

Abstract
Epilepsy with myoclonic absences is a specific seizure type characterized by bilateral rhythmic clonic jerks with impairment of consciousness. Here, we report an individual with epilepsy with myoclonic absences, mild intellectual disabilities, language disorder, and autism spectrum disorder. His interictal electroencephalogram revealed a spike‐and‐slow wave complex dominant in the frontal area. His ictal polygraphic and video‐electroencephalogram showed a characteristic diffuse synchronous 3‐Hz spike‐and‐wave burst associated with bilateral upper limb myoclonic jerks with impairment of consciousness. Using whole‐exome sequencing, we found a novel de novo variant, c.386T>G, p.(Val129Gly), in SETD1B (SET domain containing 1B). We previously reported that two individuals with a de novo SETD1B variant showed intellectual disability, epilepsy, and autism. Of note, one of those individuals and the present case showed epilepsy with myoclonic absences. Therefore, this report supports the indication that SETD1B may be a causative gene for neurodevelopmental disorders and suggests that epilepsy with myoclonic absences may be a characteristic feature of SETD1B‐related disorders.
Keywords: SETD1B, epilepsy with myoclonic absences, neurodevelopmental disorders, whole‐exome sequencing
1. INTRODUCTION
Epilepsy with myoclonic absences (EMA) is a rare epilepsy syndrome clinically characterized by absences accompanied by marked, diffuse, rhythmical myoclonias and often associated with a progressive tonic contraction.1 This seizure type is categorized as a generalized onset and nonmotor seizure and accompanied by various degrees of neurodevelopmental disorders (NDDs). Genetic etiologies of EMA remain to be largely elucidated. Previous whole‐exome sequencing (WES) studies described some epilepsy genes, including SYNGAP1,2 GLUD1,3 SCN1A,4 and SLC2A1,5 as having a possible association with EMA.
SETD1B (SET domain containing 1B) encodes a histone H3 lysine 4 (H3K4) methyltransferase, which is involved in the epigenetic control of the chromatin structure and gene expression.6 De novo microdeletions in the 12q24.3 region encompassing SETD1B were associated with syndromic NDDs,7 and two de novo variants in SETD1B have been reported in developmental disorders in a large cohort exome study.8 Recently, we reported two individuals with de novo SETD1B variants who had epilepsy (one showed EMA), developmental delay, intellectual disabilities (ID), autistic behavior, and craniofacial dysmorphic features.9 These findings suggested that de novo SETD1B variants are causative for NDDs with epilepsy.
Here, we report a new individual with a de novo SETD1B missense variant having EMA, ID, language disorder, and autism spectrum disorder (ASD). We will discuss the phenotypic features of SETD1B‐associated disorders.
2. METHODS
After receiving written informed consent, the genomic DNA of the patient and his parents was extracted from blood leukocytes. Genomic DNA of the patient was captured using a SureSelect Human All Exon V6 kit (Agilent Technologies) and sequenced by a NextSeq 500 (Illumina) with 150‐bp paired‐end reads. Exome data processing, variant calling, and variant annotation were performed as previously described.9 Candidate variants were confirmed by Sanger sequencing. Biological parentage was confirmed by analyzing 10 microsatellite markers. Experimental protocols were approved by the Institutional Review Board of Hamamatsu University School of Medicine.
3. RESULTS
3.1. Case presentation
A Japanese boy was born to nonconsanguineous healthy parents as a second child without asphyxia at 37 weeks of gestation. His birthweight and length were 3,008 g (+0.82 standard deviation [SD]) and 50 cm (+1.4 SD), respectively. There was no family history of NDDs. He required respirator support because of transient tachypnea of the newborn. Although his motor development was normal, his language development was markedly delayed. He spoked his first word at 2 years and 3 months old and was able to combine two words at 3 years old. At 5 years old, he was diagnosed with mild ID (intelligence quotient [IQ] 64, Tanaka‐Binet Intelligence Scale). At 6 years old, his full‐scale IQ (WISC‐IV) was 56. He poorly interacted with others and showed hyperactive behavior. He was then diagnosed as ASD at 6 years old based on the Pervasive Developmental Disorders Autism Society Japan Rating Scale.10
At 3 years and 7 months old, he began to show myoclonic absence seizures lasting 3‐5 seconds and occurring 4‐5 times per day. His seizures were refractory to antiepileptic drugs including valproate, ethosuximide, levetiracetam, clobazam, and topiramate. The ketogenic diet was also tried but interrupted because he did not appreciate the importance of the treatment. At 7 years and 10 months old, interictal electroencephalogram (EEG) revealed a spike‐and‐slow wave complex with dominance in the frontal area (Figure 1). In an ictal video‐EEG and polygraph (EEG and electromyogram) recording, diffuse synchronous 3‐Hz spike‐and‐wave bursts were observed synchronized with bilateral upper limb myoclonic jerks accompanied by loss of consciousness. These findings matched the criteria of myoclonic absence seizure. At the time of this writing, his myoclonic absence seizures occur 10‐20 times per day and last 30 seconds despite valproate and lamotrigine treatment.
Figure 1.
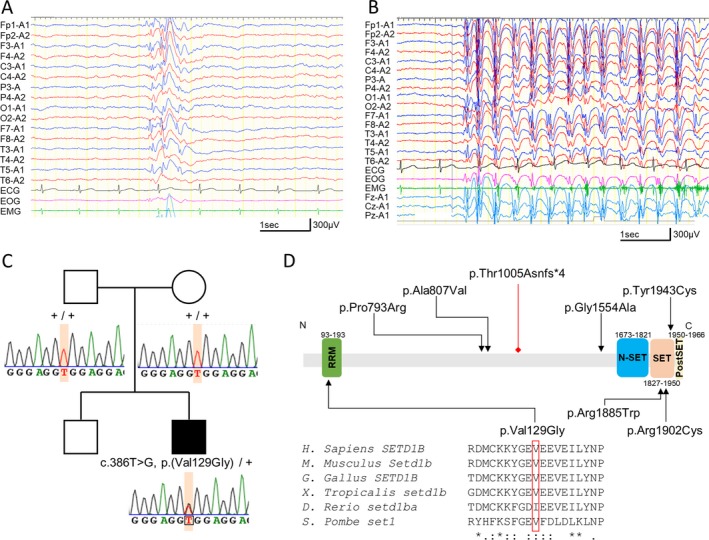
EEG findings and the SETD1B variant in the patient. A, Interictal EEG at 7 years old. EEG showed frontally dominant generalized spike‐and‐wave discharges. B, Ictal EEG at 7 years old. EEG revealed 3‐Hz diffuse spike‐and‐wave discharges with myoclonic jerks rhythmically repeated at 3 Hz. Electromyography (EMG) recorded on the left trapezius muscle. C, Familial pedigrees and SETD1B variants. Sanger sequencing showed a de novo variant in SETD1B. +, wild‐type allele. D, Schematic presentation of the SETD1B protein. The variants identified in our study are depicted below, and the variants in previous large cohort studies20, 21, 22 are shown above. Variants are represented with black triangles (missense) or a red diamond (frameshift). Locations of variants are annotated based on a new NCBI Reference Sequence: NM_001353345.1 (released on December 25, 2018). Multiple amino acid sequences of SETD1B were aligned using the ClustalW tool
Upon final examination at 8 years and 1 month old, his height was 124.6 cm (−0.23 SD), his weight was 28.8 kg (+0.49 SD), and his head circumference was 54.5 cm (+1.4 SD). He had no dysmorphic features. Brain MRI showed normal brain structure.
3.2. Variant screening
Using WES, we searched for candidate variants in known epilepsy‐ and NDD‐related genes and found a possible pathogenic variant in SETD1B (NM_001353345.1: c.386T>G, p.(Val129Gly)). Sanger sequencing confirmed that this variant occurred de novo (Figure 1C). It was absent in our 218 in‐house Japanese control exomes and public databases including the gnomAD (http://gnomad.broadinstitute.org/) and 2KJPN (https://ijgvd.megabank.tohoku.ac.jp/). This variant was predicted to be deleterious by multiple in silico pathogenicity prediction tools: The CADD PHRED score was 28.3 (http://cadd.gs.washington.edu/snv), the PP2_HVAR score was 0.994 (http://genetics.bwh.harvard.edu/pph2/), the SIFT score was 0 (http://sift.jcvi.org/), the MutationTaster score was 1.0 (http://www.mutationtaster.org/), and the Missense Tolerance Ratio was 0.503 (24.9th percentile <25th percentile; http://mtr-viewer.mdhs.unimelb.edu.au/mtr-viewer/).11 The variant was considered likely pathogenic according to ACMG Standards and Guidelines (PS2, PM2, PP3). The altered residue was located at the RNA recognition motif (RRM) domain and highly evolutionarily conserved (Figure 1D). We also examined possible pathogenic copy number variants (CNVs) using WES data with the eXome‐Hidden Markov Model (XHMM)12 and the methods developed by Nord et al13; however, no possible pathogenic CNVs were found in this case. These findings suggested that this variant is likely to be pathogenic in this case.
4. DISCUSSION
To date, we found three de novo SETD1B variants in three unrelated Japanese individuals with NDDs and epilepsy. The clinical manifestations of these individuals are summarized in Table 1. In all cases, the developmental milestones were delayed before the onset of seizures. They started to show autistic behaviors and various degrees of ID in early childhood. However, facial dysmorphic features were seen in only one of the three individuals. Epilepsy is also common to all the three individuals. Individuals with microdeletion of 12q24.31 including SETD1B also had epilepsy in two of the four cases,8 suggesting that epilepsy is strongly associated with SETD1B defects, although clinical features could be modified by defects in other genes located within the deletion interval. Interestingly, EMA was noted in two of the three individuals.9, 14 EMA is a rare and specific type of seizure that was accounts for 0.5%‐1% of total epilepsies.1 Although the myoclonic seizures of the remaining case were not thought to be myoclonic absences based on interictal EEG findings, evidence suggests that SETD1B may be a candidate gene for EMA.
Table 1.
Clinical features of cases of de novo SETD1B variants
| Individuals | This patient | Previously reported cases9, 14 | |
|---|---|---|---|
| Patient 1 | Patient 2 | ||
| SETD1B variants | c.386T>G | c.5653C>T | c.5704C>T |
| p.(Val129Gly) | p.(Arg1885Trp) | p.(Arg1902Cys) | |
| Sex, age | Male, 7 y | Female, 12 y | Male, 34 y |
| Seizure onset | 3 y and 7 mo | 2 y and 9 mo | 3 y and 11 mo |
| Seizure type at onset | Myoclonic absence seizure | Myoclonic seizures | Myoclonic seizures |
| Myoclonic absence seizure onset | 3 y and 7 mo | 2 y and 10 mo | NA |
| Course of seizures | Myoclonic absence seizure | Myoclonic absence seizure | Generalized tonic seizures |
| Initial interictal EEG | Spike‐and‐slow wave complex dominant frontal area | 2‐ to 3‐Hz diffuse spike‐and‐wave bursts with slow wave dominant background |
3‐ to 4‐Hz frontally dominant spike‐and‐slow wave complexes or polyspike‐and‐slow wave complexes |
| Ictal EEG | 3‐Hz diffuse spike‐and‐wave burst | Generalized spike‐and‐waves of 3‐4 Hz, with an anterior predominance | NA |
| Course of interictal EEG | Spike‐and‐slow wave complex dominant frontal area | No epileptiform discharge with rhythmic theta wave | Focal spike‐and‐slow wave complex |
| Response to treatment | Daily seizure with VPA and LTG | Seizure‐free from 6 ywith LTG | Monthly seizure with VPA, CBZ and TPM |
| Dysmorphic features | − | − | + |
| Intellectual disability | Mild | Mild | Profound |
| IQ | 56 (WISC‐IV) | 64 (TK‐Binet) | 10 (TK‐Binet) |
| Autism/autistic behavior | + | + | + |
| Anxiety | + | − | + |
| Language delay | + | + | + |
| Motor development | Normal | Walk without support | Walk without support |
| Brain image | Normal | Normal | Normal |
| Cancer | NA | NA | Sigmoid cancer |
Abbreviations: CBZ, carbamazepine; LTG, lamotrigine; NA, not assessed or not available; TPM, topiramate; VPA, valproate.
Previous work suggested that histone H3 lysine 4 trimethylation (H3K4me3) was involved in learning and memory consolidation.15 Animal models showed that the level of H3K4me3 increased in the CA1 region of the dorsal hippocampus during memory retrieval.16 Moreover, it was revealed that the variants in genes constituting components of the H3K4 regulatory machinery caused rare congenital dysmorphic syndromes with NDDs involving ID, ASD, and schizophrenia.17 These findings indicated that the dysfunction of the enzymatic activity of SETD1B may be likely to reduce H3K4me3 in the neuronal system and cause disabilities of normal learning and consolidation of memories. Meanwhile, involvement of epilepsy was variable in cases with variants in histone H3K4 methyltransferases. Our data suggest that epilepsy is strongly associated with SETD1B defects. In regulating the H3K4 trimethylation pattern in mammalian cells, WDR82 is a component of the SETD1A and SETD1B complexes that influence target selection, but is not associated with other SET1‐family complexes such as KMT2A‐KMT2D.18 In addition, it has been reported that SETD1B has more robust trimethylase activities than KMT2A‐KMT2D.18 Therefore, it might be possible that a strong defect of trimethylase activity caused by SETD1B defects is closely associated with the epilepsy phenotype.
SETD1B belongs to the SET1 family of histone H3K4 methyltransferases. It is an important component of the histone methyltransferase complex that generates mono‐, di‐, and trimethylated histone H3 at Lys4 and has been implicated in multiple biological processes.6 SETD1B has four evolutionarily conserved functional domains: the RRM domain, the N‐SET domain, the catalytic SET domain, and the post‐SET domain. The SET domain plays a principal role in catalyzing histone methylation, and loss‐of‐function variants in this domain may cause impairment of enzymatic activity of H3K4 methyltransferase. The RRM domain is commonly shared by the many RNA binding proteins used for interaction with RNA bases. It acts as a positive regulator of trimethylation by SET1‐family methyltransferases, and dysfunction in the RRM domain may bring about inadequate trimethylation leading to an abnormal transcriptional regulation of genes.19 Therefore, loss‐of‐function variants in the RRM domain may also induce dysregulation of enzymatic activity of H3K4 methyltransferase. In previous and present studies, eight de novo variants in SETD1B had been identified in individuals with neurodevelopmental or psychiatric diseases20, 21, 22 (Table S1). Four missense SETD1B variants were located in the SET domain or RRM domain, and these four variants were predicted to be deleterious by multiple in silico tools. While the p.(Thr1005Asnfs*4) is a null variant and is very likely to be pathogenic, the pathogenicity of the other missense variants located outside the domain was less significant (Table S1). All three patients in our work showed very similar phenotypes although their variants were located in the different domains. The clinical details of the previous studies were not available, but both missense and protein‐truncating variants may cause developmental disorders. Therefore, loss‐of‐function variants in the functional domains of SETD1B may induce dysregulation of enzymatic activity of H3K4 methyltransferase and have association with the etiology of neurodevelopmental disorders. However, further accumulation of additional cases will need to clarify the pathogenicity of variants and phenotype‐genotype correlations.
In conclusion, we described an additional case of a de novo SETD1B variant with EMA, developmental delay, ID, and ASD. This case supports the indication that SETD1B may be a causative gene for syndromic NDD with epilepsy, especially its association with EMA. However, further accumulation of cases and functional analyses are necessary for clarifying the phenotypic spectrum and the pathogenesis of SETD1B‐related disorders.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
We would like to thank the patient's family for participating in this work. We also thank K. Shibazaki, M. Tsujimura, and A. Kitamoto for their technical assistance. This work was supported by a Grant‐in‐Aid for Scientific Research (B) from the Japan Society for the Promotion of Science, Grant Number: JP16H05160; the Initiative on Rare and Undiagnosed Diseases (IRUD) Beyond from the Japan Agency for Medical Research and Development, Grant Number: JP18ek0109297; and a HUSM Grant‐in‐Aid from Hamamatsu University School of Medicine.
Hiraide T, Hattori A, Ieda D, et al. De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open. 2019;4:476–481. 10.1002/epi4.12339
REFERENCES
- 1. Bureau M, Tassinari CA. Epilepsy with myoclonic absences. Brain Dev. 2005;27:178–84. [DOI] [PubMed] [Google Scholar]
- 2. Mignot C, von Stulpnagel C, Nava C, Ville D, Sanlaville D, Lesca G, et al. Genetic and neurodevelopmental spectrum of SYNGAP1‐associated intellectual disability and epilepsy. J Med Genet. 2016;53:511–522. [DOI] [PubMed] [Google Scholar]
- 3. Bahi‐Buisson N, El Sabbagh S, Soufflet C, Escande F, Boddaert N, Valayannopoulos V, et al. Myoclonic absence epilepsy with photosensitivity and a gain of function mutation in glutamate dehydrogenase. Seizure. 2008;17:658–64. [DOI] [PubMed] [Google Scholar]
- 4. Myers KA, Scheffer IE. Myoclonic absence seizures in Dravet syndrome. Pediatr Neurol. 2017;70:67–69. [DOI] [PubMed] [Google Scholar]
- 5. Gökben S, Yılmaz S, Klepper J, Serdaroğlu G, Tekgül H. Video/EEG recording of myoclonic absences in GLUT1 deficiency syndrome with a hot‐spot R126C mutation in the SLC2A1 gene. Epilepsy Behav. 2011;21:200–202. [DOI] [PubMed] [Google Scholar]
- 6. Lee JH, Tate CM, You JS, Skalnik DG. Identification and characterization of the human Set1B histone H3‐Lys4 methyltransferase complex. J Biol Chem. 2007;282:13419–28. [DOI] [PubMed] [Google Scholar]
- 7. Labonne JDJ, Lee KH, Iwase S, Kong IK, Diamond MP, Layman LC, et al. An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet. 2016;135:757–71. [DOI] [PubMed] [Google Scholar]
- 8. Faundes V, Newman WG, Bernardini L, Canham N, Clayton‐Smith J, Dallapiccola B, et al. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am J Hum Genet. 2018;102:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hiraide T, Nakashima M, Yamoto K, Fukuda T, Kato M, Ikeda H, et al. De novo variants in SETD1B are associated with intellectual disability, epilepsy and autism. Hum Genet. 2018;137:95–104. [DOI] [PubMed] [Google Scholar]
- 10. Ito H, Tani I, Yukihiro R, Adachi J, Hara K, Ogasawara M, et al. Validation of an interview‐based rating scale developed in Japan for pervasive developmental disorders. Res Autism Spectr Disord. 2012;6:1265–72. [Google Scholar]
- 11. Traynelis J, Silk M, Wang Q, Berkovic SF, Liu L, Ascher DB, et al. Optimizing genomic medicine in epilepsy through a gene‐customized approach to missense variant interpretation. Genome Res. 2017;27:1715–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. Am J Hum Genet. 2012;91:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nord AS, Lee M, King MC, Walsh T. Accurate and exact CNV identification from targeted high‐throughput sequence data. BMC Genomics. 2011;12:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ikeda H, Imai K, Ikeda H, Matsuda K, Takahashi Y, Inoue Y. Ictal single photon emission computed tomographic study of myoclonic absence seizures. Brain Dev. 2018;40:126–9. [DOI] [PubMed] [Google Scholar]
- 15. Gupta S, Kim SY, Artis S, Molfese Dl, Schumacher A, Sweatt JD, et al. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Webb WM, Sanchez RG, Perez G, Butler AA, Hauser RM, Rich MC, et al. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol Learn Mem. 2017;142(Pt A):66–78. [DOI] [PubMed] [Google Scholar]
- 17. Collins BE, Greer CB, Coleman BC, Sweatt JD. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics Chromatin. 2019;12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu M, Wang PF, Lee JS, Martin‐Brown S, Florens L, Washburn M, et al. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28:7337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schlichter A, Cairns BR. Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J. 2005;24:1222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deciphering Developmental Disorders Study . Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg EJ, de Vries P, et al. Meta‐analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6. [DOI] [PubMed] [Google Scholar]
- 22. Wang Q, Li M, Yang Z, Hu X, Wu HM, Ni P, et al. Increased co‐expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci Rep. 2015;5:18209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials