entire aorta measured by Image J. Images presented were from a composite of 2–3 images from the same aorta. (C) Representative Oil Red O–stained aortic sinus cryosections from male JCAD+/+; ApoE−/− (n = 6) and JCAD−/−; ApoE−/− (n = 5) mice. Quantification of aortic sinus lesion area between two groups. Data were represented as percent Oil Red O positive area of the entire sinus measured by Image J. (D–F), Analysis of plaque composition in aortic sinus. The contents of macrophages (CD68 positive, green, D), smooth muscle cells (α-SMA positive, red, E) were analyzed by immunostaining-based quantification of the positive signal (mean fluorescence intensity in arbitrary units) was provided in right panels, n = 6 per each group, scale bar = 100 μm. Collagen content (blue, F) was determined by Masson’s trichrome staining. Quantification data were presented as % lesion area in the right panel, n = 8 for JCAD+/+; ApoE−/− and n = 7 for JCAD−/−; ApoE−/− group. α-SMA, alpha-smooth muscle actin.
Endothelial cell-specific JCAD deficiency reduces atherosclerosis in ApoE−/− mice fed a western diet
To assess whether endothelial JCAD plays a major role in atherosclerosis, we determined the localization and expression abundance of JCAD in vascular cells from mouse and human tissues. By confocal immunofluorescent staining, we observed that JCAD colocalized with markers of adhesion junction (VE-cadherin) (Figure 4A), further confirming the junctional localization of JCAD in ECs as described by other studies.22–24 Notably, we also observed JCAD is expressed in perinuclear vesicles (Figure 4A and Supplementary material online, Figure S17). In addition, we observed that JCAD is mainly expressed in ECs isolated from mouse and human tissues (Figure 4B–C).
Figure 4.

Endothelial cell-specific JCAD deficiency reduces atherosclerosis in ApoE−/− mice fed a western-type diet. (A) Confocal microscopy shows the colocalization of JCAD (red) with adherens junction protein VE-cadherin (green) in HUVECs. DAPI was used for counterstaining of cell nuclei. Enlarged images with arrows showing both membrane and perinuclear vesicular localization of JCAD. Bar = 40 µm, n = 4. (B) JCAD gene expression in mouse vascular cells. Relative expression of JCAD gene was compared in primary mouse lung endothelial cells (EC), mouse aortic smooth muscle cells (SMC), and mouse peritoneal macrophages (MPM), n = 3. (C) JCAD gene expression in human vascular cells. Relative expression of JCAD gene was compared in human coronary artery endothelial cells (HCAEC), human coronary artery smooth muscle cells (HCASMC), and human THP1-derived macrophages (THP1 monocytes stimulated with 100 nM PMA for 48 h), n = 2. (D-E) En face Oil Red O–staining of whole aorta from male JCADecWT; ApoE−/− (n = 7) and JCADecKO; ApoE−/− (n = 7) mice after 12 weeks of western-type diet feeding. Quantification of en face lesion area. Data were represented as percent Oil Red O-positive area of the entire aorta measured by Image J. Images presented were from a composite of 2–3 images from the same aorta. DAPI, 4′,6-diamidino-2-phenylindole; HUVECs, human umbilical vein endothelial cells; PMA, phorbol 12-myristate 13-acetate.
To better understand the role of endothelial JCAD in atherosclerosis, we generated EC specific JCAD knockout (JCADecKO) mice bred against ApoE−/− mice (Supplementary material online, Figure S18). We observed that JCADecKO; ApoE−/− mice displayed a significant reduction of atherosclerotic plaque sizes (Figure 4D and E) without altering serum levels of total cholesterol and triglycerides (Supplementary material online, Table S4).
JCAD deficiency attenuates vascular inflammation by acquiring an atheroprotective transcriptomic program
To gain mechanistic insights into the effects of JCAD on endothelial function and atherosclerosis, we performed RNA-sequencing (RNA-seq) based transcriptomic profiling in human coronary artery ECs (HCAECs) with depleted JCAD by siRNA. Gene ontology and pathway analysis of the differentially expressed genes indicated that JCAD regulates multiple biological processes (such as inflammatory responses) and important pathways (including TNFα signaling pathway) in ECs (Figure 5A and B, Supplementary material online, Tables S5–S7). Notably, JCAD depletion decreased the expression of atherogenic genes (such as CTGF, Cyr61, CCL2, BMP4, SELE, VCAM1, and ADAMTS7), while upregulating multiple atheroprotective genes (such as HMOX1, KLF2, KLF4, and GCH1) (Figure 5C). Our real-time PCR analysis also validated the altered genes revealed by RNA-seq (Figure 5D). To investigate the function of JCAD in vitro, we determined whether JCAD regulates monocyte adhesion to ECs, a key step in the initiation and progression of atherosclerosis. We observed that ECs isolated from JCAD knockout mice had fewer monocyte adhesion than those isolated from JCAD wild-type mice, which was associated with decreased protein expression of VCAM1 (Figure 5E and F).
Figure 5.

JCAD deficiency attenuates vascular inflammation by acquiring an atheroprotective transcriptional program. (A–B) Gene ontology (A) and KEGG2016 pathway (B) analysis of differentially expressed genes in JCAD depleted HCAECs by RNA-seq. RNA-seq data were deposited in NCBI Gene Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession number of GEO: GSE102498. (C) Selected atherosclerosis-relevant genes regulated by JCAD revealed by RNA-seq. * indicates established downstream target genes of YAP/TAZ. For a whole list of differentially expressed genes, please refer to Supplementary material online, Table S7. (D) Validation of RNA-seq data by real time PCR. Select downregulated and upregulated genes were presented, n = 4. (E–F) JCAD deficiency attenuates LPS-induced vascular inflammation. JCAD+/+ (WT) and JCAD−/− (KO) primary mouse lung endothelial cells were stimulated with LPS for 6 h before monocyte (THP1) adhesion assay was performed (E, n = 6). In parallel, whole cell lysates were collected for Western blot analysis to detect protein expression of VCAM1 and ICAM1 (F, n = 4), ***P < 0.001 vs. WT, ##P < 0.01 vs WT + LPS. Log2FC, Log2 fold change; siNC, scramble control siRNA; siJCAD, JCAD siRNA; LPS, lipopolysaccharide.
JCAD promotes YAP activation in human endothelial cells
Based on our RNA-seq data, we observed that JCAD depletion reduced the expression of CTGF, and Cyr61, two well-known downstream genes of the pro-atherogenic Hippo/YAP/TAZ pathway. We reasoned whether JCAD depletion could inhibit YAP activation in ECs. Using a YAP/TEAD-based dual-luciferase reporter assay, we showed that JCAD depletion reduced YAP/TAZ/TEAD transcriptional activity (Figure 6A). Confocal immunofluorescent microscopy analysis also showed that JCAD depletion decreased the percentage of nuclear-localized YAP and its homologue TAZ, in human ECs (Figure 6B) and reduced protein expression of CTGF and Cyr61 by promoting phosphorylation of YAP (Figure 6C and D). Studies in JCAD-deficient mouse lung ECs recapitulated the findings from JCAD siRNA-mediated depletion (Supplementary material online, Figure S19), which supports that JCAD deficiency reduces the nuclear-localized YAP/TAZ staining. Conversely, adenoviral-mediated JCAD overexpression increased YAP/TAZ/TEAD transcriptional activity and protein expression of CTGF and Cyr61 (Supplementary material online, Figure S20). Consistent with the notion that F-actin stress fiber drives YAP/TAZ activation, we observed measureable decreased F-actin staining in JCAD depleted ECs (Figure 6E). These data collectively suggest that JCAD promotes YAP activation in ECs.
Figure 6.

JCAD promotes YAP/TAZ activation in endothelial cells. (A) JCAD depletion decreased YAP/TAZ/TEAD luciferase activity (8XGTIIc-lux). Human umbilical vein endothelial cells (HUVECs) were transfected with control siRNA (siNC, 100 nM) or JCAD siRNA (siJCAD, 100 nM) for 48 h before transfection with 8XGTIIc-lux for 24 h, n = 3. YAP/TAZ siRNA (siYAP/TAZ, 20 nM) transfection was used as the positive control. (B) JCAD depletion inhibits YAP/TAZ nuclear staining in HUVECs (left panel). Quantification of YAP/TAZ subcellular localization in siNC (196 cells) and siJCAD (207 cells) treated HUVECs (right panel). C, cytoplasm; N, nucleus. Mean % from four independent experiments were used for quantification. (C) JCAD depletion decreased YAP/TAZ downstream protein expression (CTGF and Cyr61) in HUVECs, n = 3. (D) JCAD depletion promotes YAP phosphorylation at Ser127 in HUVECs, n = 3. (E) JCAD depletion decreased F-actin stress fiber formation in HUVECs. Mean fluorescence intensity was calculated by dividing total fluorescent intensity by cell numbers, n = 4. (F) JCAD interacting proteins identified by immunoprecipitation-based mass spectrometry. Averaged number of peptides from three independent experiments were presented. For a complete list of JCAD interacting proteins, please refer to Supplementary material online, Table S8. Abbreviations: ARMC8: Armadillo repeat-containing protein 8; FLNC: Filamin-C; CKAP4: Cytoskeleton-associated protein 4; PPP2R1A: Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha; TRIOBP: TRIO and F-actin-binding protein; WDR26: WD repeat-containing protein 26. (G) Validation of JCAD interaction with TRIOBP in HUVECs. HUVECs were infected with adenoviral HA-tagged (Ad-HA) JCAD for 48 h before whole cell lysate was collected for HA-beads pulldown assay, n = 3.
Next, we asked whether JCAD regulates stress fiber formation by interacting with important protein binding partners. To identify the JCAD interactome in ECs, we performed immunoprecipitation-based mass spectrometry analysis. From these data, we observed that JCAD interacted with multiple actin-binding proteins such as Filamin C, CKAP4, and TRIOBP (Figure 6F and Supplementary material online, Table S8). We further validated the interaction of JCAD with TRIOBP by pulldown assays (Figure 6G). Similar to JCAD depletion, TRIOBP depletion by siRNA also led to reduced stress fiber formation and decreased expression of genes downstream YAP/TAZ in ECs (Supplementary material online, Figure S21), suggesting that the interaction of JCAD with TRIOBP is likely to contribute to the effect of JCAD on stress fiber formation in ECs.
Mechanosensitive and pharmacological regulation of JCAD in human endothelial cells
To understand the regulatory factors for JCAD, we analyzed the effect of pro-atherogenic factors (LPS, TNFα, LDL, oxidized LDL, and cholesterol crystals), pro-angiogenic factor (VEGF) and biomechanical factors on JCAD expression in ECs. Our data showed that LPS, TNFα, and VEGF did not affect JCAD gene expression (Supplementary material online, Figure S22), while oxidized LDL and cholesterol crystal treatment increased JCAD gene expression (Supplementary material online, Figure S23). However, we observed that the atheroprotective unidirectional laminar flow (UF, 10 dyne/ 24 h) decreased, while atheroprone disturbed flow (DF, 0.5 dyne/ 24 h) increased JCAD expression in human ECs (Figure 7A–D). JCAD expression was also lower in mouse aortic endothelium exposed to UF (compared with DF) (Figure 7E and F), indicating that JCAD is a mechanosensitive CAD-associated gene. mRNA stability experiments showed that UF did not affect JCAD mRNA stability (Supplementary material online, Figure S24), indicating that UF may regulate JCAD gene expression transcriptionally. KLF2 is an important regulator of flow-sensitive genes; however, KLF2 depletion or overexpression did not affect JCAD gene expression in ECs (Supplementary material online, Figure S24), indicating that UF possibly regulates JCAD expression independent of KLF2. Furthermore, in an effort to identify potential JCAD inhibitors, we performed an in-house screening of compounds with vasoprotective actions. We identified (+)-JQ1 (a BRD4 inhibitor that attenuates experimental atherosclerosis and leucocyte adhesion),27 as a pharmacological inhibitor of JCAD in ECs (Supplementary material online, Figure S25).
Figure 7.

JCAD expression is decreased by unidirectional laminar flow in vitro and in vivo. (A) Scheme of a cone-plate viscometer to generate unidirectional laminar flow (UF) or disturbed flow (DF). Image was manually drawn using powerpoint. (B) JCAD gene expression is decreased by unidirectional laminar flow. HUVECs were exposed to unidirectional laminar flow for 24 h before RNA was collected for real time PCR analysis. KLF2 was used the positive control, n=4. (C) JCAD protein expression is decreased by unidirectional laminar flow. HUVECs were exposed to unidirectional laminar flow for 24 h before confocal microscopy was performed, n = 4, bar = 60 μm. (D) JCAD gene expression is increased in response to disturbed flow. VCAM1 was used as the positive control, n = 3. (E) JCAD gene expression in intimal RNA lysate isolated from aortic arch (AA) and thoracic aorta (TA) of ApoE−/− mice fed a normal diet, n = 12. (F) JCAD expression in C57BL/6J mice undergoing partial ligation of left carotid artery (LCA). Intimal RNA lysate was collected for real-time PCR analysis, n = 4. HUVECs, human umbilical vein endothelial cells; RCA, right carotid artery.
JCAD expression is increased in mouse and human atherosclerotic plaques
Finally, we assessed the expression of JCAD in mouse and human atherosclerotic plaques. We observed that JCAD gene expression in intimal ECs was increased in ApoE−/− mice fed a HFD (Figure 8A). Moreover, JCAD gene expression was significantly increased in advanced atherosclerotic plaques from human patients (Figure 8B). Intriguingly, data mining in circulating monocytes from patients with familial hypercholesterolaemia and healthy control individuals28 suggested that JCAD expression was not significantly different (Supplementary material online, Figure S26). However, using high-resolution confocal microscopy, we observed that JCAD expression in the intimal endothelium layer was increased in human atherosclerotic plaques (Figure 8C), providing additional relevance of JCAD to CAD development.
Figure 8.

JCAD expression is increased in mouse and human atherosclerotic plaques. (A) JCAD expression is increased in aortic endothelium of ApoE−/− mice fed a high fat diet (HFD) vs. normal diet (ND), n = 5. (B) JCAD expression is increased in advanced human atherosclerotic plaques, n = 9. (C) Intimal endothelial JCAD expression is increased in human atherosclerotic plaques, n = 4. A, atheroma; I, intima; L, lumen; M, media.
Discussion
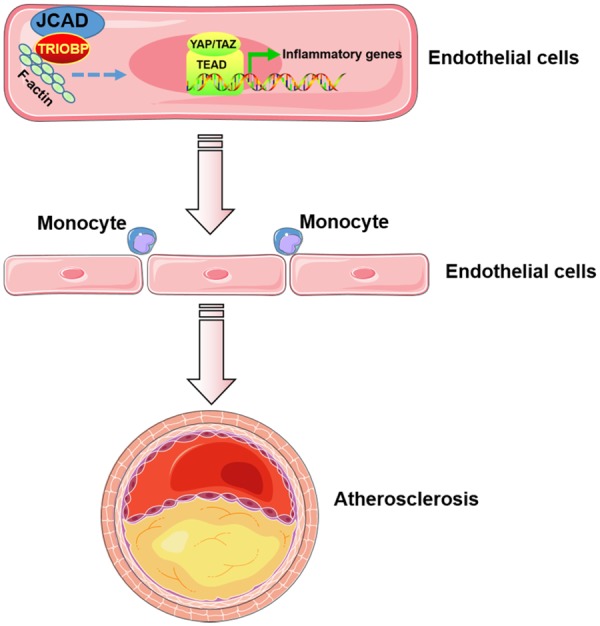
Atherosclerosis is a leading cause of cardiovascular diseases such as CAD and MI. Understanding the expression, regulation, and function of GWAS-identified CAD associated genes and their disease-causing genetic variants will advance our understanding of the pathogenesis of CAD and yield novel therapeutic strategies.10 Using genetically engineered mice and systems biology approaches, we provide direct functional evidence showing that the CAD risk gene JCAD promotes atherosclerosis in mice, and one of the underlying mechanism is through activating the Hippo/YAP/TAZ pathway in ECs. To the best of our knowledge, this is the first study that shows the causal role of JCAD in endothelial dysfunction and atherosclerosis in vivo (Take home figure). Our findings advance our understanding of the association of JCAD with increased CAD risk.
Take home figure.
JCAD promotes endothelial dysfunction and atherosclerosis. Junctional protein JCAD promotes the activation of YAP/TAZ/TEAD by interacting with TRIOBP and thus stabilizing F-actin stress fiber. By doing so, JCAD trigger the expression of inflammatory genes in endothelial cells, driving an inflammatory process via the recruitment of monocytes and resulting in the formation of atherosclerotic plaques.
Recent GWAS have identified genetic variants at the JCAD locus associated with CAD.9,19–21 There are now five reported JCAD SNPs (including rs3739998, rs2505083, rs2487928) associated with CAD/MI in multi-ethnic populations. However, the direct causal link between JCAD and atherosclerosis is lacking. In the present study, we showed that CAD GWAS risk variants at the JCAD locus are strongly associated with increased expression of JCAD in human aortic artery tissues, suggesting that JCAD may promote atherosclerosis. Using a well-established diet-induced mouse model of atherosclerosis, we further showed that global and endothelial cell-specific knockout of JCAD in mice attenuates the development of atherosclerosis. JCAD deficiency in mice also displayed improved vasodilation under conditions of high fat-feeding and an atheroprotective phenotype. These results implicate JCAD as a candidate causal gene for atherosclerosis, not only explaining the association of this gene with human CAD, but also pointing to future directions to prevent and treat atherosclerosis based on pharmacological or genetic inhibition of JCAD.
The Human Protein Atlas (https://www.proteinatlas.org) show high expression of JCAD in endothelial cell-enriched brain and lung tissues. These data are consistent with our analysis of gene expression of JCAD in mouse and human tissues. In light of the fact that endothelial dysfunction and ensuing inflammation are the hallmarks of atherosclerosis, we demonstrated that JCAD depletion reduced endothelial inflammation and improved endothelial dysfunction. JCAD deficiency also attenuated monocyte adhesion to ECs by reducing the expression of pro-adhesive cytokines/factors, while upregulating the expression of atheroprotective factors. These data are consistent with our observations of decreased staining of macrophage marker CD68 in the aortic sinus of JCAD−/−; ApoE−/− mice. JCAD deletion also ameliorated HFD-induced impaired vasorelaxation ex vivo. Thus, our study provides direct evidence linking JCAD to endothelial dysfunction. By examining the effects of various CAD risk factors on JCAD expression, we found that JCAD expression was not affected by stimulation with pro-inflammatory cytokines/stimuli (TNFα and LPS) or pro-angiogenic stimuli (VEGF). However, JCAD expression was increased in ECs exposed to oxLDL, cholesterol crystals, and DF, but was decreased by UF, suggesting that a broad regulation of JCAD expression by lipid products and local hemodynamic forces. Moreover, we observed increased JCAD expression upon challenging with the atherogenic diet. Since feeding with atherogenic diet increases levels of oxidized LDL and cholesterol crystals, our results indicate that abnormal lipid levels could be a potential cause of JCAD up-regulation and impaired vasodilation.
The Hippo pathway effector YAP/TAZ have recently been shown to regulate endothelial dysfunction and atherosclerosis in mice.29,30 Our group31 and others29,30 have shown that UF promotes YAP phosphorylation, thereby inhibiting YAP activation in ECs through different mechanisms. Our genome-wide RNA-seq profiling data suggest that JCAD links YAP/TAZ-dependent genes in ECs. Gain- and loss-of-function studies in cultured cells implicated JCAD as an upstream molecule that drives YAP/TAZ activation. Three potential mechanisms could be involved in the processes. First, JCAD has a functionally conserved domain that resembles ROCK1/2 which is critical for F-actin stress fiber formation.22 Second, JCAD has recently been shown to associate with and inhibit Hippo kinase LATS2, and positively regulate YAP activation in cancer cells32 and ECs24; Third, in our study, we show that JCAD interacts with several actin-binding proteins (e.g. TRIOBP), to regulate F-actin dependent YAP/TAZ activation. We also demonstrate that JCAD depletion/deficiency inhibits, while JCAD overexpression promotes, YAP/TAZ activation in ECs, further consolidating the role of JCAD in YAP/TAZ activation. Using small-scale compound screening, we identified (+)-JQ1 as a pharmacological inhibitor that reduces JCAD expression in ECs, suggesting that JCAD is pharmacologically targetable and amenable to therapeutic intervention.
High-risk rupture-prone atherosclerotic plaques are associated with acute coronary syndrome. Histologically, features of plaque instability/vulnerability include: large necrotic core; thin fibrous cap; increased collagen breakdown, increased infiltration of inflammatory cells (such as macrophages); increased neovascularization by vasa vasorum; and intraplaque haemorrhage.33–35 In our study, we observed that JCAD deletion decreased macrophage accumulation and increased collagen content, without significant effect on other parameters. Therefore, the pathological role of JCAD in plaque instability need to be addressed in future studies. A potential limitation of the current study is that we mainly addressed the major role of endothelial JCAD in atherosclerosis, however, comparable studies in smooth muscle cell and macrophage specific knockout mice are warranted to evaluate the precise role of JCAD in regulating atherogenic cell functions in other vascular cell types. In addition, we recognize that sample size in mass spectrometry experiment is close to borderline, thus the JCAD interactome data should be verified with additional methodologies.
Conclusions
The present study demonstrates that GWAS-identified CAD/MI risk gene JCAD promotes endothelial dysfunction and atherosclerosis in mice, highlighting the possibility of designing innovative cardiovascular drugs by targeting JCAD.
Supplementary Material
Acknowledgements
We are grateful to Prof. Paul M. Vanhoutte (Department of Pharmacology and Pharmacy, The University of Hong Kong) and Prof. Charles J. Lowenstein (Department of Medicine, University of Rochester) for a critical review and helpful suggestions on improving the manuscript; We thank Dr Lin Gan from the University of Rochester Medical Center (URMC) Mouse Genome Editing Resource for generating the JCAD floxed mice for this study. The authors would like to extend their gratitude to the URMC Mass Spectrometry Resource Laboratory (MSRL) for performing and guiding analysis of mass spectrometry data. We are grateful to the University of Rochester Genomics Research Center (URGRC) for performing RNA-sequencing analysis.
Funding
This study was supported by National Institutes of Health (NIH) grants (HL114570, HL128363, HL130167) to Z.G.J.; and the American Heart Association (17GRNT33660671) to Z.G.J.; (18CDA34110359) to S.X. Y.X. is supported by grants from the Health and Medical Creation Program of CAMS (2016-I2M-1-011) and the National Natural Science Foundation of China (81621064). S.Y.S. was supported by grants from the National Natural Science Foundation of China (81621064) and the Creative Research Groups of the National Natural Science Foundation of China (81621064). C.L.M. is supported by a NIH Pathway to Independence Award (HL125912) and Leducq Foundation Transatlantic Network of Excellence Award.
Conflict of interest: none declared.
References
- 1. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C, Stroke Statistics S. Heart Disease and Stroke Statistics-2017 update: a report from the American Heart Association. Circulation 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atlas Writing G, Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, Wilkins E, Wright L, Vos R, Bax J, Blum M, Pinto F, Vardas P.. European Society of Cardiology: cardiovascular Disease Statistics 2017. Eur Heart J 2018;39:508–579. [DOI] [PubMed] [Google Scholar]
- 3. Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwoger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ.. ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2873–2926. [DOI] [PubMed] [Google Scholar]
- 4. Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, Cooney MT, Corra U, Cosyns B, Deaton C, Graham I, Hall MS, Hobbs FDR, Lochen ML, Lollgen H, Marques-Vidal P, Perk J, Prescott E, Redon J, Richter DJ, Sattar N, Smulders Y, Tiberi M, van der Worp HB, van Dis I, Verschuren WMM, Binno S.. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016;37:2315–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwak BR, Back M, Bochaton-Piallat ML, Caligiuri G, Daemen MJ, Davies PF, Hoefer IE, Holvoet P, Jo H, Krams R, Lehoux S, Monaco C, Steffens S, Virmani R, Weber C, Wentzel JJ, Evans PC.. Biomechanical factors in atherosclerosis: mechanisms and clinical implications. Eur Heart J 2014;35:3013–20, 3020a-3020d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts R. Genetics of coronary artery disease. Circ Res 2014;114:1890–1903. [DOI] [PubMed] [Google Scholar]
- 7. Zhao Y, Chen J, Freudenberg JM, Meng Q, Rajpal DK, Yang X.. Network-based identification and prioritization of key regulators of coronary artery disease loci. Arterioscler Thromb Vasc Biol 2016;36:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.CARDIoGRAMplusC4D ConsortiumDeloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, König IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikäinen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do RDIAGRAM ConsortiumCARDIOGENICS ConsortiumDoney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Müller-Nurasyid MMuTHER ConsortiumNikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schäfer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJWellcome Trust Case Control ConsortiumWells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrières J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kähönen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Trégouët DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvänen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimäki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O'Donnell C, Reilly MP, März W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikainen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, Konig IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimaki T, Loos RJF, Melander O, Metspalu A, Marz W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O'Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M.. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kessler T, Vilne B, Schunkert H.. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med 2016;8:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller CL, Pjanic M, Quertermous T.. From Locus Association to mechanism of gene causality: the devil is in the details. Arterioscler Thromb Vasc Biol 2015;35:2079–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, Ou K, Paschos GK, Zheng XL, Parmacek MS, Rader DJ, Reilly MP.. Knockout of Adamts7, a novel coronary artery disease locus in humans, reduces atherosclerosis in mice. Circulation 2015;131:1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kessler T, Zhang L, Liu Z, Yin X, Huang Y, Wang Y, Fu Y, Mayr M, Ge Q, Xu Q, Zhu Y, Wang X, Schmidt K, de Wit C, Erdmann J, Schunkert H, Aherrahrou Z, Kong W.. ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation 2015;131:1191–1201. [DOI] [PubMed] [Google Scholar]
- 14. Zhao G, Yang W, Wu J, Chen B, Yang X, Chen J, McVey DG, Andreadi C, Gong P, Webb TR, Samani NJ, Ye S.. Influence of a coronary artery disease-associated genetic variant on FURIN expression and effect of furin on macrophage behavior. Arterioscler Thromb Vasc Biol 2018;38:1837–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krause MD, Huang RT, Wu D, Shentu TP, Harrison DL, Whalen MB, Stolze LK, Di Rienzo A, Moskowitz IP, Civelek M, Romanoski CE, Fang Y.. Genetic variant at coronary artery disease and ischemic stroke locus 1p32.2 regulates endothelial responses to hemodynamics. Proc Natl Acad Sci USA 2018;115:E11349–E11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, Leed A, Weale ME, Spencer CCA, Aguet F, Segre AV, Ardlie KG, Khera AV, Kaushik VK, Natarajan P, Kathiresan S.. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet 2017;49:1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, Khera AV, Ryan RJH, Engreitz JM, Issner R, Shoresh N, Epstein CB, de Laat W, Brown JD, Schnabel RB, Bernstein BE, Kathiresan S.. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell 2017;170:522–533.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kessler T, Wobst J, Wolf B, Eckhold J, Vilne B, Hollstein R, von Ameln S, Dang TA, Sager HB, Moritz Rumpf P, Aherrahrou R, Kastrati A, Bjorkegren JLM, Erdmann J, Lusis AJ, Civelek M, Kaiser FJ, Schunkert H.. Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 2017;136:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coronary Artery Disease Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 2011;43:339–344. [DOI] [PubMed] [Google Scholar]
- 20. Erdmann J, Willenborg C, Nahrstaedt J, Preuss M, Konig IR, Baumert J, Linsel-Nitschke P, Gieger C, Tennstedt S, Belcredi P, Aherrahrou Z, Klopp N, Loley C, Stark K, Hengstenberg C, Bruse P, Freyer J, Wagner AK, Medack A, Lieb W, Grosshennig A, Sager HB, Reinhardt A, Schafer A, Schreiber S, El Mokhtari NE, Raaz-Schrauder D, Illig T, Garlichs CD, Ekici AB, Reis A, Schrezenmeir J, Rubin D, Ziegler A, Wichmann HE, Doering A, Meisinger C, Meitinger T, Peters A, Schunkert H.. Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur Heart J 2011;32:158–168. [DOI] [PubMed] [Google Scholar]
- 21. Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, Giannakopoulou O, Jiang T, Hamby SE, Di Angelantonio E, Assimes TL, Bottinger EP, Chambers JC, Clarke R, Palmer CNA, Cubbon RM, Ellinor P, Ermel R, Evangelou E, Franks PW, Grace C, Gu D, Hingorani AD, Howson JMM, Ingelsson E, Kastrati A, Kessler T, Kyriakou T, Lehtimaki T, Lu X, Lu Y, Marz W, McPherson R, Metspalu A, Pujades-Rodriguez M, Ruusalepp A, Schadt EE, Schmidt AF, Sweeting MJ, Zalloua PA, AlGhalayini K, Keavney BD, Kooner JS, Loos RJF, Patel RS, Rutter MK, Tomaszewski M, Tzoulaki I, Zeggini E, Erdmann J, Dedoussis G, Bjorkegren JLMConsortium E-C; CARDIoGRAMplusC4D Group; UK Biobank CardioMetabolic Consortium CHD Working GroupSchunkert H, Farrall M, Danesh J, Samani NJ, Watkins H, Deloukas P.. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 2017; 49:1385–1391. [DOI] [PubMed] [Google Scholar]
- 22. Akashi M, Higashi T, Masuda S, Komori T, Furuse M.. A coronary artery disease-associated gene product, JCAD/KIAA1462, is a novel component of endothelial cell-cell junctions. Biochem Biophys Res Commun 2011;413:224–229. [DOI] [PubMed] [Google Scholar]
- 23. Hara T, Monguchi T, Iwamoto N, Akashi M, Mori K, Oshita T, Okano M, Toh R, Irino Y, Shinohara M, Yamashita Y, Shioi G, Furuse M, Ishida T, Hirata KI.. Targeted Disruption of JCAD (junctional protein associated with coronary artery disease)/KIAA1462, a coronary artery disease-associated gene product, inhibits angiogenic processes in vitro and in vivo. Arterioscler Thromb Vasc Biol 2017;37:1667–1673. [DOI] [PubMed] [Google Scholar]
- 24. Jones PD, Kaiser MA, Ghaderi Najafabadi M, Koplev S, Zhao Y, Douglas G, Kyriakou T, Andrews S, Rajmohan R, Watkins H, Channon KM, Ye S, Yang X, Bjorkegren JLM, Samani NJ, Webb TR.. JCAD, a gene at the 10p11 coronary artery disease locus, regulates hippo signaling in endothelial cells. Arterioscler Thromb Vasc Biol 2018;38:1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. von Scheidt M, Zhao Y, Kurt Z, Pan C, Zeng L, Yang X, Schunkert H, Lusis AJ.. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab 2017;25:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van der Harst P, Verweij N.. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, Yang T, Wang H, Luscinskas FW, Croce K, Bradner JE, Plutzky J.. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 2014;56:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mosig S, Rennert K, Buttner P, Krause S, Lutjohann D, Soufi M, Heller R, Funke H.. Monocytes of patients with familial hypercholesterolemia show alterations in cholesterol metabolism. BMC Med Genomics 2008;1:60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, Chien S.. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci USA 2016;113:11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S, Ai D, Mak KK, Tong KK, Kwan KM, Wang N, Chiu JJ, Zhu Y, Huang Y.. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016;540:579–582. [DOI] [PubMed] [Google Scholar]
- 31. Xu S, Koroleva M, Yin M, Jin ZG.. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl Res 2016;176:18–28.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ye J, Li TS, Xu G, Zhao YM, Zhang NP, Fan J, Wu J.. JCAD promotes progression of nonalcoholic steatohepatitis to liver cancer by inhibiting LATS2 kinase activity. Cancer Res 2017;77:5287–5300. [DOI] [PubMed] [Google Scholar]
- 33. Htun NM, Chen YC, Lim B, Schiller T, Maghzal GJ, Huang AL, Elgass KD, Rivera J, Schneider HG, Wood BR, Stocker R, Peter K.. Near-infrared autofluorescence induced by intraplaque hemorrhage and heme degradation as marker for high-risk atherosclerotic plaques. Nat Commun 2017;8:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Libby P, Pasterkamp G.. Requiem for the ‘vulnerable plaque’. Eur Heart J 2015;36:2984–2987. [DOI] [PubMed] [Google Scholar]
- 35. Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R.. Update on acute coronary syndromes: the pathologists' view. Eur Heart J 2013;34:719–728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.