


Abstract
With the emergence of genome editing technologies and synthetic biology, it is now possible to engineer genetic circuits driving a cell's phenotypic response to a stressor. However, capturing a continuous response, rather than simply a binary ‘on’ or ‘off’ response, remains a bioengineering challenge. No tools currently exist to identify gene candidates responsible for predetermining and fine-tuning cell response phenotypes. To address this gap, we devised a novel Regulostat Inferelator (RSI) algorithm to decipher intrinsic molecular devices or networks that predetermine cellular phenotypic responses. The RSI algorithm is designed to extract gene expression patterns from basal transcriptomic data in order to identify ‘regulostat’ constituent gene pairs, which exhibit rheostat-like mode-of-cooperation capable of fine-tuning cellular response. Our proof-of-concept study provides computational evidence for the existence of regulostats and that these networks predetermine cellular response prior to exposure to a stressor or drug. In addition, our work, for the first time, provides evidence of context-specific, drug–regulostat interactions in predetermining drug response phenotypes in cancer cells. Given RSI-inferred regulostat networks offer insights for prioritizing gene candidates capable of rendering a resistant phenotype sensitive to a given drug, we envision that this tool will be of great value in bioengineering and medicine.
INTRODUCTION
To survive, a living cell must constantly respond and adapt to extracellular perturbations or stressors – toxins, drugs, heavy metals, heat, and physical forces, capable of inducing cell damage or death. Cellular response phenotypes, or the characteristic traits of cellular responses to diverse types of stressors, range from sensitive to resistant depending on a given stressor. Of note, many cellular response phenotypes, especially those pertaining to adaptive responses, exhibit spectrum-like graded response traits ranging from extremely sensitive to highly resistant phenotypes (1,2) rather than simple on-or-off, all-or-none, or response-or-no-response binary states (3). As such, it is of paramount importance to understand the molecular milieu (i.e. the molecular constituents that makeup the cellular context within cells) in predetermining the extent of cellular response to a stressor.
Although genetic studies have driven current understanding of how genetic factors determine cellular response phenotypes (4–6), emerging evidence indicates that the dynamics of organisms are governed by phenotypic, not genotypic, interactions with environmental selection forces (7). In fact, recent studies indicate that the heterogeneity of cellular response phenotypes is predetermined by the molecular milieu within cells (8,9). The molecular milieu is thought to govern cellular response phenotypes much like defined atomic arrangements in the 3D structure of an antibody molecule, which predetermine its recognition towards an antigen even though the antibody has not previously encountered the antigen (10,11).
Despite the contribution of the molecular milieu in determining cellular response phenotype, conventional bioinformatics tools that rely on differential gene expression (12) and mutation-based approaches (13), rather than network- or systems-based approaches, fail to fully capture it. Although correlation-based methods are powerful approaches to decipher gene–gene associations that are altered under different conditions (14–17), none of these approaches consider changes in mode-of-cooperation (MOC) between genes across the spectrum of cellular response phenotypes, from sensitive to resistant. As a result, they lack the ability to reveal a given cell's intrinsic molecular networks that are ‘pre-built’ to predetermine the extent of response to a stressor.
To address this gap in knowledge, we devised a novel computational algorithm called Regulostat Inferelator (RSI). We hypothesized that in biological systems there exist cooperative genes that operate like rheostats to predetermine and fine-tune how cells respond to a stressor such as a drug. Here, we develop the RSI algorithm to identify networks consisting of cooperative gene pairs that operate like rheostats. We termed these intrinsic molecular devices consisting of rheostat-like gene pair networks that predetermine and fine-tune cellular response phenotype in a dynamic rather than an ‘on’ or ‘off’ manner as ‘regulostats’. We used a systems biology analytical approach on transcriptomic data because non-linear interplay between genetic, epigenetic, and environmental factors can be reflected in the transcriptome. In principle, this approach casts a wider net than genetic-based approaches such as genome-wide association studies (GWAS) and other mutation/variant-centric studies to uncover molecular factors that modulate cellular response to a stressor. Using cancer cells as a proof-of-concept study, we demonstrate RSI is a novel algorithm capable of uncovering rheostat-like gene pairs, the minimum component of regulostats that modulate the extent of phenotypic response from sensitive to resistant or vice versa.
In sum, our analyses provides computational evidence of regulostats in cancer cells and their role in predetermining drug response phenotypes. Furthermore, we demonstrate that the RSI algorithm enables researchers to dissect regulostats capable of predetermining phenotypic responses from different individual cell lines (or individual organisms) to a specific stressor or drug. Such capability may facilitate gene prioritization to engineer cellular phenotypes at the individual cell line or organismal levels. The computed results in this study are provided in a web-based resource using the Shiny package of R at http://rsi.hulilab.org/ and the source code of RSI is freely available for academic use.
MATERIALS AND METHODS
Definitions of concepts and terminology
Given a gene never acts alone but rather cooperates with a number of other genes to exert a particular function, it is therefore pleiotropic (i.e. involved in affecting multiple cellular phenotypes) (18) and nonlinear (i.e. the activity of a gene is not always linearly proportional with one or more genes in all cellular states) (19) in nature. Thus, the activity of a pair of genes (termed a gene pair hereafter) forms the basic functional unit that dictates the phenotype of a cell.
To decipher the molecular machinery that predetermines a cellular response phenotype, it is necessary to identify rheostat-like gene pairs whose collective activities constitute a molecular network that we term a ‘regulostat’ (Figure 1). The fine-tuning of a cellular phenotypic response by a gene pair in a regulostat is analogous to a sophisticated electronic device whose cooperative action among rheostats in the whole system determines the quantity of the generated current, which in turn fine-tunes the quality of the device output (e.g. pitch of the output sound, Figure 1A). In contrast to gene regulatory networks, with hardwired connections between transcriptional regulators to defined regulatory sites on target genes that control cellular phenotypes such as cell fate determination (20–22), the basic units of a regulostat network are composed of gene pairs that do not necessarily directly interact (e.g. indirect regulation via a number of transcriptional or signaling events), but rather exhibit ‘rheostat-like’ mode-of-cooperation (MOC) to fine-tune (Figure 1B and C), instead of turn on or off, the cellular response phenotype to a particular stressor (23).
Figure 1.

The conceptual overview of regulostat networks. (A) Regulostats mimic the workings of a sophisticated electronic device equipped with multiple control sites; the amount of output current at each control site is controlled by the cooperative action among rheostats that dictate the quality of the final output, e.g. the pitch displayed by a speaker. (B) Hypothetical architecture of a regulostat corresponding to a cellular response phenotype A for a given cell type. Relative amounts of gene 1 (g1) to gene 7 (g7), which comprise the regulostat, represented by pointers in the meter. Rheostat gene pairs’ modes-of-cooperation (MOC) is indicated as positive (red) or negative (blue) gene–gene co-expression correlations. Note that associations between the regulostat constituent genes are the same for sensitive (AS) and resistant (AR) phenotypes except for their opposite directionality indicating rheostat-like behavior of these gene pairs in both extreme response phenotypes. (C) Hypothetical individual cell line-specific regulostats representing sensitive (S1 and S2) and resistant (R1 and R2) cell lines harboring only a subset of rheostat-like gene pairs.
Because a regulostat is a molecular network that predetermines a cellular response phenotype, it is therefore our goal to identify gene pairs that constitute a regulostat from basal cellular state (i.e. a state before a stressor is encountered). The basal transcriptome reflects gene activities in terms of gene expression prior to stressor exposure and therefore captures the molecular factors responsible for predetermining cellular response phenotypes. Basal transcriptomic data from cancer cell lines with known phenotypic responses to a specific drug (measured in log-transformed IC50 values) are used in this study to identify rheostat-like gene pairs that constitute a regulostat prior to drug exposure.
As the 3D structure of an antibody molecule dictates its binding specificity to an antigen prior to encountering the antigen, we propose rheostat-like gene pairs inferred from basal transcriptome data (i.e., prior to exposure to a drug) play a role in predetermining drug response phenotypes. While conventional understanding of drug–gene interactions focuses on how genetic variations of a drug target affect the action of drugs (24–26), our novel approach demonstrates it is the network, rather than an individual genetic variation, driving drug response. We show it is the activities of rheostat-like gene pairs, which collectively constitute a regulostat in the basal transcriptome, that shape context-specific interactions between drug and gene pairs, thereby driving drug–network interactions and predetermining drug response phenotypes.
Datasets
Raw microarray data (*.cel files) from 1000 Cell Line (1000CL) data (27) were downloaded from ArrayExpress repository using accession number E-MTAB-3610 (The data can also be downloaded from Gene Expression Omnibus by using accession number GSE36139) generated on Affymetrix Human Genome U133 Plus 2.0 Array. This dataset contains 1001 molecularly annotated human cancer cell lines derived from 29 tissues and correlated with 265 anticancer drugs measured with IC50 values. Affymetrix Human Genome 219 Plate annotation data, as well as RMA method, were used to extract and normalize the intensity values of 18 564 genes (G) as described in our previous study (28).
The regulostat inferelator (RSI) algorithm
Overall design
The RSI algorithm is designed to dissect the repertoire of gene pairs that constitute the molecular network that we termed a regulostat; these gene pairs behave like rheostats in modulating a stressor-induced cellular response phenotype. The basic assumption is that gene pairs are the smallest constituent units of a regulostat. To behave as a ‘rheostat’ in modulating the extent of a cellular response phenotype, a gene pair should exhibit opposite mode-of-cooperation (MOC) measured by gene–gene coexpression correlation coefficients in sensitive and resistant response phenotypes, (e.g. positive coexpression correlation in sensitive phenotypes but negative coexpression correlation in resistant phenotypes). This feature allows a gene pair to ‘flip’ from a positive to negative correlation or vice versa as it tunes the extent of a cellular response to a perturbation. In principle, the RSI algorithm can be applied to infer rheostat-like gene pairs that predetermine any continuous spectrum-like cellular response phenotype, whether the response is concerned with therapeutics or environmental stressors. In this work, we used drug response phenotypes of approximately 1000 cancer cell lines from 1000CL data (27) as illustrative examples. The technical aspects of our RSI algorithm are subdivided into the following 8 stages with a schematic illustration provided in Figure 2 to enhance readability.
Figure 2.

Outline of the Regulostat Inferelator (RSI) algorithm. (A) Deciphering rheostat-like gene pairs for a given drug-cancer case and reconstruction of generic regulostat. k-Mean method is used to categorize cancer cell lines with drug responses measured in IC50 into four bins, ranging from 1 (sensitive) to 4 (resistant) with bins 2 and 3 representing transitional response phenotypes between sensitive and resistant. The data points on XY-coordinate planes from bins 1 to 4 correspond to expression values of genes i and j in cancer cell lines. To obtain gene pairs showing authentic strong gene–gene coexpression correlations, linear regression modelling was performed. Standard error and confidence intervals were computed to determine the fittest regression line. The phenotype flipping coefficient (PLC) was computed from gene–gene coexpression correlation coefficients across bins 1 to 4 for a given gene pair. Gene pairs showing absolute values of PLC > 0.8 were deemed rheostat-like candidates. Positive PLC indicated positive rheostat-like mode-of-cooperation (MOC) between a pair of genes and vice versa for a gene pair showing negative PLC. Rheostat-like gene pairs were subsequently ranked using RSI scores. Internal-assignment recovery rates were determined for the top 2000 ranked gene pairs. The top 200 gene pairs that achieved stable recovery rates in at least 5 out of 1160 drug-cancer cases were used to reconstruct the generic regulostat network. (B) Reconstruction of cancer cell line-specific regulostat that predetermined drug response phenotype. Cancer cell line #1 was used as an illustrative example. Gene–gene coexpression correlation data points of N gene pairs corresponding to cell line #1 are chosen if they show maximum or minimum point on the correlation lines of computed PLCs. The selected gene pairs were used to reconstruct regulostat network corresponding to cell line #1. Red nodes: genes with higher expression values; blue nodes: genes with lower expression values; red edges: gene pairs with positive PLCs; blue edges: gene pairs with negative PLCs. Plots show at bottom right illustrate correlations of gene pairs with data points on the XY-coordinate plane corresponding to expression values of respective genes.
Stage 1: Assignment of cells to their respective drug response phenotypes
Given a human cancer type 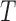 with m cell lines (CLs),
with m cell lines (CLs), 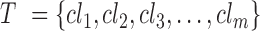 , where responses of CLs for n different anticancer drugs/compounds (given in log-transformed IC50),
, where responses of CLs for n different anticancer drugs/compounds (given in log-transformed IC50), 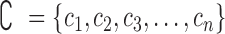 , one can cluster CLs into k different bins based on log-transformed IC50 values. k-Mean clustering, a multivariate method that is usually used in unsupervised machine learning problems, can be applied to cluster univariate data such as IC50 scores in this study. The minimum number of clusters (or bins) that linearly recapitulate the transition from sensitive to resistant phenotypes is 4, with bins 1 and 4 representing sensitive and resistant phenotypes respectively, and bins 2 and 3 representing transitional phenotypes between the two extreme phenotypes (see Figure 2A). The default parameter k is set as 4 and
, one can cluster CLs into k different bins based on log-transformed IC50 values. k-Mean clustering, a multivariate method that is usually used in unsupervised machine learning problems, can be applied to cluster univariate data such as IC50 scores in this study. The minimum number of clusters (or bins) that linearly recapitulate the transition from sensitive to resistant phenotypes is 4, with bins 1 and 4 representing sensitive and resistant phenotypes respectively, and bins 2 and 3 representing transitional phenotypes between the two extreme phenotypes (see Figure 2A). The default parameter k is set as 4 and 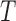 is therefore divided into four discrete bins as follows:
is therefore divided into four discrete bins as follows:
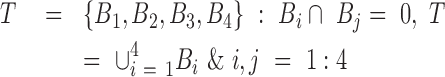 |
(1) |
In order to obtain more robust gene–gene coexpression correlation coefficients, the minimum number of cell lines within each bin for a given drug-cancer case (e.g. FK866-SBC-3 refers to treatment of drug FK866 on SBC-3 cancer cell line) is set to 4 (see Stage 2). This is because the results of coexpression correlation are sensitive to the number of data points (samples). In particular, reducing the number of data points of well correlated gene pairs might yield a poorly correlated outcome (Supplementary Figure S1). We therefore used a minimum of four data points in each bin as a default parameter/matter of standardization for a more stable result. However, we recommend a higher number of data points whenever a large sample size is available. Out of 3840 drug-cancer cases in the 1000CL data, 1169 cases had at least four cell lines within each bin that satisfied our criterion (Supplementary Data 1). Given any gene pair 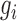 and
and 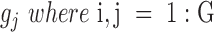 , there are at least four data points, with one data point corresponding to the gene expression values of a gene pair in a drug-cancer case.
, there are at least four data points, with one data point corresponding to the gene expression values of a gene pair in a drug-cancer case.
Stage 2: Computing gene–gene coexpression correlation coefficients in each bin
While randomly scattered data points on the XY-coordinate plane show no relation between two variables, centralized points around a linear line with a slope close to ±1 on the XY-coordinate plane illustrate strong positive/negative correlation between two variables. In this study, for any gene pair 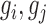 , and x and y as their gene expression values on XY-coordinate plane, where
, and x and y as their gene expression values on XY-coordinate plane, where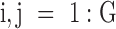 over
over 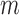 cell lines in
cell lines in 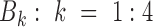 (i.e.
(i.e. 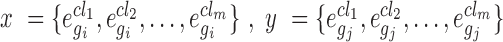 and
and 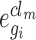 is the gene expression value of
is the gene expression value of 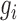 in mth cell line in bin
in mth cell line in bin 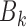 ). Pearson correlation (equation 2) and linear regression coefficients (equation 3) for the fitted model on x and y was computed as follows:
). Pearson correlation (equation 2) and linear regression coefficients (equation 3) for the fitted model on x and y was computed as follows:
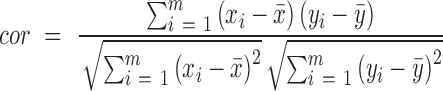 |
(2) |
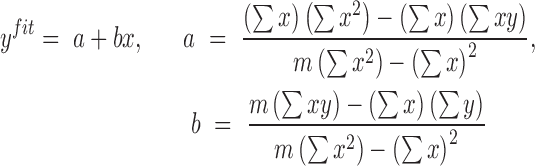 |
(3) |
Here, a linear regression model is applied to select authentic gene pairs that behaved linearly along the correlation line, as shown in Figure 2A. This is because a high correlation coefficient between two variables might have poor distribution of data points along the correlation line. To select those gene pairs that are strictly (or closely) aligned with the correlation lines, the Standard Error of the Estimate (SEE) for the fitted regression lines as well as 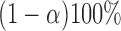 confidence interval (CI) for the slope of regression lines in bin
confidence interval (CI) for the slope of regression lines in bin  (sensitive) and bin
(sensitive) and bin  (resistant) were computed:
(resistant) were computed:
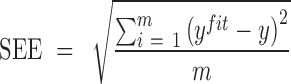 |
(4) |
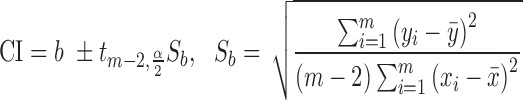 |
(5) |
The above mentioned scores were computed for all gene–gene pairwise combinations, resulting in 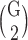 possible pairs out of a total of G = 18 564 genes. Additional scores such as the absolute values of differences of correlations in sensitive and resistant bins,
possible pairs out of a total of G = 18 564 genes. Additional scores such as the absolute values of differences of correlations in sensitive and resistant bins, 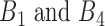 , and k bins centers were also computed by the k-mean algorithm.
, and k bins centers were also computed by the k-mean algorithm.
Stage 3: Computing phenotype flipping coefficients (PLCs) to infer rheostat-like gene pairs
As described above, a rheostat-like gene pair shows opposite modes-of-cooperation (MOC) in sensitive and resistant phenotypes in response to a perturbation. Because MOC of a gene pair is computed in a gene–gene coexpression coefficient, we termed changes of gene–gene coexpression coefficients over bins 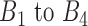 as ‘phenotype flipping coefficients’ (PLC) in the range of −1 to 1. Each gene pair has its own PLC score as well as other scores as described in Stage 2. The PLC for a gene pair was computed as follows:
as ‘phenotype flipping coefficients’ (PLC) in the range of −1 to 1. Each gene pair has its own PLC score as well as other scores as described in Stage 2. The PLC for a gene pair was computed as follows:
For a given a gene pair under the default 4-bin scenario, suppose corB1, corB2, corB3 and corB4 are the coexpression correlations of a given gene pair over bin 1 to bin 4, and cB1, cB2, cB3 and cB4 are the centers of four respective bins computed by k-mean, the PLC is the correlation of those correlation scores and bins center:
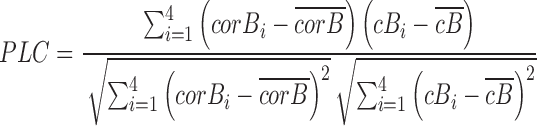 |
(6) |
where 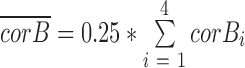 and
and 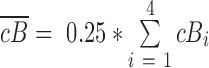 .
.
Stage 4: Filtering rheostat-like gene pairs
There are 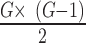 possible gene pairs of G genes to be considered. In this study, there are 18 564 gene expressions over 1018 cell lines with
possible gene pairs of G genes to be considered. In this study, there are 18 564 gene expressions over 1018 cell lines with 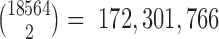 possible pairwise gene combinations. A filtering threshold is therefore needed in order to limit the final number of selected gene pairs acting as rheostat-like pairs. The filtering steps were applied as follows: First, gene pairs with correlation coefficients showing opposite signs in bin 1 and bin 4, and absolute values of PLC greater than 0.8 were kept. The reason for applying this criterion was to select rheostat-like gene pairs showing opposite MOC in sensitive and resistant phenotypes. Second, gene pairs with correlation coefficients across bins
possible pairwise gene combinations. A filtering threshold is therefore needed in order to limit the final number of selected gene pairs acting as rheostat-like pairs. The filtering steps were applied as follows: First, gene pairs with correlation coefficients showing opposite signs in bin 1 and bin 4, and absolute values of PLC greater than 0.8 were kept. The reason for applying this criterion was to select rheostat-like gene pairs showing opposite MOC in sensitive and resistant phenotypes. Second, gene pairs with correlation coefficients across bins 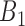 to
to 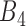 that do not follow linear ascending or descending order (e.g. when a pair's smallest correlation score is in
that do not follow linear ascending or descending order (e.g. when a pair's smallest correlation score is in 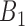 , then its correlation in
, then its correlation in 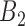 must be smaller than
must be smaller than 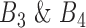 , and its correlation in
, and its correlation in 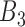 , must be smaller than
, must be smaller than 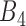 and so on, see also Figure 2A) were removed. This criterion ensures the selected rheostat-like gene pairs show linear change of coexpression correlation coefficients across bins
and so on, see also Figure 2A) were removed. This criterion ensures the selected rheostat-like gene pairs show linear change of coexpression correlation coefficients across bins 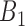 to
to 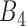 . Gene pairs are subsequently filtered based on their SEE (equation 4) and in-bound (IB) scores in
. Gene pairs are subsequently filtered based on their SEE (equation 4) and in-bound (IB) scores in 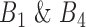 , where IB is the percent of cell lines (data points) between confidence interval CI (equation 5) bounds divided by the total number of cell lines in that bin. This step is to ensure only gene pairs showing close fitting of data points to the correlation line are selected. Those pairs with SEE scores above the third quantile (mean) and IB scores less than third quantile of all SEE scores in that bin were discarded.
, where IB is the percent of cell lines (data points) between confidence interval CI (equation 5) bounds divided by the total number of cell lines in that bin. This step is to ensure only gene pairs showing close fitting of data points to the correlation line are selected. Those pairs with SEE scores above the third quantile (mean) and IB scores less than third quantile of all SEE scores in that bin were discarded.
Stage 5: Computing the RSI Score and ranking rheostat-like gene pairs
Each gene pair has its own RSI score, which itself is composed of scores described in Stage 2 together with PLC scores described in Stage 3. All scores have roughly normal distributions but in different ranges. To normalize and standardize these scores, all scores were divided by their corresponding maximum values to transform them into a common range of [0, 1]. The RSI score for each rheostat-like gene pair is the unweighted sum of IB, 1/SEE scores in 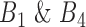 , plus the PLC and the absolute value of the difference between correlations in
, plus the PLC and the absolute value of the difference between correlations in 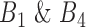 for the given gene pair as follows: RSI score = Σ[IB + 1/SEE + PLC + abs(cor Bin1 – cor Bin4)]. Finally, these rheostat-like gene pairs were ranked based on their RSI scores and the top
for the given gene pair as follows: RSI score = Σ[IB + 1/SEE + PLC + abs(cor Bin1 – cor Bin4)]. Finally, these rheostat-like gene pairs were ranked based on their RSI scores and the top 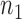 pairs were selected. In this study,
pairs were selected. In this study, 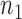 was set to be 2000.
was set to be 2000.
Stage 6: Assessing the recovery rates of RSI-inferred gene pairs comprising regulostats
We reason that if a gene pair is a bona fide rheostat-like pair, in principle, its coexpression profile should be able to recover (or retrieve) the specific drug phenotypic response in a cell line. The percentage of cell lines whose drug response phenotype is correctly recovered by a given gene pair is defined as the recovery rate and is assessed via internal accuracy of correctly classifying a given cell line according to its specific drug phenotypic response based on the IC50 value of a drug. The computation procedure for the recovery rate is performed for each drug-cancer case one at a time to evaluate the capability of rheostat-like gene pairs to modulate response phenotype for a given drug on cell lines derived from a particular cancer type. To provide the highest internal-assignment recovery rate, we condensed 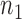 gene pairs to
gene pairs to 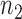 (
(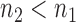 ) gene pairs. Our results for more than 1169 drug-cancer cases revealed that ∼200 gene pairs provided a stabilized recovery rate, where adding additional gene pairs does not further improve nor deteriorate the recovery rate.
) gene pairs. Our results for more than 1169 drug-cancer cases revealed that ∼200 gene pairs provided a stabilized recovery rate, where adding additional gene pairs does not further improve nor deteriorate the recovery rate.
To obtain the minimal number of gene pairs with stable recovery rate performance, 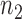 was set to be 200 in this study. To select top
was set to be 200 in this study. To select top 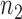 pairs, linear regression (lm) lines, with at least four data points (i.e. cell lines) corresponding to each gene pair, were used. Given a gene pair and its corresponding lm lines, the distance of all cell lines from those lm lines was computed with the expectation that a sensitive cell line should exhibit the smallest distance from lm line in
pairs, linear regression (lm) lines, with at least four data points (i.e. cell lines) corresponding to each gene pair, were used. Given a gene pair and its corresponding lm lines, the distance of all cell lines from those lm lines was computed with the expectation that a sensitive cell line should exhibit the smallest distance from lm line in 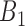 (Recall that a cell line is a point on XY-plane where its x and y coordinates is the expression values of
(Recall that a cell line is a point on XY-plane where its x and y coordinates is the expression values of 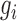 and
and 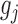 respectively). Similar criterion also applied for resistant cell lines in
respectively). Similar criterion also applied for resistant cell lines in 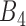 . The first pair in
. The first pair in 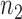 pairs is the one that provides the highest recovery rate, followed by second, third and the 200th pair added to the final list in a way that every time the recovery rate must be either improved or stay the same when adding a new gene pair to the list. To demonstrate how the maximal stable recovery rate is calculated, we provide a specific example in the Supplementary File.
pairs is the one that provides the highest recovery rate, followed by second, third and the 200th pair added to the final list in a way that every time the recovery rate must be either improved or stay the same when adding a new gene pair to the list. To demonstrate how the maximal stable recovery rate is calculated, we provide a specific example in the Supplementary File.
Stage 7: Constructing the generic regulostat network
The 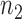 gene pairs that achieved a stable recovery rate in Stage 6 were then plotted in network form with nodes of the graph representing genes and the edge colors between the nodes were determined by PLC values. R packages igraph v1.1.2 and RCy3 1.0.1, as well as the Cytoscape 3.5.1 network visualization tool, were used to reconstruct the regulostat networks. For each drug-cancer case, there is a list of
gene pairs that achieved a stable recovery rate in Stage 6 were then plotted in network form with nodes of the graph representing genes and the edge colors between the nodes were determined by PLC values. R packages igraph v1.1.2 and RCy3 1.0.1, as well as the Cytoscape 3.5.1 network visualization tool, were used to reconstruct the regulostat networks. For each drug-cancer case, there is a list of 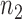 pairs which provides the highest internal recovery rate for a given drug-cancer case. To reconstruct the generic (i.e. common) regulostat network corresponding to a given cancer type of interest across all drugs, a set of gene pairs that are common in at least 5 different drug-cancer cases among
pairs which provides the highest internal recovery rate for a given drug-cancer case. To reconstruct the generic (i.e. common) regulostat network corresponding to a given cancer type of interest across all drugs, a set of gene pairs that are common in at least 5 different drug-cancer cases among  lists were selected.
lists were selected.
Stage 8: Reconstructing drug-cell line-specific regulostat networks
A modified network reconstruction approach as described in Stage 7 was used to obtain drug-cell line-specific regulostat networks. Given a cell line of interest with respect to a drug, e.g.  , from the
, from the  list of corresponding drug-cancer cases, a set of rheostat-like gene pairs in which at least one of the genes in each pair has the maximum or minimum coexpression point in comparison to all other cell lines was selected (see Figure 2B).
list of corresponding drug-cancer cases, a set of rheostat-like gene pairs in which at least one of the genes in each pair has the maximum or minimum coexpression point in comparison to all other cell lines was selected (see Figure 2B).
Assessment of the performance of the RSI algorithm
We designed the following test schemes to assess the performance of the RSI algorithm. Here, we first chose drug-cancer cases with at least 4 cell lines assigned by k-mean with k = 4, 6 or 8 depending on a 4-, 6- or 8-bin scenario. Our data survey indicated that 15 drug-cancer cases fulfilled this criterion (Supplementary Table S1 in the Supplementary File). These 15 drug-cancer cases were used as standard test data for all test schemes described below.
Test Scheme 1: Assess the effects of bin number on detecting the presence of rheostat-like gene pairs
Since computing phenotypic flipping coefficients (PLC) involves evaluating changes in gene pair coexpression coefficients across different bins, increasing the number of bins to more than four might affect whether rheostat-like gene pairs can still be observed. As such, we expanded our studies to include 6- and 8-bin scenarios, with bin 1 always containing sensitive cell lines and bin 6 (for 6-bin scenario) and bin 8 (for 8-bin scenario) always containing resistant cell lines. The intermediate bins were those that showed transitional changes between a sensitive and resistant phenotype. We used the RSI algorithm to perform studies of the selected 15 drug-cancer cases with 6- and 8-bin scenarios.
Test Scheme 2: Permutation tests to assess the robustness of the identified rheostat-like gene pairs
To determine whether rheostat-like gene pairs identified in the default 4-bin scenario are robust with respect to real data, we devised the following two permutation strategies:
Permutation strategy 1: Permutate the cell line labels, which had the same effect as permutating the IC50 values
Permutation strategy 2: Permutate the gene names
We performed 100 permutations for each strategy using the selected 15 drug-cancer cases as test data. The top 200 rheostat-like gene pairs identified from each of these permutation tests were used to recover cancer cell lines from real data according to their respective specific drug phenotypic response.
Test Scheme 3: Evaluate the performance of RSI score and its derived scoring schemes
We assessed the performance of our original RSI scoring scheme (Stage 5 of the RSI algorithm) in comparison to nine other modified versions:
Original RSI score = Σ[IB + 1/SEE + PLC + abs(cor Bin1 – cor Bin4)]
Scheme 1: RSI score = Σ[1/SEE + PLC + abs(cor Bin1 – cor Bin4)]
Scheme 2: RSI score = Σ[IB + PLC + abs(cor Bin1 – cor Bin4)]
Scheme 3: RSI score = Σ[IB + 1/SEE + abs(cor Bin1 – cor Bin4)]
Scheme 4: RSI score = Σ[IB + 1/SEE + PLC]
Scheme 5: RSI score = log[(IB)*(1/SEE)*(PLC)*abs(cor Bin1 – cor Bin4)]
Scheme 6: RSI score = log[(1/SEE)*(PLC)*abs(cor Bin1 – cor Bin4)]
Scheme 7: RSI score = log[(IB)*(PLC)*abs(cor Bin1 – cor Bin4)]
Scheme 8: RSI score = log[(IB)*(1/SEE)*abs(cor Bin1 – cor Bin4)]
Scheme 9: RSI score = log[(IB)*(1/SEE)*(PLC)]
Computations were performed for the selected 15 drug-cancer cases, and the top 100 rheostat-like gene pairs ranked by each of these modified RSI scoring schemes was compared to those of the original RSI scoring scheme. The top 100 rheostat-like gene pairs from each modified RSI scoring scheme were also used to assess drug response phenotype recovery rates for the 15 drug-cancer cases.
Test Scheme 4: Assess the effect of sample size on PLC and RSI scores
To determine to what extent changing sample size might affect the distribution of PLC and RSI scores, we employed N-fold tests akin to N-fold cross-validation tests used in machine learning methods. We tested the effect of PLC and RSI score distribution by reducing the selected 15 drug-cancer cases to 90%, 80%, and 50% of their original data size using the default 4-bin scenario and original RSI scoring scheme. 10-fold, 5-fold and 2-fold tests were performed. Here, the 5-fold test for the SB-715992-LUNG case is used as an illustrative example. The number of data points (cell line samples) for this drug-cancer case in bins 1 to 4 is as follows: Bin 1 [45], Bin 2 [44], Bin 3 [37], and Bin 4 [48]. For the first fold (fold 1), 20% of cell lines in each bin were randomly removed and the remaining 80% of cell lines were subjected to the RSI algorithm where both PLC and RSI scores were computed. The whole process was repeated 4 more times (folds 2–5) by returning the previously removed 20% cell lines back to their respective bins and randomly removing another 20%-set of cell lines from each bin. The whole procedure was the same for 10-fold and 2-fold tests, with 10% and 50% of data points (cell lines) respectively removed from each bin.
Test Scheme 5: Evaluate the overall performance of the RSI algorithm via cross-validation tests
Here, we used the 15 selected drug-cancer cases with 50% (2-fold cross-validation), 80% (5-fold cross validation) and 90% (10-fold cross-validation) of data for training the model and the remaining 50%, 20% and 10% as unseen data, respectively. Similarity of data features (i.e. gene pairs) selected at different folds during cross-validation is a general measure of the robustness of prediction. Here, Tanimoto distance (29) that measures similarity between two subsets of selected gene pairs X and Y from two respective folds from N-fold cross-validation (where N is 10, 5 and 2) is used:
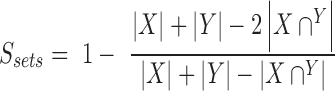 |
In each fold, ∼2000–2500 gene pairs are selected from 172,301,766 possible gene pairs. Similarities of gene pairs over different training sets on different folds are compared. Using the recovery rate computational procedure, we next compared the internal accuracy obtained from the whole dataset with the accuracy obtained for 10-, 5- and 2-fold cross-validations on these 15 drug-cancer cases.
Pathway enrichment analysis
KEGG canonical pathway enrichment analysis was performed using WebGestaltR v 0.1.1 R package (30) with gene symbol as input gene ID type against all human genes. This analysis included 924 genes most frequently found as a component of 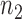 top rheostat-like gene pairs in at least 50 out of 1169 drug-cancer cases (Supplementary Data 2). Default parameters using hypergeometric overlap statistics, BH multiple test adjustment, and significance level with P-value < 0.05 are deemed enriched from this 924-gene list. Similar criteria are used for enrichment analysis of genes residing in a generic regulostat.
top rheostat-like gene pairs in at least 50 out of 1169 drug-cancer cases (Supplementary Data 2). Default parameters using hypergeometric overlap statistics, BH multiple test adjustment, and significance level with P-value < 0.05 are deemed enriched from this 924-gene list. Similar criteria are used for enrichment analysis of genes residing in a generic regulostat.
Analysis on the proportion of pooled regulostat-constituent genes with molecular functions pertaining to drug response phenotype and drug targets
The top 200 gene pairs identified using recovery rate procedure for regulostat networks across 1169 drug-cancer cases were pooled together. Genes that occurred in only one drug-cancer case were removed; the resulting 3366 genes were designated pooled regulostat-constituent genes. The selected molecular functions pertaining to drug response phenotypes in cancer cells were evaluated from the following databases: transcription factors (http://www.tfcheckpoint.org/), kinases (https://www.uniprot.org/), metabolic enzymes (https://www.genome.jp/kegg/), drug metabolizing enzymes (https://www.genome.jp/kegg/), transporters (http://www.tcdb.org/), cell cycle (https://www.genome.jp/kegg/), DNA repair (https://www.mdanderson.org/documents/Labs/Wood-Laboratory/human-dna-repair-genes.html), apoptosis (https://www.genome.jp/kegg/), and cellular stress (http://software.broadinstitute.org/gsea/msigdb/index.jsp). In addition, the proportion of known drug targets (http://www.broadinstitute.org/repurposing) in the selected top 200 gene pairs was also assessed. Full lists of genes encoding these functional categories are provided in Supplementary Data 3. 5 × 105 permutations of randomized gene sets with equal size of pooled regulostat-constituent genes (i.e. 3366 genes) were generated followed by Fisher's exact test (31) where significance of over-representation for each functional category can be computed. The reference gene size used is 18564.
Computing chemical similarity of drugs
The PubChem 881-bit substructure molecular fingerprint (ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt) were calculated for each compound (Supplementary Data 8) using the cdk package version 1.5.11 (https://pubs.acs.org/doi/10.1021/ci025584y). Fingerprints for these compounds were clustered using hierarchical clustering method with Tanimoto distance (29) and complete linkage.
Clustering of regulostat-constituent gene pairs vs. drug-cancer cases
A subset of 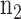 lists of 1169 drug-cancer cases, which achieve a stable internal-assignment recovery rate common among at least five different drug-cancer cases, were selected for clustering analysis. The clustering analysis was performed by using hclust function of stat package in R, which uses the complete linkage method for hierarchical clustering with default parameters.
lists of 1169 drug-cancer cases, which achieve a stable internal-assignment recovery rate common among at least five different drug-cancer cases, were selected for clustering analysis. The clustering analysis was performed by using hclust function of stat package in R, which uses the complete linkage method for hierarchical clustering with default parameters.
WEB-BASED RESOURCE AND SOURCE CODE
We developed a web-based resource using the Shiny package of R to enable researchers to explore and visualize our computed results (http://rsi.hulilab.org/). Detailed descriptions of this web-based resource's features are provided in the Supplementary File and the online web-based tutorial. The RSI source code is freely available to academic researchers.
RESULTS
Deciphering regulostat-constituent gene pairs that predetermine drug response phenotypes in cancer cell lines
As proof-of-concept examples for our algorithm, we used 1000 Cell Line (1000CL) data containing 1001 molecularly annotated human cancer cell lines with drug response data corresponding to 265 anti-cancer drugs (27). The RSI algorithm first assigns cells into four consecutive bins, or clusters, via k-mean method according to their response phenotypes (measured in log-transformed IC50 values). Bins 1 and 4 correspond to sensitive and resistant phenotypes at either ends of the spectrum, with bins 2 and 3 representing transitional response phenotypes in between. We requested that at least 4 different cell lines be presented in each bin to compute gene–gene coexpression correlation coefficients, which are necessary to ensure a robust result. The final, cleaned up data set that meets our criteria contains a total of 1169 drug-cancer cases (Supplementary Data 1).
We computed the flipping of coexpression correlations for a gene pair across two extreme phenotypic ends as the phenotype flipping coefficient (PLC) (Figure 2A and Materials and Methods RSI algorithm, Stages 2 to 4). Gene pairs with absolute values of PLCs greater than 0.8 were deemed to be potential rheostat-like units of a regulostat for a given cellular phenotype. A linear regression model was then applied to select authentic, strongly coexpressed gene pairs with coexpression profiles close to correlation lines for sensitive (bin 1) and resistant (bin 4) phenotypes (Figure 2A and Materials and Methods RSI algorithm, Stages 2). The standard error of the estimate for the fitted regression lines and confidence interval for the slope of regression lines in bins 1 and 4 were then computed. Under these selection criteria, the resulting filtered gene pairs exhibited strong but opposite coexpression correlations in sensitive (bin 1) and resistant (bin 4) phenotypes, but only showed weak coexpression correlations across transitional phenotypes (bins 2 and 3) (Figure 2A). Gene pairs showing MOC characterized by such flipping of coexpression correlations across sensitive (bin 1) to resistant (bin 4) phenotypes are referred to as ‘rheostat-like gene pairs’.
Recovery rates of top rheostat-like gene pairs reveal evidence of regulostats in modulating drug responses in cancer cells
Using RSI scores of the top 2000 ranked gene pairs, we examined to what extent these gene pairs can recover a cancer cell line's phenotypic response to a specific drug, given that drug's corresponding IC50 value. A step-by-step procedure to compute the recovery rate with an illustrative example is provided in the Supplementary File. We assessed the recovery rate, or percentage of cell lines correctly recovered to their drug response phenotypes. We found ∼200 gene pairs were capable of achieving stable maximal recovery rates out of 1169 drug-cancer cases (Figure 3A and B) and across tissue-specific cases (Figure 4A). As shown in Figure 3A and B, the overall maximal recovery rates for all 1169 drug-cancer cases are between 60 and 80%, which is comparable with drug response recovery rates corresponding to each cancer type (Figure 4A). The recovery rate for whether a gene pair can correctly recover a specific drug phenotypic response in a cell line is evaluated based on the distance between expression profiles of genes constituting a rheostat-like gene pair to the lm line in each bin. As such, the more cell lines of a given cancer type that can be recovered to the known drug response phenotype (including both sensitive and resistant cells), the stronger the relatedness of this gene pair in acting as a ‘rheostat-like’ switch in determining the extent of drug response. Although at this stage RSI does not quantify rheostat-like gene pairs in terms of recovery for both sensitive and resistant cell lines, the recovery rate can nonetheless be perceived as an approximate measure for the involvement of a gene pair in predetermining and fine-tuning drug response phenotype in a given cancer type. Our study indicates that the top-ranked 200 rheostat-like gene pairs are constituents of regulostats and their coexpression profiles can recover known phenotypic responses specific to a given drug in cancer cell lines.
Figure 3.

Assessment and characterization of RSI-inferred rheostat-like gene pairs for their potential constituency in a regulostat. (A) Assessment of capabilities of RSI-inferred rheostat-like gene pairs to recover cell lines with known drug response phenotypes across 1169 drug-cancer cases by means of internal-assessment recovery rate. Majority of drug-cancer cases show stable maximal recovery rates at the range of 60–80%. (B) Boxplot for the overall recovery rate across 1169 drug-cancer cases. (C) Top 40 most commonly observed regulostat-constituent genes across 1169 drug-cancer cases. Frequency indicates the number of times a given gene presents in a rheostat-like gene pair across the 1169 drug-cancer cases. (D) Enrichment analysis for canonical pathways of 924 regulostat-constituent genes that are most frequently found in at least 50 out of 1169 drug-cancer cases (frequency ≥ 50).
Figure 4.

Assessment and characterization of RSI-inferred rheostat-like gene pairs for their potential constituency in a regulostat derived from specific tissue of origin. (A) Boxplot for the overall recovery rate across drug-cancer cases correspond to specific tissue of origin by means of internal-assessment recovery rate. (B) Top 15 most commonly observed regulostat-constituent genes for cancer cells derived from specific tissue of origin. Frequency indicates the number of occurrences a given gene appears as a constituent of rheostat-like gene pairs across a particular cancer type.
Assessment of the RSI algorithm via different test schemes revealed robust existence of rheostat-like gene pairs in predetermining drug response phenotypes
We sought to determine whether these rheostat-like gene pairs that recovered known drug response phenotypes in cell lines are indeed gene candidates within a regulostat that predetermine cellular response phenotypes. First, we evaluated how changing the bin number—from 4 to 6 and 8—affected the conclusion for the existence of rheostat-like gene pairs. As shown in Supplementary Figure S2A and B, we found cancer cell lines in the 6- and 8-bin scenarios exhibit drug response phenotype recovery rates comparable to the 4-bin scenario. Such findings demonstrate changing bin numbers from 4 to 6 or 8 does not negate the conclusion for the existence of regulostats in predetermining drug response phenotypes. Supplementary Figure S3 illustrates PCBP4-ELK3 as an example of such a rheostat-like gene pair using 4-, 6- and 8-bin scenarios.
To assess the robustness of the identified rheostat-like gene pairs in recovering specific drug phenotypic responses in cancer cell lines, we tested the top 200 gene pairs identified using two different permutation schemes. The rationale for our approach was that if the top-ranked rheostat-like gene pairs inferred from real data are indeed biologically relevant to drug response phenotypes, they should exhibit good recovery rates in identifying cancer cell lines with defined drug response phenotypes compared with gene pairs inferred from permutated data. Supplementary Figure S4A shows substantially better recovery rates of gene pairs inferred from real data compared with gene pairs identified from permutations on cell line labels (equivalent to permutating the IC50 values), indicating biological relevance of rheostat-like gene pairs inferred from real data in predetermining drug response phenotypes. However, gene pairs selected from gene name permutated data showed comparable recovery rates with gene pairs inferred from real data (Supplementary Figure S4B). This is because in the scenario of permutating gene names, the same corresponding pairs with rheostat-like MOC inferred from real data will always be identified except only the names of gene pairs are different, therefore the selected pairs from permutated data always exhibit comparable recovery rates with gene pairs inferred from real data. In summary, our permutation tests showed that the identified rheostat-like gene pairs from real data are constituents of regulostats that are relevant in predetermining drug response phenotypes in cancer cell lines.
To evaluate how modifications of RSI scoring schemes affect the ranking of rheostat-like gene pairs, we examined how the rank order of the top 100 gene pairs are affected by each of nine modified RSI scoring schemes (see Materials and Methods). As shown in Supplementary Figure S5, we found Schemes 3, 4, 5, 8 and 9 in general show high linear correlations to the top 100 rheostat-like gene pairs identified using the original RSI scoring scheme. The consistency of gene pair ranking correlation between the original and Scheme 5 indicates that both additive and multiplicative schemes rank top gene pairs equally well. Interestingly, our results suggest excluding PLC and absolute values of correlation coefficients between Bins 1 and 4 had no major impact on the ranking of rheostat-like gene pairs as with the original RSI scoring scheme. We reason that this is due to the small value differences these two terms contribute to affect the ranking of rheostat-like gene pairs. In comparing the recovery rates of the top 100 gene pairs identified by each of the nine modified RSI scoring schemes to the original RSI scoring scheme, we found the original RSI scoring scheme exhibits the best recovery rates for 7, and near best recovery rates for 4, out of 15 drug-cancer case (Supplementary Figure S6). Our results therefore suggest that the original RSI scoring scheme performs well in identifying top-ranked rheostat-like gene pairs that contribute to predetermining drug response phenotypes.
To test how changing the sample size can affect the distribution of PLC and RSI scores, we designed N-fold tests akin to N-fold cross-validation tests used in machine learning methods (see Materials and Methods). Using SB-715992-LUNG cancer case as an illustrative example (Supplementary Figure S7), we found comparable RSI score distribution for all of the tested folds (10-, 5-, 2-fold performed at 90%, 80% and 50% of original data size; see Supplementary Data 4 for all tested 15 drug-cancer cases). These results suggest that the computed RSI scores are robust, at least up to a 50% reduction of the original sample size.
Finally, we evaluated the overall performance of the RSI algorithm in detecting rheostat-like gene pairs of a regulostat via cross-validation tests. We employed 10-, 5- and 2-fold cross-validations on the selected 15 drug cancer cases. In each fold ∼2000–2500 gene pairs were selected from 172,301,766 possible gene pairs. Similarity heatmaps for the identity of ∼2000–2500 gene pairs selected from each of these folds using Tanimoto distance are provided in Supplementary Data 5. Due to the large number of possible gene pair combinations (172,301,766 gene pairs), the probability of selecting any random set size of 2500 pairs out these possible combinations will be: 100 × (2500/172301766) ∼ 0.001451%. Thus, the probability of selecting exactly two similar sets out of 10-fold cross-validation is almost 0. The worst performance is at the 2-fold cross-validation with similarity distance 0.09, with P-value <1.0e-6 computed from 1,000,000 permutation test indicates statistical significance in finding rheostat-like gene pairs in each of these folds.
Using the recovery rate computation procedure, we then assessed the RSI performance by comparing the internal accuracy (i.e. recovery rate obtained by using whole dataset) with the accuracies obtained from 10-, 5- and 2-fold cross-validation tests. Plots for performance of each cross-validation fold corresponding to the 15 drug-cancer cases are provided in Supplementary Data 6. Supplementary Figure S8A–C summarizes the distribution of accuracies cumulated at selected top 200 gene pairs that recovered known drug response phenotypes in cell lines for 10-, 5- and 2-fold cross-validation tests, respectively. As shown in Supplementary Figure S8, both internal and cross-validation tests show high distribution of accuracies greater than 80% of performance for 10-, 5- and even 2-fold cross-validations, indicating that the top 200 gene pairs selected by the RSI algorithm are indeed rheostat-like gene candidates involved in predetermining drug response phenotypes of cancer cells.
Although lower fold cross-validations using a smaller number of samples for training processes often yield lower performance rates, we investigated whether the drop of internal accuracies in lower fold cross-validations, in particular 2-fold cross-validations, may be due to specific drug signals embedded in the selected 15 drug-cancer validation models. Since our cross-validations were performed for one drug-one cancer type for a total of 15 selected drug-cancer cases (i.e. 15 drug-cancer models with respect to 2-, 5- and 10-fold cross-validations), surveying the change of internal accuracies across different folds of cross-validations based on drug similarity might provide clues for the existence of specific drug signals. We therefore performed drug similarity analysis on 15 drugs whose chemical structures (in SMILES formats) are available from PubChem and HMS LINCS DB (Supplementary Data 7 and 8). Chemical similarity of these 15 drugs was computed using PubChem 881-bit substructure molecular fingerprint and clustered based on Tanimoto distance (29) and the result is represented as a dendogram in Supplementary Figure S9A. Heatmaps for average internal accuracies for 15 drug-cancer cases corresponding to 2-, 5- and 10-fold cross-validations was then organized based on chemical similarities (Supplementary Figure S9B-D). As shown in Supplementary Figure S9A, drugs within sister branches of mitomycin C (13 drugs including mitomycin C) in general show high average internal accuracies in 10-fold cross-validations (>93%) but the average internal accuracies generally drop in lower fold cross-validations and 5 of these drugs drop to <80% of average internal accuracies at 2-fold cross validations. However, IPA-3 and elesclomol, which belong to different chemical families than mitomycin C and its sister branches exhibit much more stable internal accuracies across all 2-, 5- and 10-fold cross-validations. The finding that specific drug signals affect internal accuracies in lower fold cross-validation models indicates the existence of drug–network interactions, which warrant further investigation.
Frequently observed regulostat-constituent genes are key components in cancer-associated pathways
Having uncovered rheostat-like gene pairs for each drug-cancer case, we sought to identify the top 40 most frequently observed genes comprising the pairs across 1169 drug-cancer cases (Figure 3C). Closer inspection of the biological functions played by these top 40 genes revealed striking functional relatedness to the hallmarks of cancer progression and survival (32) (Supplementary Data 9). Further enrichment analysis for canonical pathways involving 924 genes most frequently observed as a component of the top 200 rheostat-like gene pairs in at least 50 out of 1169 drug-cancer cases (frequency ≥ 50) (Supplementary Data 2) also revealed striking relatedness to cancer-associated pathways (Figure 3D). Of particular interest, the Hippo signaling pathway, which is responsible for controlling organ size during development via regulation of cell proliferation and apoptosis and has been found to be elevated in liver cancer (33), was among the top enriched pathways. Yes-associated protein 1 (YAP1), which is among the top 40 most frequently observed regulostat-constituent genes (Figure 3C), is the component of the Hippo signaling pathway that functions as a tumor suppressor by promoting apoptosis (Supplementary Data 9). In addition to the Hippo signaling pathway, other highly enriched canonical pathways also strikingly correlated with cancer biology (Figure 3D). These pathways regulate cancerous signaling (PI3K-Akt signaling pathway, MAPK signaling pathway, p53 signaling pathway, TGFβ signaling pathway, Rap1 signaling pathway), cell remodeling (focal adhesion, tight junction, ECM-receptor interaction, proteoglycan in cancer, cell adhesion molecules, regulation of actin cytoskeleton, adherens junction), cell death (apoptosis), protein processing, and folate biosynthesis that is crucial for DNA replication and cell division. Of note, the top 15 most commonly observed regulostat-constituent genes were specific to their tissue of origin (Figure 4B). Taken together, our analyses revealed regulostats that predetermine drug response in cancer cells are composed of genes driving cancer cell survival.
Transcription factors, kinases, drug targets and genes with molecular functions related to drug responses are over-represented among pooled regulostat-constituent genes
Given the molecular milieu affects signaling events and the fine-tuning of cellular response phenotypes, we sought to elucidate the molecular functions of key components within regulostats. In particular, we sought to determine the extent to which regulostat-constituent genes pooled from 1169 drug-cancer cases (called pooled regulostat-constituent genes hereafter) consist of transcriptional regulators, kinases, and metabolic enzymes, as well as other functional categories related to drug response phenotypes, including drug metabolic enzymes, transporters, DNA repair, cell cycle, apoptosis and cellular stress, and the proportion of these pooled regulostat-constituent genes that are known drug targets.
Our survey indicated transcription factors (18.38%), drug targets (12.92%), metabolic enzymes (8.08%), and kinases (3.12%) represent four major functional categories found in pooled regulostat-constituent genes (Supplementary Figure S10 and Supplementary Table S2). Apoptosis (1.04%), cell cycle (1.01%), and cellular stress (0.65%) were additional molecular functions represented in the data. Results from Fisher's exact test reveal that 7 out of 10 of these selected drug response-related molecular functions are significant at the threshold level of P-value < 0.05 (Supplementary Table S2).
Further analysis of the top 20 most frequently observed genes from these drug response phenotype-related genes suggest their central roles in regulating processes such as cell growth, repair, homeostasis, and apoptosis (Supplementary Figure S11), which collectively predetermine drug response phenotype under specific molecular contexts. For instance, transcription factors YAP1 (Yes Associated Protein 1), ESRRG (Estrogen Related Receptor Gamma), and kinases TGFBR1/2 (Transforming Growth Factor Beta Receptor 1/2), ERBB4 (Erb-B2 Receptor Tyrosine Kinase 4), CDK6/7 (Cyclin Dependent Kinase 6/7) are known to play important roles in cell cycle. Of note, a number of the top 20 metabolic enzymes such as AOX1 (Aldehyde Oxidase 1), ADH7 (Alcohol Dehydrogenase 7), HSD17B1 (Hydroxysteroid 17-Beta Dehydrogenase 1), and MAOB (Monoamine Oxidase B) are involved in xenobiotic metabolic processes related to drug metabolism such as cytochrome P450 and dopamine metabolism. In addition, metabolic enzymes such as ATP5G2 (ATP Synthase Membrane Subunit C Locus 2) and SUCLG2 (Succinate-CoA Ligase GDP-Forming Beta Subunit) are important in energetic processes required to sustain cellular activities.
Generic regulostat network indicates core genes that regulate diverse drug responses
We next sought to identify and characterize rheostat-like gene pairs observed in multiple drug response phenotype by reconstructing a network we termed a generic regulostat. To examine the molecular signals underpinning gene pairs observed in regulostat networks of at least five drug-cancer cases, the top 200 gene pairs that achieved stable internal-assignment recovery rates as described above were clustered according to their respective PLCs. The overall clustered heatmap (Supplementary Data 10) featured data from cancers derived from blood, lung, and the digestive tract. As shown in Figure 5A, clustering results showed that cancer cell lines derived from the same tissue of origin clustered together with a number of defined drugs, suggesting the role of cellular context in shaping drug–gene pair interactions and the mode-of-action of drugs. For instance, BMS-708163.1 (γ-secretase inhibitor), vinorelbine (microtubule inhibitor), bleomycin (DNA damage), docetaxel (microtubule inhibitor), and mitomycin C (DNA cross-linker) clustered together for cancer cell lines derived from the digestive tract via gene pairs PPP3CA (Protein Phosphatase 3 Catalytic Subunit Alpha)-ESRRG (Estrogen Related Receptor γ) and PPP3CA-GCG (glucagon) (Figure 5A). Our results therefore indicate the existence of drug–gene pair interactions under specific cellular contexts.
Figure 5.

Characterization of rheostat-like gene pairs in a generic regulostat. (A) Selected clustering results featuring cancer cells derived from lung, digestive tract, and blood based on PLC values for gene pairs from  lists for at least 5 of 1169 drug-cancer cases achieving a stable internal-assignment recovery rate. Colors indicate PLC values. Grey: no PLC available for a gene pair in a given drug-cancer case. (B) Regulostat network comprised of rheostat-like gene pairs observed in at least 5 of 1169 drug-cancer cases. Node size represents the frequency (i.e. number of occurrences) for a given gene and edge thickness represents the frequency a given gene pair normalized by 5 across 1169 drug-cancer cases.
lists for at least 5 of 1169 drug-cancer cases achieving a stable internal-assignment recovery rate. Colors indicate PLC values. Grey: no PLC available for a gene pair in a given drug-cancer case. (B) Regulostat network comprised of rheostat-like gene pairs observed in at least 5 of 1169 drug-cancer cases. Node size represents the frequency (i.e. number of occurrences) for a given gene and edge thickness represents the frequency a given gene pair normalized by 5 across 1169 drug-cancer cases.
Next, pathway enrichment analysis for genes residing in the generic regulostat network shown in Figure 5B reveals a number of metabolic pathways (drug metabolism - cytochrome P450 and metabolism of xenobiotics by cytochrome P450, TCA cycle, sphingolipid metabolism, tryptophan and tyrosine metabolism, glycerolipid metabolism, and retinol metabolism) are enriched (Supplementary Data 11). In addition, numerous cancer-related pathways (MAPK signaling pathway, endocytosis, VEGF signaling pathway, calcium signaling pathway, Adherens junction, phosphatidylinositol signaling system, ECM-receptor interaction, TGF-beta signaling pathway, and ErbB signaling pathway) as well as immune response-related pathways (T- and B-cell receptor signaling pathway, natural killer cell mediated cytotoxicity and cytokine–cytokine receptor interaction) are also enriched (Supplementary Data 11). Because the generic regulostat is composed of gene pairs observed in at least five different drug-cancer cases, we anticipate that these gene pairs and their associated pathways, in principle, play important roles in drug–network interactions predetermining the extent of cancer cell response phenotypes to a broad range of drugs.
Closer examination of the generic regulostat network (Figure 5B) revealed genes such as PPM1A (protein phosphatase, Mg2+/Mn2+ dependent, 1A), TMEM260 (a transmembrane protein), ABCA4 (ATP-binding cassette, sub-family A (ABC1), member 4), LOC100507480 (an uncharacterized non-coding RNA gene), MAPK1 (mitogen-activated protein kinase 1), KRT222 (keratin 222), PPP3CA (protein phosphatase 3, catalytic subunit, alpha isozyme), and FGF20 (fibroblast growth factor 20) appear most frequently among rheostat-like gene pairs in different drug-cancer cases (Figure 5B). We posit that these genes play a role in determining the extent of diverse cancer drug response phenotypes and associations of these ‘high frequency’ genes to other genes in the generic regulostat imply novel functional crosstalk in determining common drug response phenotypes in diverse types of cancer cells. In particular, MAPK1 appears as a hub in connecting DIRAS3 (DIRAS family, GTP-binding RAS-like 3), CT55 (Cancer/Testis Antigen 55), KRT8 (keratin 8) and NAA11 (N(alpha)-acetyltransferase 11, NatA catalytic subunit), suggesting a key role of functional crosstalk mediated by these genes in modulating broad drug response phenotypes in cancer cells. Equally important is the ‘high frequency’ gene pairs, in particular PPAP2A (phosphatidic acid phosphatase type 2A) with RALYL (RALY RNA binding protein-like), ZNF322 (zinc finger protein 322) with TKTL1 (transketolase-like 1), FGF20 (fibroblast growth factor 20) with THADA (thyroid adenoma associated), ZFP42 (zinc finger protein 42 homolog (mouse)) with APOOL (apolipoprotein O-like), PPP3CA (protein phosphatase 3, catalytic subunit, alpha isozyme) with ESRRG (estrogen-related receptor gamma), and PPP3CA with GCG (glucagon). Although these genes are known to play key roles in cellular homeostasis and survival, our work reveals their novel functional crosstalk within a regulostat to determine drug response phenotypes in cancer cells. Intriguingly, we noticed a relatively high number of zinc finger proteins (ZC3H8, ZDHHC23, ZFP42, ZNF217, ZNF239, ZNF322, ZPLD1) are present in the generic regulostat network (Figure 5B), highlighting the potential key roles of transcriptional regulation by these zinc finger proteins in determining general drug response phenotypes in cancer cells.
Construction of regulostat networks corresponding to single lung cancer cell lines provides insights to engineer drug response phenotypes
Next, we sought to illustrate that RSI is capable of reconstructing regulostat networks with respect to specific cancer cell lines (Figure 2B). By comparing regulostat networks from different cell lines showing similar response phenotypes to a given stressor, one can delineate common molecular constituents or mechanisms that are responsible for convergent response phenotypes. In addition, cell line-specific regulostats allow one to determine genes that are essential for survival in the presence of a stressor and which genes may be perturbed to rescue the potentially harmful effects caused by the stressor. Finally, cell line-specific regulostats for a broad spectrum of stressors facilitate systematic comparative analyses with sensitive or resistant phenotypes, which can be important in prioritizing drugs for individualized medicine.
To reconstruct a cell line-specific regulostat, gene pairs from 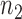 were chosen if they showed maximum or minimum point on the correlation lines of computed PLCs (Figure 2B). The selected gene pairs were then used to reconstruct a regulostat network corresponding to a given cell line. We reconstructed regulostats for selected lung cancer cell lines corresponding to drug FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase as illustrative examples (Figure 6). Networks of regulostats corresponding to FK866-sensitive lung cancer cell lines SBC-3 (Figure 6A) and NCI-H1876 (Figure 6B) and FK866-resistant lung cancer cell lines SW1573 (Figure 6C) and SK-LU-1 (Figure 6D) were constructed.
were chosen if they showed maximum or minimum point on the correlation lines of computed PLCs (Figure 2B). The selected gene pairs were then used to reconstruct a regulostat network corresponding to a given cell line. We reconstructed regulostats for selected lung cancer cell lines corresponding to drug FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase as illustrative examples (Figure 6). Networks of regulostats corresponding to FK866-sensitive lung cancer cell lines SBC-3 (Figure 6A) and NCI-H1876 (Figure 6B) and FK866-resistant lung cancer cell lines SW1573 (Figure 6C) and SK-LU-1 (Figure 6D) were constructed.
Figure 6.

Cell line-specific regulostats of lung cancer cells. (A) Regulostat specific to an SBC-3 lung cancer cell line that is sensitive to drug FK866 (bin 1). (B) Regulostat specific to NCI-H1876 lung cancer cell line that is sensitive to drug FK866 (bin 1). (C) Regulostat specific to SW1573 lung cancer cell line that is resistant to drug FK866 (bin 4). (D) Regulostat specific to SK-LU-1 lung cancer cell line that is resistant to drug FK866 (bin 4). The node size shows the gene expression level for that specific cell line, and its color is red if that gene has the highest expression value in the correlation line of PLC or blue vice versa (see Figure 2B). Yellow nodes indicate the gene has both the highest and lowest expression when combined with different genes in a given number of pairs. Red edges: positive PLCs; blue edges: negative PLCs.
Here, the signs (i.e. positive and negative) of PLCs are important to infer MOC of drug–gene pair interactions in predetermining the response phenotype corresponding to a cell line and how to manipulate it. As shown in Figure 2A, positive PLC for a given gene pair indicates a flip of MOCs from negative (bin 1) to positive gene–gene coexpression correlation for the resistant (bin 4) phenotype and vice versa for the negative PLC scenario. For instance, the regulostat for FK866-sensitive cell line SBC-3 (Figure 6A) shows that ADAMTS12-TMC5 and ADAMTS12-MLPH are negative PLC pairs (indicated by blue edges). These gene pairs are at the minimal side of the correlation line of positive gene–gene coexpression correlations (indicated by blue nodes) in bin 1. This means suppressing the expression of these genes can confer sensitivity of the SBC-3 cell line to FK866. On the contrary, CD52-PCDHGC3 and TMEM109-TPM1 are positive PLC pairs (indicated by red edges) of the sensitive cell line NCI-H1876 (Figure 6B), with one gene at the maximal value (red node) and its counterpart gene at the minimal value (blue node) of the correlation line of negative gene–gene coexpression correlation in bin 1. For such a scenario, expressing genes shown in red nodes and suppressing genes shown in blue nodes will strengthen the negative MOCs of these pairs and in principle will promote sensitivity of the NCI-H1876 cell line to FK866. Similar principles of MOC are also applied to resistant cell lines (Figure 6C and D).
Thus, regulostat network models of single cell lines (or of an individual organism) provide context-dependent mechanistic insights to prioritize key gene pairs capable of modulating cellular response phenotypes. Moreover, regulostat networks suggest directionality for potential intervention, either activation or inhibition of selected gene pairs depending on their MOC.
DISCUSSION
Understanding how molecular contexts predetermine the extent of cellular responses has important implications in many aspects of biology and medicine. Of note, molecular phenotyping and the use of cellular response phenotypes are emerging as important considerations to improve how biological responses are engineered as well as drug discovery efforts (34–38). Given a cell likely responds to a stressor in a continuous manner, new tools capable of capturing that continuous response rather than simply a binary ‘on’ or ‘off’ response are needed. As such, we devised the RSI algorithm to identify gene pairs showing rheostat-like mode-of-cooperation that predetermine and modulate the cell's response to a given stressor in a continuous manner. In essence, the algorithm searches for gene pairs that act like a rheostat, or an adjustable dimmer switch for a light bulb (rather than a simple on-off light switch). The RSI algorithm reconstructs networks composed of rheostat-like gene pairs involved in predetermining cellular response phenotypes; we termed such networks regulostats.
Our proof-of-concept study together with a series of in silico validations provides computational evidence for the existence of regulostats capable of predetermining drug response phenotypes in cancer cells. We reconstruct regulostat networks corresponding to a given cancer type of interest across all drugs and also regulostats for specific drug phenotypic responses to identify rheostat-like gene pairs. For the first time, our work provides evidence of drug–regulostat interactions, where rheostat-like mode-of-cooperative action of gene pairs, the basic molecular units of a regulostat, predetermine the extent of drug responses in cancer cells. This kind of context-specific drug–gene pair interaction is conceptually different from conventional drug–gene interactions where genetic variations of a drug target are the major factors causing altered drug actions (24–26). The RSI algorithm therefore lays the conceptual foundation to enable researchers to dissect rheostat-like gene pairs collectively constituting a ‘regulostat’ in large-scale omics data, thus honing in on the networks driving drug response phenotypes. Of note, our analyses indicate over-representation of transcription factors, kinases, drug targets, as well as genes with molecular functions related to drug responses, such as apoptosis and cellular stress, in pooled regulostat-constituent genes. Such findings indicate that transcriptional regulation and signaling via phosphorylation events are key modulatory modes shaping the activities of a regulostat in terms of predetermining and modulating how a cell responds to a particular stressor.
Our work shows regulostats are present in most if not all cellular contexts corresponding to a given stressor. In contrast to the conventional gene-based model where cellular response phenotypes are mainly explained by genetic mutations (39,40) or polymorphisms (41,42), the regulostat model provides an alternative, systems biology analytical framework to indicate how cellular response phenotypes and phenotypic selection are predetermined by bigger-picture networks or regulostats in a cell capable of fine-tuning a response. Importantly, by identifying key rheostat-like gene pairs responsible for conferring drug resistance as candidates for perturbation, the regulostat model will enable researchers to escape the vicious cycle of discovering a new drug only to have cancer cells acquire resistance to it (43,44). In principle, rheostat-like genes within inferred regulostats can be prioritized to modulate drug response, thus rendering a resistant phenotype sensitive to a given drug.
The utility of RSI is not restricted to studying drug response phenotypes. We reason that regulostat models also have important implications in the area of gene essentiality (i.e. the dependency of cells on a particular gene for survival/fitness under specific conditions). For instance, genes whose activities that are responsible for resisting the pressure exerted by a given stressor will confer fitness advantages for cell adaptation and survival and are essential for these cells. Therefore, gene essentiality is context-dependent by nature (45). Deciphering the regulostat with respect to a stressor therefore can provide insight for essential genes acting as rheostat-like pairs that confer resistance to the pressure exerted by the stressor.
Based on its wide implications and the importance of regulostat models in bioengineering and medicine, we anticipate that the RSI algorithm will create a paradigm shift. By providing mechanistic insights in terms of how gene pairs cooperate to determine cellular phenotypes, in health and disease, it is possible to prioritize specific gene target candidates. As such, novel biological phenotypes can be engineered to offer the promise of individualized therapy for reversing resistance to anticancer drugs.
Supplementary Material
ACKNOWLEDGEMENTS
Author's contributions: C.Y.U., M.G.B. and C.Z. contributed equally to this work. C.Y.U., M.G.B., C.Z. and H.L. contributed to the conception and design of the study. C.Y.U., M.G.B., C.Z. and H.L. contributed to the acquisition of data. C.Y.U., M.G.B., C.Z., J.L., C.C. and H.L. contributed to the analysis and interpretation of data. C.Y.U., M.G.B., C.C. and H.L. drafted the manuscript. H.L. supervised the study.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01CA196631, R01CA208517, R01AG056318, R01AG61796, P50CA136393]; Glenn Foundation for Medical Research; W.M. Keck Foundation; Mayo Clinic Center for Biomedical Discovery and Mayo Clinic Center for Individualized Medicine. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Rutkowski D.T., Kaufman R.J.. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 2007; 32:469–476. [DOI] [PubMed] [Google Scholar]
- 2. Sumner E.R., Avery S.V.. Phenotypic heterogeneity: differential stress resistance among individual cells of the yeast Saccharomyces cerevisiae. Microbiology. 2002; 148:345–351. [DOI] [PubMed] [Google Scholar]
- 3. Becskei A., Séraphin B., Serrano L.. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 2001; 20:2528–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garnett M.J., Edelman E.J., Heidorn S.J., Greenman C.D., Dastur A., Lau K.W., Greninger P., Thompson I.R., Luo X, Soares J et al.. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012; 483:570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nelson M.R., Johnson T., Warren L., Hughes A.R., Chissoe S.L., Xu C.F., Waterworth D.M.. The genetics of drug efficacy: opportunities and challenges. Nat. Rev. Genet. 2016; 17:197–206. [DOI] [PubMed] [Google Scholar]
- 6. Relling M.V., Evans W.E.. Pharmacogenomics in the clinic. Nature. 2015; 526:343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gatenby R. Perspective: finding cancer's first principles. Nature. 2012; 491:S55. [DOI] [PubMed] [Google Scholar]
- 8. López-Maury L., Marguerat S., Bähler J.. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008; 9:583–593. [DOI] [PubMed] [Google Scholar]
- 9. Avery S.V. Microbial cell individuality and the underlying sources of heterogeneity. Nat. Rev. Microbiol. 2006; 4:577–587. [DOI] [PubMed] [Google Scholar]
- 10. Burnet F.M. The Clonal Selection Theory of Acquired Immunity. 1959; Cambridge: The University Press. [Google Scholar]
- 11. Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996; 381:751–758. [DOI] [PubMed] [Google Scholar]
- 12. Clark N.R., Hu K.S., Feldmann A.S., Kou Y., Chen E.Y., Duan Q., Ma’ayan A.. The characteristic direction: a geometrical approach to identify differentially expressed genes. BMC Bioinformatics. 2014; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J.C., Alvarez M.J., Talos F., Dhruv H., Rieckhof G.E., Iyer A., Diefes K.L., Aldape K., Berens M., Shen M.M. et al.. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell. 2014; 159:402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stuart J.M., Segal E., Koller D., Kim S.K.. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003; 302:249–255. [DOI] [PubMed] [Google Scholar]
- 15. Wu G., Stein L.. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012; 13:R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clarke C., Madden S.F., Doolan P., Aherne S.T., Joyce H., O’Driscoll L., Gallagher W.M., Hennessy B.T., Moriarty M., Crown J. et al.. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013; 34:2300–2308. [DOI] [PubMed] [Google Scholar]
- 17. Yang Y., Han L., Yuan Y., Li J., Hei N., Liang H.. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014; 5:3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solovieff N., Cotsapas C., Lee P.H., Purcell S.M., Smoller J.W.. Pleiotropy in complex traits: challenges and strategies. Nat. Rev. Genet. 2013; 14:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steinacher A., Bates D.G., Akman O.E., Soyer O.S.. Nonlinear dynamics in gene regulation promote robustness and evolvability of gene expression levels. PLoS One. 2016; 11:e0153295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davidson E.H. Emerging properties of animal gene regulatory networks. Nature. 2010; 468:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. et al.. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006; 38:431–440. [DOI] [PubMed] [Google Scholar]
- 22. Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G. et al.. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005; 122:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rossi F.M., Kringstein A.M., Spicher A., Guicherit O.M., Blau H.M.. Transcriptional control: rheostat converted to on/off switch. Mol. Cell. 2000; 6:723–728. [DOI] [PubMed] [Google Scholar]
- 24. Cotto K.C., Wagner A.H., Feng Y.Y., Kiwala S., Coffman A.C., Spies G., Wollam A., Spies N.C., Griffith O.L., Griffith M.. DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2018; 46:D1068–D1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tannenbaum C., Sheehan N.L.. Understanding and preventing drug-drug and drug–gene interactions. Expert. Rev. Clin. Pharmacol. 2014; 7:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ryan C.J., Cimermančič P., Szpiech Z.A., Sali A., Hernandez R.D., Krogan N.J.. High-resolution network biology: connecting sequence with function. Nat. Rev. Genet. 2013; 14:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H. et al.. A landscape of pharmacogenomic interactions in cancer. Cell. 2016; 166:740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghanat Bari M., Ung C.Y., Zhang C., Zhu S., Li H.. Machine learning-assisted network inference approach to identify a new class of genes that coordinate the functionality of cancer networks. Sci. Rep. 2017; 7:6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duda R.O., Peter E. H., David G. S.. Pattern Classification. 2012; John Wiley & Sons. [Google Scholar]
- 30. Wang J., Vasaikar S., Shi Z., Greer M., Zhang B.. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017; 45:W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fisher R.A. On the interpretation of χ2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 1922; 85:87–94. [Google Scholar]
- 32. Hanahan D., Weinberg R.A.. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 33. Lu L., Li Y., Kim S.M., Bossuyt W., Liu P., Qiu Q., Wang Y., Halder G., Finegold M.J., Lee J.S et al.. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng W., Thorne N., McKew J.C.. Phenotypic screens as a renewed approach for drug discovery. Drug Discov. Today. 2013; 18:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moffat J.G., Vincent F., Lee J.A., Eder J., Prunotto M.. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov. 2017; 16:531–543. [DOI] [PubMed] [Google Scholar]
- 36. Xie L., Xie L., Kinnings S.L., Bourne P.E.. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu. Rev. Pharmacol. Toxicol. 2012; 52:361–379. [DOI] [PubMed] [Google Scholar]
- 37. Ritchie M.D., Holzinger E.R., Li R., Pendergrass S.A., Kim D.. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015; 16:85–97. [DOI] [PubMed] [Google Scholar]
- 38. Drawnel F.M., Zhang J.D., Küng E., Aoyama N., Benmansour F., Araujo Del Rosario A., Jensen Zoffmann S., Delobel F., Prummer M., Weibel F. et al.. Molecular phenotyping combines molecular information, biological relevance, and patient data to improve productivity of early drug discovery. Cell Chem. Biol. 2017; 24:624–634. [DOI] [PubMed] [Google Scholar]
- 39. Hartwell L.H., Szankasi P., Roberts C.J., Murray A.W., Friend S.H.. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997; 278:1064–1068. [DOI] [PubMed] [Google Scholar]
- 40. Paez J.G., Jänne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J et al.. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497–1500. [DOI] [PubMed] [Google Scholar]
- 41. Danesi R., De Braud F., Fogli S., Di Paolo A., Del Tacca M.. Pharmacogenetic determinants of anti-cancer drug activity and toxicity. Trends Pharmacol. Sci. 2001; 22:420–426. [DOI] [PubMed] [Google Scholar]
- 42. Wilson J.F., Weale M.E., Smith A.C., Gratrix F., Fletcher B., Thomas M.G., Bradman N., Goldstein D.B.. Population genetic structure of variable drug response. Nat. Genet. 2001; 29:265–269. [DOI] [PubMed] [Google Scholar]
- 43. Holohan C., Van Schaeybroeck S., Longley D.B., Johnston P.G.. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013; 13:714–726. [DOI] [PubMed] [Google Scholar]
- 44. Garraway L.A., Jänne P.A.. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012; 2:214–226. [DOI] [PubMed] [Google Scholar]
- 45. Rancati G., Moffat J., Typas A., Pavelka N.. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018; 19:34–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.