

Abstract
Objective
Our objective was to evaluate the protective efficacy of the neurosteroid pregnanolone (3α‐hydroxy‐5β pregnan‐20‐one), a GABAA receptor‐positive allosteric modulator, as an adjunct to benzodiazepine therapy against the chemical warfare nerve agent (CWNA) sarin (GB), using whole‐body exposure, an operationally relevant route of exposure to volatile GB.
Methods
Rats implanted with telemetry transmitters for the continuous measurement of cortical electroencephalographic (EEG) activity were exposed for 60 minutes to 3.0 LCt50 of GB via whole‐body exposure. At the onset of toxic signs, rats were administered an intramuscular injection of atropine sulfate (2 mg/kg) and the oxime HI‐6 (93.6 mg/kg) to increase survival rate and, 30 minutes after seizure onset, treated subcutaneously with diazepam (10 mg/kg) and intravenously with pregnanolone (4 mg/kg) or vehicle. Animals were evaluated for GB‐induced status epilepticus (SE), spontaneous recurrent seizures (SRS), impairment in spatial memory acquisition, and brain pathology, and treatment groups were compared.
Results
Delayed dual therapy with pregnanolone and diazepam reduced time in SE in GB‐exposed rats compared to those treated with delayed diazepam monotherapy. The combination therapy of pregnanolone with diazepam also prevented impairment in the Morris water maze and reduced the neuronal loss and neuronal degeneration, evaluated at one and three months after exposure.
Significance
Neurosteroid administration as an adjunct to benzodiazepine therapy offers an effective means to treat benzodiazepine‐refractory SE, such as occurs following delayed treatment of GB exposure. This study is the first to present data on the efficacy of delayed pregnanolone and diazepam dual therapy in reducing seizure activity, performance deficits and brain pathology following an operationally relevant route of exposure to GB and supports the use of a neurosteroid as an adjunct to standard anticonvulsant therapy for the treatment of CWNA‐induced SE.
Keywords: neurosteroids, benzodiazepine, chemical warfare nerve agent, refractory status epilepticus, thalamus
Key Points.
Sarin whole‐body exposure in rats results in benzodiazepine‐pharmacoresistant status epilepticus when diazepam treatment is delayed.
Performance deficits in the Morris water maze and severe brain pathology are present in the months after whole‐body exposure to sarin in diazepam‐treated rats.
Delayed pregnanolone and diazepam dual therapy rapidly terminates status epilepticus in sarin‐exposed rats compared with diazepam monotherapy.
Delayed pregnanolone and diazepam dual therapy provides functional and neuronal protection against sarin exposure.
Neurosteroid treatment may be a promising adjunct to benzodiazepines in treating refractory status epilepticus.
1. INTRODUCTION
Chemical warfare nerve agents (CWNA) are highly toxic organophosphorus (OP) chemicals that irreversibly inhibit the enzyme acetylcholinesterase (AChE), leading to the accumulation of acetylcholine (ACh) at peripheral and central synapses, which may result in an acute cholinergic crisis, including miosis, hypersecretions, muscle fasciculations, tremors, seizures, and death from respiratory failure (reviewed in Cannard1). The current treatment strategy for CWNA exposure includes a muscarinic ACh receptor antagonist (eg, atropine sulfate), an oxime to reactivate nonfunctional synaptic AChE (eg, 2‐PAM or HI‐6), and a benzodiazepine (eg, diazepam or midazolam) as an anticonvulsant. Rapid administration of these medical countermeasures following CWNA exposure can promote survival and prevent seizure development.2 However, with delayed treatment, glutamatergic mechanisms may sustain seizure activity,3 contributing to the development of seizures that progress to a prolonged and self‐sustained activity level (status epilepticus; SE); these seizures are pharmacoresistant to benzodiazepine therapy.4, 5 Early treatment with midazolam is effective at terminating seizures; however, treatment with midazolam delayed to 30 minutes after G‐agent sarin (GB) exposure in rats does not terminate seizures or prevent performance deficits or seizure‐induced brain damage.6
During a mass casualties event (eg, terrorist attack with the volatile GB), civilians will not carry the antidotes to GB, thus resulting in a delayed time to treatment and likely increase in the severity of symptoms of exposure, including prolonged and treatment‐resistant SE. Identification of effective therapeutics against CWNA‐induced seizures that become refractory to current medical countermeasures is critical.
Prolonged SE leads to the internalization of γ‐aminobutyric acid (GABA) type A (GABAA) receptors7, 8 and externalization of N‐methyl‐D‐aspartate (NMDA) receptor subunits (reviewed in Niquet, et al5 and Wasterlain, et al9). This reduction in the availability of synaptic GABAA receptor targets may contribute to the decrease in benzodiazepine efficacy when treatment of CWNA exposure is delayed. Furthermore, there is a positive correlation with the initial seizure duration and epileptogenesis and more extensive brain pathology.10 Ultimately, the inability to treat SE results in epileptogenesis, performance deficits, and extensive neuronal loss, including loss of GABAergic interneurons.2, 10, 11, 12, 13, 14, 15
One means to treat pharmacoresistant seizures is through the use of neurosteroids, which are positive allosteric modulators of synaptic and extrasynaptic GABAA receptors, and have the potential to produce anticonvulsant effects similar to diazepam but with fewer side effects (reviewed in Birzniece, et al16, and Reddy17). Neurosteroids, such as progesterone and its metabolites, bind to the GABAA receptor at binding sites distinct from benzodiazepines and potentiate GABA‐induced Cl‐ influx.18, 19, 20 Neurosteroids also bind to extrasynaptic GABAA receptors, which mediate tonic inhibition and are considered to have an important role in the pathophysiology of epilepsy (reviewed in Reddy21).
The endogenous activity of neurosteroids is important in regulating seizure activity and may be antiepileptic in the progression of epilepsy.20, 22 The upregulation of cytochrome P450 cholesterol side‐chain cleavage enzyme, important in neurosteroid synthesis, increases in hippocampal glial cells during the latent period following pilocarpine‐induced SE and delays the appearance of spontaneous recurrent seizures (SRS).22 Furthermore, the 5‐alpha‐reductase blocker finasteride, which decreases neurosteroid synthesis, terminates the latent period and promotes seizure activity in epileptic‐prone rats.22 Exogenous administration of the neurosteroid pregnanolone and its analogues is protective in seizure and traumatic brain injury models, including NMDA‐, picrotoxin‐, or pentylenetetrazol‐induced seizures,23, 24 ischemia25, amygdala‐kindled seizures, and models of SE (reviewed in Rogawski26).
We currently report on acute administration of pregnanolone in combination with diazepam in treating seizures that develop following whole‐body exposure to GB, a highly volatile G‐agent with a narrow window of opportunity for therapeutic intervention (reviewed in Munro27). Sarin is primarily an inhalation threat with the main route of entry being the respiratory tract and to a lesser extent skin absorption.28, 29 Although the majority of animal experiments use injection as means of exposure, in the present study, we implemented an operationally relevant model of whole‐body exposure to a lethal concentration of GB to assess the acute and long‐term protective effects of pregnanolone as an adjunct to diazepam when treatment was delayed to 30 minutes after GB‐induced SE. Delayed pregnanolone and diazepam dual therapy quickly terminated GB‐induced SE, prevented the development of performance deficits, and reduced brain pathology.
2. METHODS
2.1. Animals
Male Sprague Dawley rats (350‐400 g; Charles River Laboratories) were housed individually and maintained on a normal 12:12 hours light‐dark cycle (lights on at 0600) at a room temperature of 21 ± 2°C and a humidity level of 60% + 20% at the U.S. Army Combat Capabilities Development Command Chemical Biological Center (USACCDCBC). Rats were moved to the USAMRICD 3‐4 days after GB exposure and gradually shifted (4 h/d) to a reverse 12:12 hours light‐dark cycle (lights on 2100). After 3 weeks of acclimation to the reverse light‐dark cycle, the rats were evaluated during their dark (active) cycle in the Morris water maze test. Food and water were available ad libitum. Rats were weighed daily Monday through Friday. The experimental protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the United States Army Medical Research Institute of Chemical Defense and at USACCDCBC, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89–544), as amended.
2.2. Surgeries
Rats were anesthetized with isoflurane (3%‐5% induction, 1%‐3% maintenance), secured in a Kopf stereotaxic apparatus (David Kopf Instruments) and surgically implanted with a subcutaneous wireless transmitter (F40‐EET; Data Sciences International [DSI], Inc.) to record a 2‐channel (bi‐hemispheric) electrocortigram. Four cortical electrodes made of stainless steel (cortical screws) were placed in the frontal and parietal cortices (2 mm mediolateral to the sagittal suture, 1.6 mm anterior and 4 mm posterior to bregma). Stainless steel wires from the transmitters were implanted subcutaneously (s.c.), wrapped around the electrodes, and secured in place using dental acrylic. The incision sites were sutured, and bacitracin was applied. Buprenorphine (0.03 mg/kg, s.c.) was given for analgesia immediately after surgery and again 6‐12 hours after surgery for up to two days after surgery. Rats then received two weeks of surgical recovery prior to agent exposure.
2.3. Chemicals
Sarin (isopropyl methylphosphonofluoridate; CAS 107‐44‐8; GB) was obtained from USACCDCBC chemical agent standard analytical reagent material stock (certified purity > 97%) and prior to use verified by quantitative 31P‐NMR (nuclear magnetic resonance) and stored in sealed ampules until vapor dissemination into the whole‐body chamber. All external standards for GB vapor quantification were prepared daily. Atropine sulfate, pregnanolone, and 30% 2‐Hydroxypropyl‐β‐cyclodextrin were purchased from Sigma‐Aldrich. HI‐6 [4‐carbamoyl‐2′‐hydroxyiminomethyl‐1,1′‐oxydimethylen‐di(pyridinium chloride)] was prepared by Starkes Associates. Diazepam (United States Pharmacopeia, USP) was purchased from Hospira Inc. Buprenorphine hydrochloride was purchased from Reckitt Benckiser Pharmaceuticals Inc. Chemicals used for transcardial perfusion (4% paraformaldehyde in 0.1 M phosphate buffer; PB), and 20% sucrose in 0.1 MPB, were purchased from FD Neurotechnologies Inc.
2.4. Sarin whole‐body exposures and treatments
During the middle of the light cycle, rats were placed in individual perforated boxes and exposed to GB vapor by whole‐body exposure in a 1000 L dynamic airflow inhalation chamber (Figure 1) as previously described.30 The Rochester style chamber was constructed of stainless steel, with plexiglass windows on each of its six sides. Butyl rubber gloves were attached to the plexiglass windows and a passport existed to enable treatments during the exposure period (see Whalley, et al31 for methods on ports). Chamber airflow and temperature were monitored continuously, and relative humidity was measured at the beginning and end of the exposure. A Harvard Pump 11 Elite syringe pump (Harvard Apparatus) was used to deliver liquid GB into a spray atomizer, where it was mixed with compressed air to form vapor. Two sampling methods were used to monitor and analyze GB vapor concentration in the chamber.32 Receivers placed outside the plexiglass recorded EEG activity from up to 3 rats per exposure (Figure 1).
Figure 1.

Whole‐body exposure chamber is shown on the left image, and a close‐up of setup inside the chamber (red circle) is shown on the right image. The chamber is equipped with telemetry receivers covered in foil outside the chamber and a plastic‐perforated enclosure inside the chamber to hold the rat. Access ports enabled treatment of animals via a catheter while in the chamber, as well as drawing of blood for analysis of agent levels in blood
Rats were exposed for 60 minutes to 3.0 LCt50 of GB using the 60 minutes LCt50 = 453 mg‐min/m3 and the LC50 = 7.55 mg/m3 as determined previously.33 Rats were treated intramuscularly (i.m.) with an admix of atropine sulfate (2 mg/kg) and HI‐6 (93.6 mg/kg) at onset of toxic signs and were then treated with diazepam (10 mg/kg, s.c.) and pregnanolone [(4 mg/kg intravenously, (i.v.) via a jugular catheter] or diazepam and vehicle (30% 2‐Hydroxypropyl‐β‐cyclodextrin in sterile water; iv) 30 minutes after seizure onset. Dose and route of pregnanolone were in part selected based on pharmacokinetic studies in which 4 mg/kg/min administered i.v. via tail vein was the threshold intravenous dose of pregnanolone needed to obtain a “silent second” on the EEG and is a deep level of anesthesia34. Air control (no agent) rats were exposed to air only in a similar chamber and administered atropine sulfate and HI‐6 followed 30 minutes later by diazepam. Based on prior studies in which untreated animals did not survive exposure to 2LCt50 GB (Lumley, unpub data), a GB only group was not included in this study. During the exposures, rats were observed for toxic signs (tremors, salivation, and behavioral seizures) and real‐time EEG was monitored. Prior to drug administration, 30 minutes after seizure onset and again 24 hours after exposure, blood was drawn from the catheter for GB analysis in red blood cells and plasma (see Whalley, et al.31, and Jakubowski, et al.35 for detailed methods). After exposure, rats were placed in a clean outgassing box to ensure that the rats did not receive a low dose from equipment outgassing and to allow for safe handling and transport. The chamber was purged with air for 10 minutes. The GB t99, defined as the time for the chamber to attain 99% of its equilibrium concentration, ranged from 2.32 to 5.2 minutes.
2.5. EEG recording and analysis
Each F40‐EET telemetry device transmits biopotential data and was associated to an individual model RPC‐1 PhysioTel receiver (DSI) placed under each rat's home cage for EEG acquisition (24 h/d). Following 24 hours of baseline collection, rats were moved to an exposure chamber where receivers outside of the chamber recorded real‐time EEG during the 60‐minute exposure period (Figure 1). EEG was recorded for up to 90 days after exposure. Data were digitized at 250 Hz, 60 Hz notch filter, 0.1 Hz hi‐pass filter, and 1 KHz low‐pass filter and recorded using Dataquest ART 4.1 (Acquisition software; DSI). Seizure onset was recorded and treatment administered 30 minutes after seizure onset within the chamber. Initial seizure duration was blindly determined using NeuroScore (DSI) and was defined as rhythmic high‐amplitude spikes (>2 × baseline) that lasted at least 10 seconds (based on Nissinen, et al36). Scoring of epileptiform activity for the full dataset was identified using a customized MATLAB (Release 2010b, MathWorks) algorithm,37 and the steps were as follows. EEG files were converted to European Data Format (.edf)38 format using Neuroscore (Data Sciences International [DSI], Inc.). The left parietal channel signal was chosen arbitrarily for all the animals to be analyzed by the customized algorithm since bi‐hemispheric channels displayed similar pattern. The data were pre‐processed extracting 2‐second epochs and conditioning the signal using a Butterworth filter (passband of 0.1‐125 Hz; notch filter of 60 Hz). The EEG power was averaged (24‐hour baseline) to determine individual power spectra detection threshold for seizures. Another threshold was set by the slope of a linear robust fit applied to a fast Fourier transform (0.1 Hz resolution with Nyquist frequency of 125 Hz). A visual screening of each event was performed by an observer blind to the treatment conditions to reject false positives in a list of candidate seizures. Measures included SE onset and duration, latency to SRS, number of SRS, and the number of rats that developed SRS. This method of EEG analysis is enough to distinguish isolated spike‐wave discharges from hypersynchronous seizures that occur during acute periods (eg, SE and early recurrent seizures) and chronic stages (eg, SRSs). The different types of detected seizures were similar to those described by Rossetti et al14 and de Araujo Furtado et al39. Power analysis was evaluated during and after exposure and normalized to baseline. The power was averaged in 60‐second epochs with 50% overlap to get the power spectrum density, and the median of power in 60‐minute intervals for up to 90 days after exposure was determined for five frequency bands: delta (0.1‐4 Hz), theta (4.1‐8 Hz), alpha (8.1‐12 Hz), beta (12.1‐25 Hz), and gamma (25.1‐50 Hz). Data were normalized (ratio to baseline) to compensate for individual variations. SRS events were identified by the algorithm and the observer by oscillations in the delta band during seizure that progressed to frequencies in the theta band, culminating with frequencies in the alpha band and the theta band. Due to system limitations at the time of this study, long‐term continuous video‐integration was not feasible.
2.6. Morris water maze
Behavioral testing was conducted at least 3 hours into the dark cycle, when animals are active and spend more time awake. Three groups of rats (Air control, GB + delayed diazepam monotherapy, and GB + delayed pregnanolone and diazepam dual therapy) were evaluated at 1 month after exposure for spatial memory acquisition in the Morris water maze (MWM). A hidden platform (10 × 10 cm) was in a fixed position below the water surface in a 170 cm diameter × 60 cm high pool with paint blackened water (26°C; for detailed methods see Schultz, et al15). Briefly, rats received four 60‐second trials per session, 2 training sessions per day and a 30‐minute rest period between sessions for a total of 8 trials/d. Latency to escape, swim speed, distance travelled (path length), and thigmotaxis (perimeter swim) were measured using a video tracking program (HVS Watermaze 2100, HVS Image). After three training days, the platform was removed, and two 60‐second probe trials were conducted. Then, visual acuity was evaluated in four successive trials measuring latency to locate a visible platform. In a subset of rats, tests were repeated 3 months after exposure with new visual cues and target quadrant.
2.7. Neuroanatomical assessments
Rats were deeply anesthetized with sodium pentobarbital [(75 mg/kg, intraperitoneal (i.p.)] and perfused with 0.1 M phosphate buffer (PB) followed by 4% paraformaldehyde in 0.1 MPB (FD Neurotechnologies, Inc.). Heparin sodium injection (0.1 mL per L of 1000 Units per mL; Henry Schein Animal Health) was added to the PB. Brains were post‐fixed for 6 hours in 4% paraformaldehyde at 4‐8°C and cryoprotected in 20% sucrose in 0.1 MPB, and tissue was rapidly frozen and stored at −75°C. Histological sectioning and staining were completed by FD Neurotechnologies, Inc. Coronal 50 μm sections were stained with proprietary FD NeuroSilver™ stain to identify degenerating neuronal fibers. Select brain regions were scored blindly for the neuropathology on a scale of 0‐4 as follows: 0—normal tissue; 1—minimal (up to 10% damage); 2—mild (above 10% up to 25% damage); 3—moderate (above 25% up to 45% damage); 4—severe (above 45% damage) (as previously described in Myhrer, et al13, and McDonough, et al40). The groups were compared for damage in the fiber tracts, hippocampus, thalamus, amygdala, and piriform cortex based on recognition of important nuclei within these structures as previously reported.14 For neuronal density measures, coronal 30 µm sections were stained for NeuN immunoreactivity using a monoclonal mouse anti‐NeuN IgG (1:10 000; Millipore) and the avidin‐biotin complex method.41 NeuN profile density was quantified using Image Pro v7.0 (Media Cybernetics Inc.). Brain regions from coronal sections (bregma range of −2.92 to −3.12 mm 42) were traced for regions in the thalamus, amygdala, and piriform cortex using anatomical landmarks; cell density was calculated and averaged in three replicates per region. Total number of cells was divided by the size of the respective region of interest.
2.8. Statistical analysis
Statistical analyses were performed using SPSS (IBM Inc.) and graphs compiled using SigmaPlot (Systat Software Inc.). The initial seizure duration was compared between GB‐exposed treatment groups using a t test. Repeated measures two‐way ANOVA was used to analyze MWM, body weight, and temperature data. Significant interaction effects were analyzed using a one‐way ANOVA. The ordinal data attained from qualitative silver staining analysis were analyzed using a Kruskal‐Wallis test and Mann‐Whitney U test. NeuN immunoreactivity was analyzed using a one‐way ANOVA followed by Tukey's test. Test result values of P < 0.05 were considered statistically significant.
3. RESULTS
3.1. Acute EEG and behavioral seizure and development of spontaneous recurrent seizures following whole‐body GB exposure
Mean latency to toxic signs onset in rats exposed to 3 LCt50 GB was 11.2 minutes (±2.76 (SD)) and to seizure onset was 14.9 minutes (±5.9) into the 60 minutes GB exposure. Two rats died prior to diazepam treatment, and five rats did not develop seizure and were excluded from analysis. EEG tracings prior to SE, during SE and following treatment are shown in Figure 2. Seizure duration during the first 24 hours following GB exposure was significantly reduced in the delayed pregnanolone and diazepam dual therapy rats compared with delayed diazepam monotherapy (Figure 3A; P < 0.01). In addition, delta power increased beginning at 10 minutes into the GB exposure and preceded seizure onset. Treatment with pregnanolone and diazepam reduced delta power within minutes, whereas diazepam monotherapy rats had prolonged increase in delta power (Figure 3B). However, the reversal of the GB‐induced delta power increase in the pregnanolone and diazepam‐treated rats was transient and delta power increase returned within 2 hours of treatment (Figure 3B,C). We observe EEG power in theta power decreases following diazepam treatment in air control group, while the GB‐exposed rats treated with diazepam show an increase in theta power. The diazepam and pregnanolone dual therapy prevents the GB‐induced increase in theta power, albeit only for the first 10 minutes following treatment. A gradual reduction in gamma power is observed so that by 50 minutes into the GB exposure values for the diazepam monotherapy group are significantly different from the air control group; diazepam failed to restore normal gamma power for at least the first 4 hours. Gamma power for the diazepam pregnanolone dual therapy group did not show a significant reduction in gamma power until 2.5 hours after GB exposure. Only three rats exposed to GB developed SRS, two that were treated with diazepam (latency 28 and 36 days; duration 14 and 90 seconds, respectively) and one treated with combination pregnanolone and diazepam (latency 29 days; duration 45 seconds), with each rat showing only one SRS.
Figure 2.
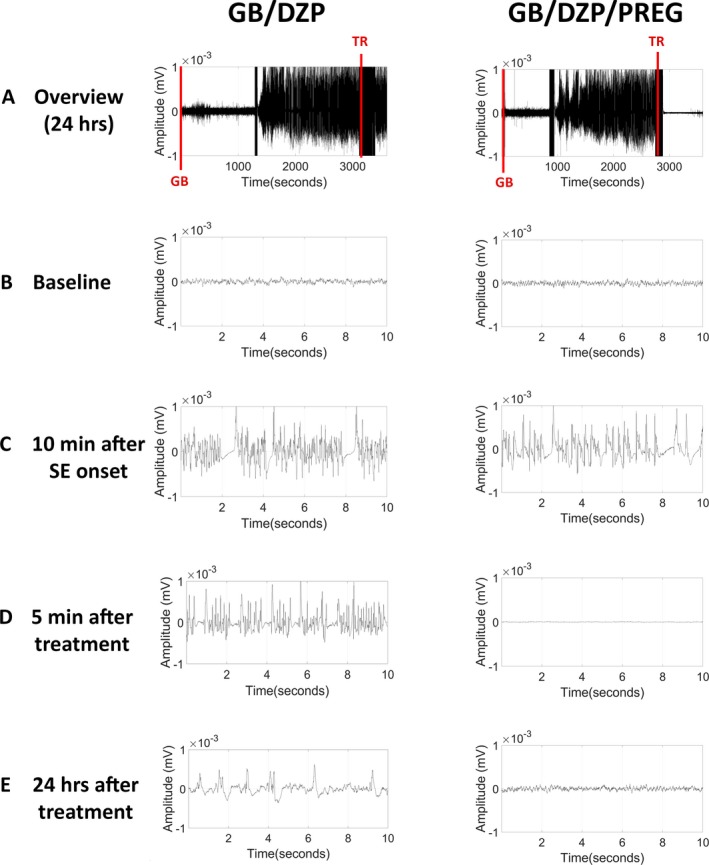
Representative EEG tracings depicting the progression of EEG graphic activity before, during and after GB exposure and treatment (TR). A, The overview image corresponds to compressed EEG for a 24 h period. Note that GB‐exposed rats that received diazepam and pregnanolone (GB/DZP/PREG) 30 minutes after seizure onset had reduced overall amplitude of the signal faster than those that received diazepam monotherapy (GB/DZP). B, A 10‐s representative image of baseline EEG is shown. C, Ten min after SE onset, both groups presented high‐amplitude hypersynchronous epileptiform discharges. D, Five minutes after treatment, only the GB/DZP/PREG group showed total blockade of seizures. E, Twenty‐four hours after treatment the GB/DZP group still displayed high‐amplitude spikes, while the GB/DZP/PREG group had EEG similar to the baseline
Figure 3.
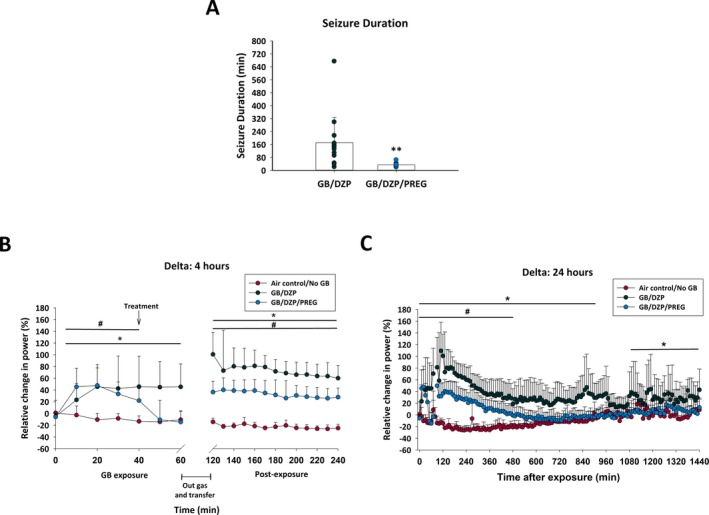
Rats exposed via whole‐body inhalation to GB and treated with atropine sulfate and HI‐6 at onset of toxic signs and with diazepam with or without pregnanolone 30 min after seizure onset were evaluated for initial seizure duration and EEG power. A, Dual therapy with diazepam and pregnanolone (GB/DZP/PREG; n = 16) reduced initial seizure duration, terminating seizure activity within a few minutes of treatment, compared to rats treated with diazepam monotherapy (GB/DZP; n = 15) which displayed seizure activity for several hours after exposure (P < 0.001). B and C, The percent of relative change from baseline in the power of delta (0.1‐4 Hz) band was calculated for each group up to 4 and 24 h for GB/DZP/PREG and GB/DZP groups compared with values of control (Air control/No GB; n = 10) rats. The power spectra in the delta band returned to normal levels faster in the GB/DZP/PREG group when compared to GB/DZP group. During the refractory seizures period, GB/DZP power spectra remained elevated for sustained periods of time when compared with Air control/No GB, while the GB/DZP/PREG power spectra remained elevated only for a limited time (up to 8 h) when compared to Air control/No GB. Statistically significant differences between groups at specific time points are marked by a line indicated above or below the graphed data. *P < 0.05, GB/DZP compared with Air control/No GB. #P < 0.05, GB/DZP/PREG compared with Air control/No GB
3.2. Body weights and temperature and GB
For changes in body weight, there was a significant interaction between treatment group and day (P < 0.001). An ANOVA indicated that rats exposed to GB that received diazepam weighed less than air control rats 1 and 2 days after exposure (P < 0.01) and returned to baseline by 5 days after exposure, while rats that received pregnanolone and diazepam were not different in body weight from air control or from baseline (Figure 4A). Body weight in diazepam‐treated GB‐exposed rats recovered by 7 days after exposure. Control and pregnanolone‐ and diazepam‐treated GB‐exposed rats had significant increase in body weight by 5 days after exposure (P < 0.05). Weight changes are only shown in Figure 4A for the animals in the chronic (3 months) study.
Figure 4.

Rats exposed to GB and treated with atropine sulfate and HI‐6 at onset of toxic signs and with diazepam monotherapy (GB/DZP; n = 7) 30 min after seizure onset lost significant weight by 24 h after exposure. Shown are body weight changes in rats that were evaluated daily (M‐F) over a 90‐d period after GB exposure. Body weight remained less than baseline and less than air control (Air control/No GB; n = 6) on post‐exposure days (PED) 1 and 2 (**P < 0.01). In contrast, rats exposed to GB that received pregnanolone and diazepam dual therapy (GB/DZP/PREG; n = 8) were not different from Air control/No GB initially after exposure, had significant gain above their baseline by PED 5 and by 2 mo after exposure weighed more than air control rats (P < 0.05). GB‐exposed rats tended to gain more weight than air control by 3 mo after exposure but this did not reach significance. B, Rats that received diazepam and pregnanolone dual therapy (n = 16) 30 min after GB‐induced seizure onset had greater reduction in body temperature between 1 and 6 h after exposure compared to air control rats treated with diazepam (n = 15; *P < 0.05). All groups had reduced body temperature compared with their baseline, with air control having reduced temperature 1‐4 h after air exposure, GB/DZP rats having reduced body temperature 2‐7 h after exposure and GB/DZP/PREG reducing temperature 1‐8 h after exposure
For body temperature, there was a significant interaction effect since all rats had decreased body temperature after exposure, yet rats treated with pregnanolone and diazepam had a greater decrease in body temperature, reaching moderate hypothermia, compared with air control rats from 1 to 6 hours after exposure (Figure 4B). Air control (diazepam‐treated) rats had decreased temperature 1‐4 hours after air exposure, GB‐exposed diazepam‐treated rats had decreased temperature from 2 to 7 hours, and GB‐exposed pregnanolone and diazepam‐treated rats had decreased temperature from 1 to 8 hours after GB.
At 30 minutes after seizure onset, the fluoride ion‐extracted GB averaged 64.3 ng/g in red blood cells (rbc) and 32.9 ng/g in plasma of rats. At 24 hours after exposure, plasma showed a robust decrease in GB concentration to 2.8 ng/g, while the GB in rbc remained elevated at 55.6 ng/g.
3.3. Morris water maze (MWM)
Rats exposed to 3 LCt50 GB and treated with diazepam were impaired in a MWM test compared with air control, while rats exposed to GB and treated with pregnanolone and diazepam performed similarly to air control 1 and 3 months after exposure (Figure 5). There was a significant effect of treatment group in that GB‐exposed rats treated with diazepam had longer latency to locate the submerged platform, had greater distance travelled and greater thigmotaxis compared with air control rats (P < 0.05; Figure 5A‐C), whereas those treated with pregnanolone and diazepam were not significantly different from air control. There was no significant group effect on swim speed (Figure 5D). Performance in the MWM was similar between the last session of the 1‐month test and the first session of the 3‐month test for all groups, suggesting that the rats retained their ability to locate the platform in the MWM test despite different cues and target location from the 1‐month test.
Figure 5.
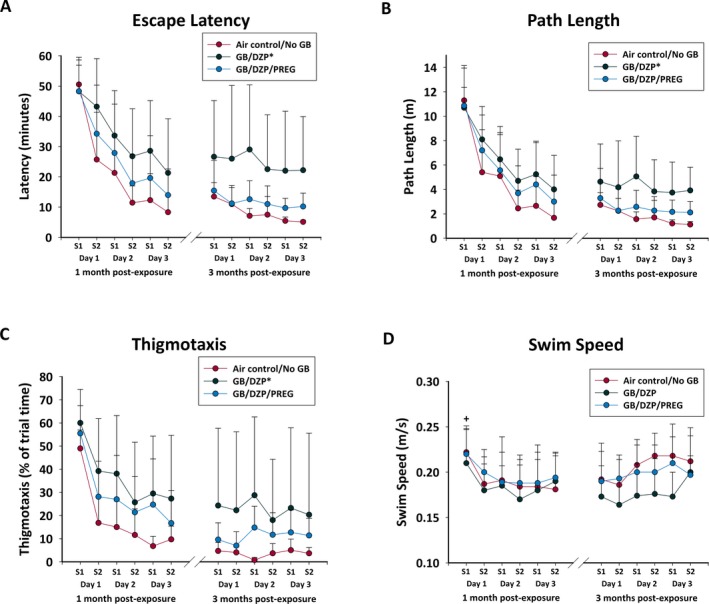
Rats exposed to GB and treated with atropine sulfate and HI‐6 at onset of toxic signs and with diazepam monotherapy (GB/DZP; n = 8) 30 min after seizure onset had impaired performance in Morris water maze test 1 mo after exposure, compared with Air control/No GB (n = 8) shown by (A) increased latency to escape, (B) increased path length (distance travelled), and (C) increased thigmotaxis (perimeter swim). Rats exposed to GB that received pregnanolone and diazepam dual therapy (GB/DZP/PREG; n = 15) were not impaired. All groups improved performance with repeated trials, but the diazepam‐treated GB‐exposed did not perform as well as the air control rats. D, There was no significant effect of GB on swim speed in any group, although all rats had greater swim speed on their first session (S1) compared with their other sessions. Re‐evaluation at 3 mo in a subset of rats showed that the GB/DZP group (n = 6) continued to show greater latency to escape and more thigmotaxis compared with Air control/NoGB (n = 5), while GB/DZP/PREG rats (n = 8) were not impaired. *P < 0.05; +P < 0.05
In the probe test, GB‐exposed rats that received delayed diazepam monotherapy spent less time in the target quad compared with those that received pregnanolone (at 1 and 3 months) and had fewer crossings of the platform (at 3 months). At 1 month after GB, diazepam‐treated rats tended to spend more time in thigmotaxis compared with air control or pregnanolone‐treated rats. During the visual acuity test 1 month after GB exposure (and trend at 3 months), rats that received diazepam after GB exposure had longer latency to locate the visible platform (36.7 ± 15.8 seconds) compared with air control (14.2 ± 5.9 seconds; P < 0.05). Latency to locate the platform in pregnanolone and diazepam‐treated rats (21.7 ± 12.4 seconds) was not significantly different from air control at either time point.
3.4. Neuropathology
In GB‐exposed rats that received delayed diazepam monotherapy, silver staining revealed brain pathology in fiber tracts throughout the brain, as well as in the thalamus, amygdala, and piriform cortex at 1 and/or 3 months after exposure (Figure 6). The combination of pregnanolone and diazepam following GB exposure prevented or reduced damage in fiber tracts, the thalamus, and the piriform cortex at 1 and 3 months after exposure, as well as in the amygdala at 3 months after exposure. GB‐exposed rats treated with delayed diazepam monotherapy also had significant neuronal loss within the medial and lateral thalamus compared with air control, which was attenuated in the pregnanolone and diazepam dual therapy‐treated group (Figure 7). GB exposure also resulted in neuronal loss in the amygdala and piriform cortex at 1 and 3 months after exposure in diazepam‐treated rats; the addition of pregnanolone did not prevent this neuronal loss.
Figure 6.
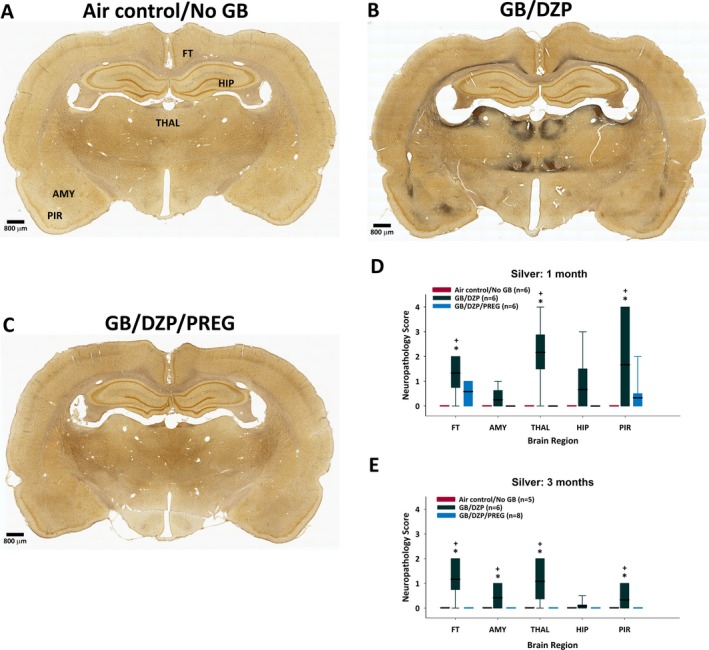
Silver stain of neuronal degeneration in brains was evaluated in rats euthanized 1 or 3 mo after whole‐body exposure to GB followed by atropine sulfate and HI‐6 at onset of toxic signs and diazepam with or without pregnanolone 30 min after seizure onset. Representative images of silver‐stained coronal sections from (A) Air control/No GB rats, (B) rats exposed to GB and treated with delayed diazepam monotherapy (GB/DZP), and (C) rats exposed to GB and treated with delayed diazepam and pregnanolone dual therapy (GB/DZP/PREG) showing neuronal fiber degeneration with darkened regions indicative of fiber and neuronal degeneration. D and E, A semi‐qualitative assessment of silver‐stained brain sections was performed, and the median neuropathology score for each group is presented in a box‐and‐whiskers graph (box: median or 50th percentile represented by bold line, with limits of box representing the 25th and 75th percentiles; whiskers: 10th and 90th percentiles). GB‐exposed rats that received diazepam had extensive neuropathology in the fiber tracts, thalamus and piriform cortex 1 mo after GB exposure (D), as well as in the amygdala 3 mo (E) after GB exposure compared with air control rats (+P < 0.05). Rats that received dual therapy of pregnanolone and diazepam were not significantly different from control and similar to control were significantly different from diazepam monotherapy (*P < 0.05). GB/DZP, GB plus diazepam; GB/DZP/PREG, GB plus diazepam and pregnanolone; FT, fiber tracts; AMY, amygdala; THAL, thalamus; HIP, hippocampus; PIR, piriform cortex
Figure 7.
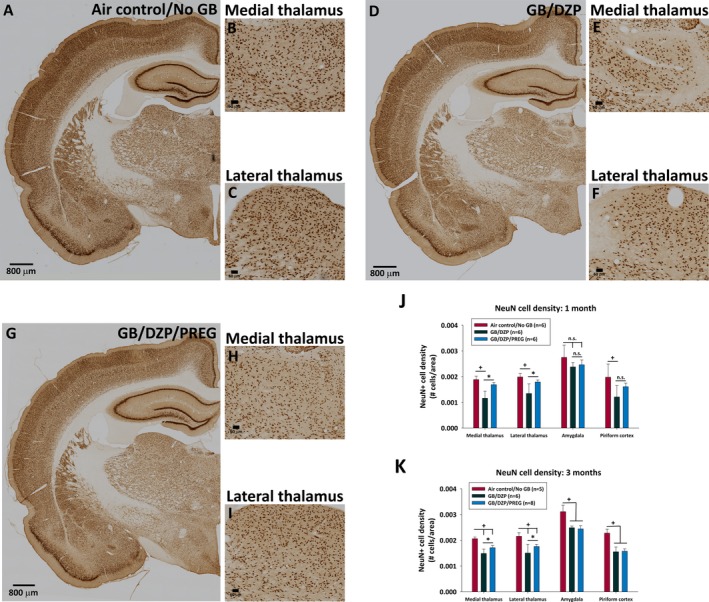
NeuN cell density in rat brains collected 1 or 3 mo after whole‐body exposure to GB followed by atropine sulfate and HI‐6 at onset of toxic signs and diazepam with or without pregnanolone 30 min after seizure onset. Representative images of NeuN‐stained coronal brain sections in control rats not exposed to GB (Air control/NoGB; A‐C), rats exposed to GB and treated with delayed diazepam monotherapy (GB/DZP; D‐F), and rats exposed to GB and treated with delayed diazepam and pregnanolone dual therapy (GB/DZP/PREG; G‐I). J, Bar graphs of average (±SD) NeuN cell density 1 mo after GB exposure showing fewer neurons in the medial and lateral thalamus and the piriform cortex of GB‐exposed rats treated with delayed diazepam monotherapy compared with air control (+P < 0.05). Rats that received delayed dual therapy of diazepam and pregnanolone did not have significant loss of neurons in the thalamus or piriform cortex compared with air control and in the thalamus had significantly more neurons compared with diazepam monotherapy (*P < 0.05). K, Bar graphs of average (±SD) NeuN cell density 3 mo after GB exposure showing that rats treated with pregnanolone and diazepam dual therapy had less neuronal loss in the medial and lateral thalamus compared with those that received diazepam monotherapy. Both GB exposure groups had significantly loss of neurons in the amygdala and piriform cortex compared with air control at 3 mo after GB exposure (+P < 0.05)
4. DISCUSSION
Our findings support the long‐term benefit of combining a neurosteroid with a benzodiazepine to rapidly terminate CWNA‐induced seizure activity and reduce brain damage that may contribute to epileptogenesis and performance deficits. Unless benzodiazepine treatment is administered shortly after exposure (eg, within 30 minutes), benzodiazepine‐refractory SE develops (reviewed in Niquet, et al5, Lallement, et al2, and 43). Effective therapies to rapidly terminate seizure activity are particularly important in the event of civilian mass casualties where a delay in treatment of initial SE is highly probable. Furthermore, considering 50% of patients treated with a first‐line antiepileptic drug therapy go into remission and another 15%‐25% do not respond with one or more additional drug therapies (reviewed in Biagini, et al22), there is a need to identify more effective drugs or drug combinations to treat refractory SE. We report that treatment with a drug combination that acts at varying subunits of GABAA receptors to target both extrasynaptic and synaptic GABAA receptors is beneficial in treating benzodiazepine‐pharmacoresistant seizures. Combination therapy with pregnanolone and diazepam delayed to 30 minutes after seizure onset quickly terminated seizure activity, prevented long‐term performance deficits, protected against weight loss, and reduced the extent of neuropathology in brain regions particularly sensitive to seizure‐induced damage. In comparison, rats treated with delayed diazepam monotherapy had prolonged seizure activity, impaired performance, and extensive brain pathology up to 3 months after exposure.
The animals treated with pregnanolone and diazepam had reduced frequency and duration of seizure activity and faster attenuation of SE when compared with rats treated with diazepam alone. Evaluation of individual power bands during exposure and following treatment revealed GB‐induced increase in delta and theta band and reduced gamma activity, which is consistent with prior studies in soman‐exposed rodents.44, 45, 46, 47 The pregnanolone plus diazepam treatment rapidly attenuated GB‐induced increase in delta band within 5 minutes. Jalilifar and co‐workers48 found that an increase in delta oscillations is associated with behavioral seizure progression in a model of amygdala kindling. The reversible increase in delta and theta bands in the pregnanolone and diazepam dual therapy rats, along with reduced brain pathology and prevention of performance deficits, indicates a superior efficacy of this combination when compared to diazepam monotherapy. However, the reversal of GB‐induced increase in delta and theta band was transient, and the dual therapy did not completely protect the brain.
Only three rats exposed to GB by whole‐body exposure developed SRS, two that were treated with diazepam, and one treated with dual therapy; latency to SRS was ~1 month with only a single SRS in each animal. This is in contrast with previous studies in our laboratory showing that following subcutaneous exposure to 1.2 LD50 soman in rats, the majority of animals that receive delayed benzodiazepine treatment develop SRS within weeks of exposure (Wasterlain et al., submitted). Similarly, nearly all serum carboxylesterase knockout (ES1−/−) mice exposed subcutaneously to a seizure‐inducing dose of soman develop SRS within a week of exposure.44 It is possible that the contrasting effects on SRS incidence and frequency between subcutaneous soman and whole‐body exposure to GB may be a result of more severe effects of soman, which undergoes a rapid dealkylation reaction (aging) which prevents reactivation by oximes,49 and may result in more prolonged SE and associated SRS, rather than effect of route of administration.
We did not include a pregnanolone monotherapy group in this study as the focus was on evaluating a neurosteroid as an adjunct to diazepam and not as a replacement for diazepam. However, we previously observed that the neurosteroids allopregnanolone and pregnanolone were less effective compared to dual neurosteroid therapy with diazepam against SE following 1.2 LD50 soman (Lumley, unpublished data). In addition to protective efficacy of neurosteroids in many seizure and traumatic brain injury models (reviewed in Rogawski et al26, and Reddy21), neurosteroids at inactive doses increase the effectiveness of diazepam above diazepam monotherapy against pentylenetetrazol‐induced seizures.24 Also, combination of the neurosteroid allopregnanolone with diazepam protects mice against tetramethylenedisulfotetramine (TETS), a potent convulsant GABA receptor blocker,50 suggesting that neurosteroids and benzodiazepine combination therapy may have broad‐spectrum effects in treating SE. One suggested benefit of neurosteroids compared with benzodiazepines is that there is no tolerance to the anticonvulsant effects of neurosteroids, as mice treated repeatedly with high doses of pregnanolone do not develop tolerance to the anticonvulsant effects of pregnenolone.51 However, tolerance to the anesthetic effects of allopregnanolone, as well as decreased expression of GABAA alpha4 receptor, occurs following both acute and chronic administration.52 If tolerance occurs, additional drugs other than those acting on the GABAA receptor may be needed to treat SE, unless only one treatment time is anticipated.
Since increasing GABA receptor function with pregnanolone and diazepam combination reduced fiber degeneration and neuronal loss in the thalamus but did not fully protect neurons in the piriform cortex or the amygdala, there may be additional need for a drug that targets the glutamatergic component. One candidate is the NMDA antagonist ketamine, which synergizes with benzodiazepines in treating cholinergic‐induced refractory SE4, 5, or caramiphen which has anti‐cholinergic and anti‐glutamatergic effects and in combination with diazepam reduces SE, SRS, and brain damage.15 Combination therapy that increases GABA activity and reduces glutamate activity provides additional benefit, and administration of lower drug doses may reduce side effects and potentially drug tolerance. Future studies are needed to determine whether neurosteroids in combination with ketamine and benzodiazepine are beneficial.
In addition to the pregnanolone and diazepam combination treatment reducing time in SE following GB exposure, the moderate hypothermia in this treatment group may be relevant to the neuroprotective findings considering that hypothermia is neuroprotective in animal models of epileptic, ischemic, and traumatic brain injury (reviewed in Motamedi, et al53). Hypothermia alters numerous mechanisms that may contribute to neuroprotection, including reduction in cerebral metabolic rate and in oxygen consumption, alteration of ion pumps and channels, and slowing of release of excitatory neurotransmitters, all of which may inhibit seizure activity.54 Mild hypothermia reduces seizure frequency following intra‐amygdalar kainate injection55 and in combination with diazepam reduces performant pathway‐induced SE.56 Hypothermia also reduces blood‐brain barrier disruption and cell‐mediated inflammation induced by traumatic brain injury57, 58 and could be protective against similar effects that follow exposure to nerve agents. In pilocarpine‐exposed rats, deep hypothermia reduces SE54 and moderate hypothermia (30‐33°C) reduces mortality, prevents the calcium plateau, and reduces hippocampal injury.59 Exposure to CWNA, including GB and GD, reduces body temperature in animal models,60, 61 as does diazepam62 and pregnanolone63 treatments. Although the diazepam‐treated GB‐exposed rats also had reduced body temperature, it was not significantly greater than the air control rats that received diazepam. In contrast, the pregnanolone and diazepam combination‐treated rats reduced temperature below 33°C and were significantly lower compared with control.
Whole‐body exposure to GB for 60 minutes followed by delayed treatment with diazepam reduced body weight during the first 2‐3 days after exposure, similarly to findings after 10 minutes whole‐body exposure to GB64 and after soman exposure.65 In the current study, GB‐exposed rats tended to gain more weight than air control weight in the months after exposure, but this did not reach significance. We observe similar weight gain in soman‐exposed rats by 3 months after exposure (unpub data). Consistent with these findings in nerve agent‐exposed rats, pilocarpine‐induced SE and epileptogenesis are associated with weight gain and increase in abdominal fat66, 67 and obesity is commonly comorbid with human epilepsy.68 Interestingly, in the pilocarpine study, changes in expression of metabolic genes in the hippocampus are observed. The CA1 region of the hippocampus, typically damaged following SE, including following whole‐body exposure to GB,64 is linked with hypothalamic regions that regulate food intake as well as regions of the thalamus.69 In addition, a small subset of neurons in the dentate gyrus are important for hippocampal regulation of the hypothalamic‐pituitary axis feedback control.70 More studies are needed to determine potential long‐term effects of GB exposure on body weight and food intake.
Delayed combination therapy with pregnanolone and diazepam, but not diazepam monotherapy, prevented the GB‐induced performance deficits in the Morris water test. A main effect of exposure to GB‐induced prolonged SE is increased thigmotaxis (perimeter swim), and longer latency to locate the submerged platform and greater distanced travelled. Although diazepam‐treated GB‐exposed rats do not perform as well as control on this test, performance improves with repeated trials. Medidorsal thalamic lesions increase thigmotaxis in the Morris water test in rats and, although their search strategies differ from intact animals, the animals demonstrate learning.71 In GB‐exposed rats treated with delayed diazepam monotherapy, the thalamus was extremely damaged, while those that received pregnanolone and diazepam did not have thalamic damage or display thigmotaxis. In a visual acuity test 30 days after GB exposure, GB‐exposed rats treated with delayed diazepam monotherapy took longer to reach the visible platform which may relate to time in thigmotaxis or may suggest impaired vision. However, 90 days after GB exposure, performance in the visual acuity test was not different among groups. A recent draft report by the National Institute of Environmental Health Sciences (NIEHS) on the long‐term follow‐up of humans exposed to GB includes reports of vision problems for weeks to years after exposure in humans, to include blurred vision and dim vision, as well as long‐term effects on learning and memory, but animal data are lacking (unpublished). The first observable toxic sign following a whole‐body exposure to a G‐agent is miosis or constriction of the pupils, which may last for days or weeks (reviewed in Munro27). The current study provides evidence of long‐term performance deficits following GB exposure in rats.
In addition to demonstrating the long‐term protective effects of combining a neurosteroid and a benzodiazepine in treating GB, this study is the first to demonstrate real‐time EEG recording of GB‐exposed rats in an operationally relevant whole‐body exposure chamber and treatment with medical countermeasures during the exposure. The majority of animal studies to assess drug efficacy against CWNA exposure use injection of agent, which is easier, safer, has better dosing accuracy with higher throughput and less cost. However, operationally relevant exposures may better model human physiological responses to G‐agent exposure. In conclusion, our findings of long‐term benefits of pregnanolone and diazepam combination therapy following GB‐induced SE suggest that neurosteroid combination with a benzodiazepine may be an effective strategy against CWNA‐induced refractory SE, but that additional therapies may also be needed.
DISCLOSURES OF CONFLICTS OF INTEREST
Author 5 was paid under a contract to process and analyze EEG data while blinded to experimental conditions. All other authors have no conflicts of interest.
ACKNOWLEDGEMENTS
This research was supported by the DTRA‐JSTO, Medical S&T Division to Lucille Lumley. The authors acknowledge Nicole Vincelli and Edward Emm for technical support in the exposures and Wuya Lumeh, Julia Morgan, Mark Schultz, Caroline Schultz, and Nathan Kelley for technical support in telemetry or behavioral data collection and/or data compilation, and Katie Walker for reference management and data entry. The authors acknowledge Cindy Kronman for editorial review.
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Lumley L, Miller D, Muse WT, et al. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long‐term effects of whole‐body sarin exposure in rats. Epilepsia Open. 2019;4:382–396. 10.1002/epi4.12344
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
REFERENCES
- 1. Cannard K. The acute treatment of nerve agent exposure. J Neurol Sci 2006;249(1):86–94. [DOI] [PubMed] [Google Scholar]
- 2. Lallement G, Dorandeu F, Filliat P, Carpentier P, Baille V, Blanchet G. Medical management of organophosphate‐induced seizures. J Physiol Paris 1998;92(5–6):369–73. [DOI] [PubMed] [Google Scholar]
- 3. McDonough JH Jr, Shih TM. Neuropharmacological mechanisms of nerve agent‐induced seizure and neuropathology. Neurosci Biobehav Rev 1997;21(5):559–79. [DOI] [PubMed] [Google Scholar]
- 4. Niquet J, Baldwin R, Norman K, Suchomelova L, Lumley L, Wasterlain CG. Midazolam‐ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia 2016;57(9):1406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Niquet J, Baldwin R, Suchomelova L, Lumley L, Naylor D, Eavey R, Wasterlain CG. Benzodiazepine‐refractory status epilepticus: pathophysiology and principles of treatment. Ann N Y Acad Sci 2016;1378(1):166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilat E, Kadar T, Levy A, Rabinovitz I, Cohen G, Kapon Y, Sahar R, Brandeis R. Anticonvulsant treatment of sarin‐induced seizures with nasal midazolam: an electrographic, behavioral, and histological study in freely moving rats. Toxicol Appl Pharmacol 2005;209(1):74–85. [DOI] [PubMed] [Google Scholar]
- 7. Goodkin HP, Yeh J‐L, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci 2005;25(23):5511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 2005;25(34):7724–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wasterlain CG, Naylor DE, Liu H, Niquet J, Baldwin R. Trafficking of NMDA receptors during status epilepticus: therapeutic implications. Epilepsia 2013;54 (Suppl 6):78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Araujo Furtado M, Lumley LA, Robison C, Tong LC, Lichtenstein S, Yourick DL. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague‐Dawley rats. Epilepsia 2010;51(8):1503–10. [DOI] [PubMed] [Google Scholar]
- 11. Apland JP, Aroniadou‐Anderjaska V, Figueiredo TH, Rossetti F, Miller SL, Braga MFM. The limitations of diazepam as a treatment for nerve agent‐induced seizures and neuropathology in rats: comparison with UBP302. J Pharmacol Exp Ther 2014;351(2):359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marrero‐Rosado Brenda, Rossetti Franco, Rice Matthew W, Moffett Mark C, Lee Robyn B, Stone Michael F, Lumley Lucille A. Age‐related susceptibility to epileptogenesis and neuronal loss in male Fischer rats exposed to Ssman and treated with medical countermeasures. Toxicol Sci 2018;164(1):142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Myhrer T, Andersen JM, Nguyen NHT, Aas P. Soman‐induced convulsions in rats terminated with pharmacological agents after 45 min: neuropathology and cognitive performance. Neurotoxicology 2005;26(1):39–48. [DOI] [PubMed] [Google Scholar]
- 14. Rossetti F, de Araujo Furtado M, Pak T, Bailey K, Shields M, Chanda S, Addis M, Robertson BD, Moffett M, Lumley LA, Yourick DL. Combined diazepam and HDAC inhibitor treatment protects against seizures and neuronal damage caused by soman exposure. Neurotoxicology 2012;33(3):500–11. [DOI] [PubMed] [Google Scholar]
- 15. Schultz MK, Wright LKM, de Araujo Furtado M, Stone MF, Moffett MC, Kelley NR, Bourne AR, Lumeh WZ, Schultz CR, Schwartz JE, Lumley LA. Caramiphen edisylate as adjunct to standard therapy attenuates soman‐induced seizures and cognitive deficits in rats. Neurotoxicol Teratol 2014;44: 89–104. [DOI] [PubMed] [Google Scholar]
- 16. Birzniece V, Bäckström T, Johansson IM, Lindblad C, Lundgren P, Löfgren M, Olsson T, Ragagnin G, Taube M, Turkmen S, Wahlström G, Wang MD, Wihlbäck AC, Zhu D. Neuroactive steroid effects on cognitive functions with a focus on the serotonin and GABA systems. Brain Res Rev 2006;51(2):212–39. [DOI] [PubMed] [Google Scholar]
- 17. Reddy DS. Neurosteroids for the potential protection of humans against organophosphate toxicity. Ann N Y Acad Sci 2016;1378(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kokate TG, Svensson BE, Rogawski MA. Anticonvulsant activity of neurosteroids: correlation with gamma‐aminobutyric acid‐evoked chloride current potentiation. J Pharmacol Exp Ther 1994;270(3):1223–9. [PubMed] [Google Scholar]
- 19. Lambert JJ, Belelli D, Peden DR, Vardy AW, Peters JA. Neurosteroid modulation of GABA A receptors. Prog Neurobiol 2003;71(1):67–80. [DOI] [PubMed] [Google Scholar]
- 20. Reddy DS, Rogawski MA. Stress‐induced deoxycorticosterone‐derived neurosteroids modulate GABA(A) receptor function and seizure susceptibility. J Neurosci 2002;22(9):3795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reddy DS. GABA‐A receptors mediate tonic inhibition and neurosteroid sensitivity in the brain. Vitam Horm 2018;107:177–91. [DOI] [PubMed] [Google Scholar]
- 22. Biagini G, Rustichelli C, Curia G, Vinet J, Lucchi C, Pugnaghi M, Meletti S. Neurosteroids and epileptogenesis. J Neuroendocrinol 2013;25(11):980–90. [DOI] [PubMed] [Google Scholar]
- 23. Członkowska AI, Krząścik P, Sienkiewicz‐Jarosz H, Siemiątkowski M, Szyndler J, Bidziński A, Płaźnik A. The effects of neurosteroids on picrotoxin‐, bicuculline‐and NMDA‐induced seizures, and a hypnotic effect of ethanol. Pharmacol Biochem Behav 2000;67(2):345–53. [DOI] [PubMed] [Google Scholar]
- 24. Gasior M, Carter RB, Goldberg SR, Witkin JM. Anticonvulsant and behavioral effects of neuroactive steroids alone and in conjunction with diazepam. J Pharmacol Exp Ther 1997;282(2):543–53. [PubMed] [Google Scholar]
- 25. Sayeed I, Guo Q, Hoffman SW, Stein DG. Allopregnanolone, a progesterone metabolite, is more effective than progesterone in reducing cortical infarct volume after transient middle cerebral artery occlusion. Ann Emerg Med 2006;47(4):381–9. [DOI] [PubMed] [Google Scholar]
- 26. Rogawski MA. Molecular targets versus models for new antiepileptic drug discovery. Epilepsy Res 2006;68(1):22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Munro N. Toxicity of the organophosphate chemical warfare agents GA, GB, and VX: implications for public protection. Environ Health Perspect 1994;102(1):18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartosova‐Sevelova L, Bajgar J. Changes of acetylcholinesterase activity after long‐term exposure to sarin vapors in rats. Hum Exp Toxicol 2005;24(7):363–7. [DOI] [PubMed] [Google Scholar]
- 29. Sterri SH, Kloster O, Valdal G. Reduction of blood cholinesterase activities following administration of soman by different routes in guinea‐pig. Acta Pharmacol Toxicol (Copenh) 1982;50(5):326–31. [DOI] [PubMed] [Google Scholar]
- 30. Muse WT, Thomson S, Crouse C, Matson K. Generation, sampling, and analysis for low‐level GB (Sarin) and GF (Cyclosarin) vapor for inhalation toxicology studies. Inhal Toxicol 2006;18(14):1101–8. [DOI] [PubMed] [Google Scholar]
- 31. Whalley CE, McGuire JM, Miller DB, Jakubowski EM, Mioduszewski RJ, Thomson SA, Lumley LA, McDonough JH, Shih TMA. Kinetics of sarin (GB) following a single sublethal inhalation exposure in the guinea pig. Inhal Toxicol 2007;19(8):667–81. [DOI] [PubMed] [Google Scholar]
- 32. Wright LK, Lumley LA, Lee RB, Taylor JT, Miller DB, Muse WT Jr, Emm EJ, Whalley CE. Younger rats are more susceptible to the lethal effects of sarin than adult rats: 24 h LC50 for whole‐body (10 and 60 min) exposures. Drug Chem Toxicol 2017;40(2):134–9. [DOI] [PubMed] [Google Scholar]
- 33. Mioduszewski R, Manthei J, Way R, Burnett D, Gaviola B, Muse W, et al. Interaction of exposure concentration and duration in determining acute toxic effects of sarin vapor in rats. Toxicol Sci 2002;66(2):176–84. [DOI] [PubMed] [Google Scholar]
- 34. Zhu D, Wang Md, Bäckström T, Wahlström G. Evaluation and comparison of the pharmacokinetic and pharmacodynamic properties of allopregnanolone and pregnanolone at induction of anaesthesia in the male rat. Br J Anaesth 2001;86(3):403–12. [DOI] [PubMed] [Google Scholar]
- 35. Jakubowski EM, McGuire JM, Evans RA, Edwards JL, Hulet SW, Benton BJ, Forster JS, Burnett DC, Muse WT, Matson K, Crouse CL, Mioduszewski RJ, Thomson SA. Quantitation of fluoride ion released sarin in red blood cell samples by gas chromatography‐chemical ionization mass spectrometry using isotope dilution and large‐volume injection. J Anal Toxicol 2004;28(5):357–63. [DOI] [PubMed] [Google Scholar]
- 36. Nissinen J, Lukasiuk K, Pitkänen A. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus 2001;11(3):299–310. [DOI] [PubMed] [Google Scholar]
- 37. de Araujo Furtado M, Zheng A, Sedigh‐Sarvestani M, Lumley L, Lichtenstein S, Yourick D. Analyzing large data sets acquired through telemetry from rats exposed to organophosphorous compounds: an EEG study. J Neurosci Methods 2009;184(1):176–83. [DOI] [PubMed] [Google Scholar]
- 38. Kemp B, Värri A, Rosa AC, Nielsen KD, Gade J. A simple format for exchange of digitized polygraphic recordings. Electroencephalogr Clin Neurophysiol 1992;82(5):391–3. [DOI] [PubMed] [Google Scholar]
- 39. de Araujo Furtado M, Rossetti F, Chanda S, Yourick D. Exposure to nerve agents: from status epilepticus to neuroinflammation, brain damage, neurogenesis and epilepsy. Neurotoxicology 2012;33(6):1476–90. [DOI] [PubMed] [Google Scholar]
- 40. McDonough JH Jr, Dochterman LW, Smith CD, Shih TM. Protection against nerve agent‐induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology 1995;16(1):123–32. [PubMed] [Google Scholar]
- 41. Hsu SM, Raine L, Fanger H. Use of avidin‐biotin‐peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 1981;29(4):577–80. [DOI] [PubMed] [Google Scholar]
- 42. Paxinos G, Watson C. The rat brain in stereotaxic coordinates: hard cover edition. Elsevier; 2006. [Google Scholar]
- 43. Shih TM, McDonough JH Jr. Organophosphorus nerve agents‐induced seizures and efficacy of atropine sulfate as anticonvulsant treatment. Pharmacol Biochem Behav 1999;64(1):147–53. [DOI] [PubMed] [Google Scholar]
- 44. Marrero‐Rosado B, de Araujo Furtado M, Schultz CR, Stone M, Kundrick E, Walker K, O'Brien S, Du F, Lumley LA. Soman‐induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia 2018;59(12):2206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carpentier P, Foquin A, Dorandeu F, Lallement G. Delta activity as an early indicator for soman‐induced brain damage: a review. Neurotoxicology 2001;22(3):299–315. [DOI] [PubMed] [Google Scholar]
- 46. Koplovitz I, Skvorak JP. Electrocorticographic changes during generalized convulsive status epilepticus in soman intoxicated rats. Epilepsy Res 1998;30(2):159–64. [DOI] [PubMed] [Google Scholar]
- 47. McDonough JH Jr, Clark TR, Slone TW Jr, Zoeffel D, Brown K, Kim S, Smith CD. Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology 1998;19(3):381–91. [PubMed] [Google Scholar]
- 48. Jalilifar M, Yadollahpour A, Moazedi AA, Ghotbeddin Z. Classifying amygdala kindling stages using quantitative assessments of extracellular recording of EEG in rats. Brain Res Bull 2016;127: 148–55. [DOI] [PubMed] [Google Scholar]
- 49. Worek F, Thiermann H, Wille T. The oximes HI‐6 and MMB‐4 fail to reactivate soman‐inhibited human and guinea pig AChE: A kinetic in vitro study. Toxicol Lett 2018;293:216–21. [DOI] [PubMed] [Google Scholar]
- 50. Bruun DA, Cao Z, Inceoglu B, Vito ST, Austin AT, Hulsizer S, Hammock BD, Tancredi DJ, Rogawski MA, Pessah IN, Lein PJ. Combined treatment with diazepam and allopregnanolone reverses tetramethylenedisulfotetramine (TETS)‐induced calcium dysregulation in cultured neurons and protects TETS‐intoxicated mice against lethal seizures. Neuropharmacology 2015;95: 332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kokate TG, Yamaguchi S, Pannell LK, Rajamani U, Carroll DM, Grossman AB, Rogawski MA. Lack of anticonvulsant tolerance to the neuroactive steroid pregnanolone in mice. J Pharmacol Exp Ther 1998;287(2):553–8. [PubMed] [Google Scholar]
- 52. Turkmen S, Backstrom T, Wahlstrom G, Andreen L, Johansson IM. Tolerance to allopregnanolone with focus on the GABA‐A receptor. Br J Pharmacol 2011;162(2):311–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Motamedi GK, Lesser RP, Vicini S. Therapeutic brain hypothermia, its mechanisms of action, and its prospects as a treatment for epilepsy. Epilepsia 2013;54(6):959–70. [DOI] [PubMed] [Google Scholar]
- 54. Niquet J, Baldwin R, Gezalian M, Wasterlain CG. Deep hypothermia for the treatment of refractory status epilepticus. Epilepsy Behav 2015;49: 313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maeda T, Hashizume K, Tanaka T. Effect of hypothermia on kainic acid‐induced limbic seizures: an electroencephalographic and 14C‐deoxyglucose autoradiographic study. Brain Res 1999;818(2):228–35. [DOI] [PubMed] [Google Scholar]
- 56. Schmitt Fc, Buchheim K, Meierkord H, Holtkamp M. Anticonvulsant properties of hypothermia in experimental status epilepticus. Neurobiol Dis 2006;23(3):689–96. [DOI] [PubMed] [Google Scholar]
- 57. Lotocki G, de Rivero Vaccari JP, Perez ER, Sanchez‐Molano J, Furones‐Alonso O, Bramlett HM, Dietrich WD. Alterations in blood‐brain barrier permeability to large and small molecules and leukocyte accumulation after traumatic brain injury: effects of post‐traumatic hypothermia. J Neurotrauma 2009;26(7):1123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oda Yasutaka, Gao Guoyi, Wei Enoch P, Povlishock John T. Combinational therapy using hypothermia and the immunophilin ligand FK506 to targtet altered pial arteriolar reactivity, axonal damage, and blood‐brain barrier dysfunction after traumatic brain injury in rat. J Cereb Blood Flow Metab 2011;31(4):1143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Phillips KF, Deshpande LS, DeLorenzo RJ. Hypothermia reduces mortality, prevents the calcium plateau, and is neuroprotective following status epilepticus in rats. Front Neurol 2018;9:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meeter E, Wolthuis OL. The effects of cholinesterase inhibitors on the body temperature of the rat. Eur J Pharmacol 1968;4(1):18–24. [DOI] [PubMed] [Google Scholar]
- 61. Clement JG. Variability of sarin‐induced hypothermia in mice: investigation into incidence and mechanism. Biochem Pharmacol 1991;42(6):1316–8. [DOI] [PubMed] [Google Scholar]
- 62. Mailliet F, Galloux P, Poisson D. Comparative effects of melatonin, zolpidem and diazepam on sleep, body temperature, blood pressure and heart rate measured by radiotelemetry in Wistar rats. Psychopharmacology (Berl) 2001;156(4):417–26. [DOI] [PubMed] [Google Scholar]
- 63. Melchior CL, Allen PM. Interaction of pregnanolone and pregnenolone sulfate with ethanol and pentobarbital. Pharmacol Biochem Behav 1992;42(4):605–11. [DOI] [PubMed] [Google Scholar]
- 64. Grauer E, Chapman S, Rabinovitz I, Raveh L, Weissman BA, Kadar T, Allon N. Single whole‐body exposure to sarin vapor in rats: long‐term neuronal and behavioral deficits. Toxicol Appl Pharmacol 2008;227(2):265–74. [DOI] [PubMed] [Google Scholar]
- 65. Schultz MK, Wright LKM, Stone MF, Schwartz JE, Kelley NR, Moffett MC, Lee RB, Lumley LA. The anticholinergic and antiglutamatergic drug caramiphen reduces seizure duration in soman‐exposed rats: synergism with the benzodiazepine diazepam. Toxicol Appl Pharmacol 2012;259(3):376–86. [DOI] [PubMed] [Google Scholar]
- 66. Ruiz N, Pacheco LF, Farrell B, Cox CB, Ermolinsky BS, Garrido‐Sanabria ER, Nair S. Metabolic gene expression changes in the hippocampus of obese epileptic male rats in the pilocarpine model of temporal lobe epilepsy. Brain Res 2011;1426: 86–95. [DOI] [PubMed] [Google Scholar]
- 67. Scharfman HE, Kim M, Hintz TM, MacLusky NJ. Seizures and reproductive function: insights from female rats with epilepsy. Ann Neurol 2008;64(6):687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ladino LD, Hernandez‐Ronquillo L, Tellez‐Zenteno JF. Obesity and its association with generalised epilepsy, idiopathic syndrome, and family history of epilepsy. Epileptic Disord 2014;16(3):343–53. [DOI] [PubMed] [Google Scholar]
- 69. Cenquizca LA, Swanson LW. Analysis of direct hippocampal cortical field CA1 axonal projections to diencephalon in the rat. J Comp Neurol 2006;497(1):101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011;476(7361):458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dolleman‐van der Weel MJ, Morris RG, Witter MP. Neurotoxic lesions of the thalamic reuniens or mediodorsal nucleus in rats affect non‐mnemonic aspects of watermaze learning. Brain Struct Funct 2009;213(3):329–42. [DOI] [PubMed] [Google Scholar]