

Abstract
Objective
Molecular genetic etiologies in epilepsy have become better understood in recent years, creating important opportunities for precision medicine. Building on these advances, detailed studies of the complexities and outcomes of genetic testing for epilepsy can provide useful insights that inform and refine diagnostic approaches and illuminate the potential for precision medicine in epilepsy.
Methods
We used a multi‐gene next‐generation sequencing (NGS) panel with simultaneous sequence and exonic copy number variant detection to investigate up to 183 epilepsy‐related genes in 9769 individuals. Clinical variant interpretation was performed using a semi‐quantitative scoring system based on existing professional practice guidelines.
Results
Molecular genetic testing provided a diagnosis in 14.9%‐24.4% of individuals with epilepsy, depending on the NGS panel used. More than half of these diagnoses were in children younger than 5 years. Notably, the testing had possible precision medicine implications in 33% of individuals who received definitive diagnostic results. Only 30 genes provided 80% of molecular diagnoses. While most clinically significant findings were single‐nucleotide variants, ~15% were other types that are often challenging to detect with traditional methods. In addition to clinically significant variants, there were many others that initially had uncertain significance; reclassification of 1612 such variants with parental testing or other evidence contributed to 18.5% of diagnostic results overall and 6.1% of results with precision medicine implications.
Significance
Using an NGS gene panel with key high‐yield genes and robust analytic sensitivity as a first‐tier test early in the diagnostic process, especially for children younger than 5 years, can possibly enable precision medicine approaches in a significant number of individuals with epilepsy.
Keywords: diagnostic genetic testing, next‐generation sequencing panel, copy number variant, precision medicine, clinical management, variant of uncertain significance
Key Points.
Using a multi‐gene panel with key high‐yield genes as a first‐tier test early in the diagnostic process may possibly support precision medicine interventions in a significant number of individuals with epilepsy
Children with epilepsy who are younger than 5 years show a high diagnostic yield from genetic testing
Intragenic deletions and duplications contribute a significant proportion of clinically significant changes in epilepsy genes
Many variants of uncertain significance can be reclassified with familial testing and have clinical management implications
1. INTRODUCTION
Epilepsy is increasingly recognized to have genetic causes, and approaches to its diagnostic workup have been described.1, 2, 3 Aside from determining prognosis and recurrence risk, the identification of a genetic etiology can guide strategies for clinical management in certain forms of epilepsy, providing powerful opportunities for precision medicine.4, 5, 6, 7, 8, 9, 10 This is particularly important in early‐onset epilepsies, several of which are good candidates for precision medicine but in current standard practice do not attract the requisite urgent attention.11
Next‐generation sequencing (NGS) gene panels and whole‐exome or whole‐genome sequencing (WES/WGS) have been used as diagnostic tools for epilepsy and identify a large variety of sequence and copy number variants.2, 12, 13, 14, 15, 16, 17 Accurate interpretation of clinically important variants can be challenging amid naturally existing variation in epilepsy‐related genes.18, 19 One recent study reports a 15% diagnostic yield from a NGS gene panel for epilepsy,12 but most previous studies have involved small cohorts that obscured detailed understanding of the complexities of molecular genetic analysis.20, 21, 22, 23 However, no studies to date have extensively investigated the proportion of individuals in a large cohort with epilepsy for whom a positive molecular genetic diagnosis invokes clinical management implications or precision medicine approaches, either through the use of therapies that ameliorate or eliminate symptoms or through the avoidance of certain contraindicated anti‐epileptic drugs (AEDs).
We analyzed the results of genetic testing in a large cohort with epilepsy to understand the proportion of individuals who receive results with possible precision medicine implications (PMIs), as determined by current literature on using specific therapies for epilepsy. Furthermore, we examined the rate of definitive molecular diagnoses, the spectrum of variants and their classifications, and diagnostic yield by age groups. These data provide deep insight that can inform clinicians on the appropriate use of and expectations from diagnostic genetic testing for epilepsy.
2. MATERIALS AND METHODS
2.1. Next‐generation sequencing assay
Invitae's epilepsy test is an NGS‐based targeted gene panel (not exome‐based) sequenced at high depth of coverage (50× minimum, 350× average) to simultaneously identify single‐nucleotide variants (SNVs), short and long indels, exon‐level deletions/duplications (copy number variants, or CNVs), rare structural rearrangements that disrupt coding sequences, and triplet repeat expansions (in the ARX gene). The 183‐gene panel contains well‐known and recently described genes associated with monogenic epilepsy. NGS panel testing was performed as previously described using DNA prepared from blood or saliva samples.17
2.2. Test referral sources
A cohort of individuals clinically diagnosed with various forms of epilepsy was referred for diagnostic genetic testing and analyzed in a consecutive series. All participants provided informed consent for the testing. The clinical specialties of referring providers were determined using National Provider Identifiers (NPIs) and the National Plan and Provider Enumeration System (NPPES) Registry. Although information provided from ordering clinicians was not standardized, we used relevant keywords to categorize healthcare centers and specialty clinics into appropriate groups. Ancestry information for referred individuals was self‐reported.
2.3. Clinical testing and variant interpretation
Clinicians requested testing for all genes on the epilepsy panel or chose subpanels for narrower clinical indications. Interpretation of observed variants in epilepsy genes was performed as described previously.24 Clinical reports included variants classified as pathogenic or likely pathogenic (LP/P) or variants of uncertain significance (VUS) but not those classified as likely benign or benign (LB/B). A definitive molecular diagnosis included either single LP/P variants in genes associated with dominant or X‐linked inheritance, or two variants (in homozygous or compound heterozygous states) in genes associated with recessive inheritance. Genes on the epilepsy panel were categorized as “solid evidence” or “preliminary evidence” depending on the strength of each gene‐disorder relationship, as conceptually described previously.25 Variants in preliminary evidence genes were classified only as VUS, LB, or B; they did not reach LP/P classifications until the gene‐disease relationship was re‐curated to a solid evidence category. Per institutional review board (IRB) approval (Western IRB #20161796), all reportable variants observed at Invitae were de‐identified and deposited in the ClinVar database and available for research studies. All variants were collected from Invitae's internal database for this study. Categories of possible precision medicine implications, as listed in Table 1 and referred to in the following sections, were determined based on existing published literature and through annotation by multiple clinicians with expertise in treating epilepsy.
Table 1.
List of positive molecular diagnoses shown with the number of individuals corresponding to each diagnosis, the affected genes, and possible or theoretical precision medicine implications as established from published literature and reviewed by clinician authors.
| Gene | # PosMDx at Invitae | Precision medicine categorya | Precision medicine evidence gradeb | Possible or theoretical positive treatment implications based on published literature | Possible or theoretical treatment contraindications based on published literature | PubMed references (PMID) |
|---|---|---|---|---|---|---|
| ALDH7A1 | 6 | Biochemical | Strong | Pyridoxine (vitamin B6) and folinic acid | 24664145, 20301659 | |
| CSTB | 0 | AED ontraindications | Strong | Valproic acid | Sodium channel blockers (phenytoin, carbamazepine, oxcarbazepine), GABAergic drugs (tiagabine, vigabatrin), and gabapentin and pregabalin may aggravate myoclonus and myoclonic seizures. Phenytoin aggravates neurologic symptoms or even accelerates cerebellar degeneration | 20301321, 20301321 |
| EPM2A | 0 | AED contraindications | Strong | Phenytoin and possibly carbamazepine, oxcarbazepine, and lamotrigine | 20301563, 20301563 | |
| FOLR1 | 0 | Biochemical | Strong | Oral folinic acid reduces severity of seizures by correcting folate deficiency | 20447151 | |
| GAMT | 1 | Biochemical | Strong | Oral creatine monohydrate corrects creatine deficiency | Supplementation of ornithine and dietary restriction of arginine or protein | 23622406, 20301745 |
| GATM (AGAT) | 0 | Biochemical | Strong | Oral creatine monohydrate corrects creatine deficiency | 23622406, 20301745, 23770102 | |
| KCNQ2 | 103 | AED indications | Strong | Phenytoin, carbamazepine | 25880994, 20437616 | |
| NHLRC1 (EPM2B) | 1 | AED contraindications | Strong | Phenytoin and possibly carbamazepine, oxcarbazepine, and lamotrigine | 20301563, 20301563 | |
| PNPO | 1 | Biochemical | Strong | Pyridoxal phosphate | 20301659, 20301659. 25639976 | |
| POLG | 5 | AED contraindications | Strong | Avoid valproic acid to prevent liver toxicity | 20301791, 20301791, 21038416 | |
| SCN1A | 236 | AED contraindications | Strong | Clobazam and valproic acid are the optimal first‐line medications. Optimal response to anti‐epileptic drugs that bind the GABA receptor. Seizure triggers should be avoided, including hot temperatures (warm baths, exercise on hot days, untreated fever) and bright lights. | Sodium channel blockers are contraindicated (phenytoin, carbamazepine, lamotrigine, etc.) | 28564577, 20301494 |
| SCN2A | 49 | AED indications | Strong | Sodium channel blockers (eg, phenytoin, carbamazepine, lamotrigine) work well with early infantile epilepsies (<3 mo old) | 28379373, 26291284 | |
| SCN8A | 16 | AED indications | Strong | Phenytoin, carbamazepine, oxcarbazepine) | 26252990, 25951352, 26029160, 27559564 | |
| SLC2A1 | 23 | Biochemical | Strong | Ketogenic diet | 20301603, 29303961 | |
| SLC6A8 | 7 | Biochemical | Strong | Oral creatine corrects creatine deficiency (useful to differentiate transporter mutations from biosynthesis mutations as oral creatine supplementation will not work if transporter is non‐functional) | 24953403, 20301745 | |
| TPP1 | 13 | Biochemical | Strong | Tripeptidyl‐peptidase I enzyme replacement therapy | 28335910, 27083890 | |
| TSC1 | 16 | AED indications | Strong | Vigabatrin for spasms, preventive anti‐epileptic treatment | 21507691, 15563014, 10073425 | |
| TSC2 | 19 | AED indications | Strong | Vigabatrin for spasms, preventive anti‐epileptic treatment | 21507691, 15563014, 10073425 | |
| ALDH5A1 | 0 | Biochemical, AED contraindications | Emerging | Valproate is contraindicated since it inhibits SSADH enzyme activity. | 20301374 | |
| ATP1A3 | 12 | AED indications | Emerging | Flunarizine, topiramate. Ketogenic diet may also be effective | Avoidance of specific stressors or triggers, using daily prophylactic medications such as flunarizine or topiramate, or implementing strategies to induce sleep as a management tactic. | 28900444, 20301294, 25447930 |
| DEPDC5 | 59 | AED indications | Emerging | Surgery may be explored early in the disease course. Early assessment of mTOR inhibitors. | 26434565, 27683934 | |
| GLRA1 | 4 | AED indications | Emerging | Clonazepam | 15365143, 20301437, 16713923 | |
| GNAO1 | 9 | AED indications | Emerging | Tetrabenazine and DBS were the most useful | 28357411, 27068059 | |
| GOSR2 | 0 | AED indications | Emerging | Phenytoin is effective; however, it is not used because these individuals are often misdiagnosed as having as Unverricht‐Lundborg disease caused by pathogenic variants in EPM2A where avoidance of phenytoin is recommended. Establishing a molecular diagnosis based on sequencing of GOSR2 and EPM2A in these individuals can resolve the need to use phenytoin as a treatment option. | 23449775, 20301321 | |
| GRIN1 | 5 | AED indications | Emerging | Memantine, NMDA receptor channel blocker | 29194067 | |
| GRIN2A | 16 | AED indications | Emerging | Memantine, dextromethorphan | 24839611, 27683935 | |
| GRIN2B | 3 | AED indications | Emerging |
Memantine treatment may offer some beneficial effects for gain‐of‐function variants in GRIN2B. Valproic acid therapy dose optimization with GRIN2B variant (−200T> G) carriers. |
28377535, 21806385, 28533163 | |
| KCNQ3 | 3 | AED indications | Emerging | Carbamazepine (CBZ) is the drug of choice in benign familial neonatal seizures in individuals with KCNQ3 variants | 27888506, 24851285 | |
| KCNT1 | 21 | AED indications | Emerging | Quinidine in early infancy | 26369628, 26740507 | |
| NGLY1 | 2 | Biochemical | Emerging | Proton pump inhibitors | 28512024, 29419975 | |
| PCDH19 | 52 | AED indications | Emerging | Phenytoin, potassium bromide, and clobazam showed high efficacy with long‐term benefits to become seizure‐free. Consider corticosteroids as an adjunctive option in acute treatment. | 26820223, 23712037, 25891919 | |
| PIGA | 1 | AED indications | Emerging | Ketogenic diet | 26597089, 27126216 | |
| PRRT2 | 106 | AED indications | Emerging | Oxcarbazepine; carbamazepine | Avoiding stress, sleep deprivation, anxiety, and other triggers | 28056630, 20301633 |
| QARS | 0 | AED indications | Emerging | Ketogenic diet | 28056632 | |
| SLC6A1 | 18 | AED indications | Emerging | Ketogenic diet | 27600546 |
This table does not reflect existing professional practice guidelines for all genetic forms of epilepsy but refers to possible precision medicine opportunities in epilepsy based on existing published literature. Physicians still need to carefully assess the selection of therapies for specific patients based on their clinical presentation and individual factors. For diagnoses of specific epilepsy syndromes, confirmation of these PMIs by clinical specialists involved in the patients' care is essential.
This column shows curation of genes associated with biochemical disorders, genes for which pathogenic variants point to contraindications for certain anti‐epileptic drugs (AEDs), and genes with indications for specific types of AEDs.
Genes are separated into two groups: those with established evidence for association with precision medicine approaches and those for which such evidence is only emerging.
3. RESULTS
3.1. Patient demographics and referrals
Clinicians requested testing for all genes on the panel (9225 individuals) or chose subpanels for early infantile epileptic encephalopathy (EIEE; 188 individuals) or the Rett/Angelman spectrum of syndromic epilepsy disorders (356 individuals). Most individuals in this clinical cohort were younger than 5 years of age (range, 0‐82 years; mean, 8.63 years; median, 6 years). Half (52%) of the individuals in this cohort were male. Most referrals originated from clinicians self‐identified as neurologists with genetics expertise (36%), followed by clinical geneticists (25%), neurologists (15%), and pediatricians (5%). The remaining 19% were associated with a variety of other specialties or did not provide this information. Regarding referral sites, nearly 28% of individuals were from neurology clinics and 15% from genetics clinics. Another 27% originated from pediatrics clinics, and the remaining 30% were from sites without a stated specialization. Most test orders (73%) were received from clinics at academic centers. Orders from genetics clinics showed a higher diagnostic yield relative to the others (17.9% vs 14.9%, P = 0.004, chi‐squared), while tests ordered from neurology clinics had a slightly lower yield (12.1% vs 16.5%, P < 0.001, chi‐squared). Lastly, with respect to geographic origin, the majority of referred individuals were from North America (88.5%), and the rest were from South America (4.6%), Asia (3.6%), Europe (1.7%), Oceania (Australia and New Zealand) (0.9%), and Africa (0.7%). Their self‐reported population origins were European Caucasian (42.3%), Hispanic (21.8%), African or African American (8.4%), Middle Eastern (0.4%), Ashkenazi Jewish (0.9%), Asian (4.6%), Native American (0.4%), Pacific Islander (0.4%), of combined ancestries (3.2%), and other or unknown ancestries (17.8%).
3.2. Yield of molecular diagnoses
A definitive positive molecular diagnosis (hereafter referred to as “PosMD”) was obtained in 1502 of 9769 individuals in this cohort, corresponding to a positive yield of 14.9% among individuals tested on the comprehensive panel and 24% each on the syndromic epilepsy and EIEE subpanels (Figure 1a). Individuals referred with autism, intellectual disability, or developmental delay in addition to epilepsy had a higher rate of PosMDs compared with that in the rest of the cohort (Figure 1b, P < 0.001). Notably, a mere eight genes accounted for 50% of PosMDs, another 22 genes increased that proportion to 80%, while 76 additional genes contributed to the rest (Figure 1c and Table S1). Thirteen individuals had a PosMD in two or more genes, with at least one gene in each instance associated with a severe early‐onset epilepsy (Table S3).
Figure 1.
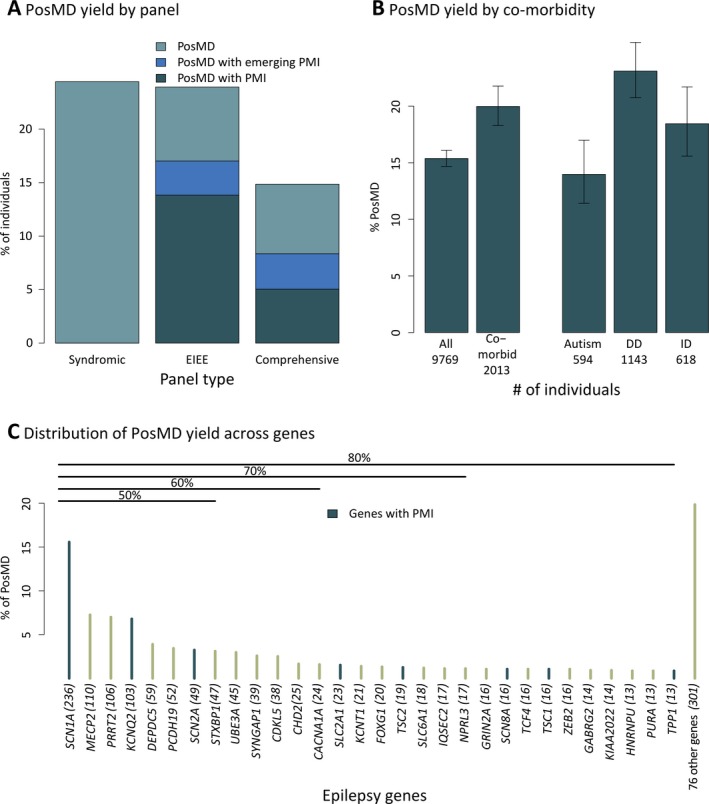
Distribution of positive molecular diagnoses (PosMDs) in a large unselected clinical cohort with epilepsy. Panel A shows a high diagnostic yield exceeding 20% in NGS panels for the Rett/Angelman spectrum of syndromic epilepsies and early infantile epileptic encephalopathies (EIEE), while the comprehensive panel showed a 14.9% yield. These represent consolidated figures derived from data from different versions of each panel. Nearly half of the solid evidence genes (see Methods) in the current panel were discovered only within the last 5‐10 years and together contributed a significant rate of PosMDs. These newer genes contributed as much as 7% alone (PRRT2) and more than 20% together to the overall diagnostic yield. Panel B shows that the diagnostic yield tends to be higher when epilepsy is accompanied by comorbidities such as intellectual impairment (ID), autism, or developmental delay (DD; P < 0.001, chi‐squared). Error bars represent 95% confidence intervals using the Wilson method. Panel C shows the number of PosMDs by individual genes on the NGS panel. Only eight genes accounted for 50% of all PosMDs, while another 22 genes raised this yield to 80%. The remaining 20% of PosMDs were spread across 76 genes. Seventy‐eight genes had no PosMDs, and 48 genes produced no LP/P at all. Genes with precision medicine implications are shown in blue
3.3. Results with possible PMIs
A subset of 491 individuals, representing 33% of all individuals with PosMDs, received results with PMIs (Figures 1a and 2 and Table 1). Fifty‐one individuals were diagnosed with biochemical disorders, mostly due to pathogenic variants in SLC2A1 and TPP1 and, occasionally, in SLC6A8, ALDH7A1, GAMT, and PNPO. Another 242 individuals had results that pointed to contraindications for certain AEDs primarily due to variants in SCN1A, but also in POLG and NHLRC1. No PosMDs were found in EPM2A or CSTB, which are also associated with disorders with certain AED contraindications. The remaining 198 individuals had results compatible with indications for using specific AEDs. PosMDs with PMI were not restricted to genes narrowly associated with a few specific forms of epilepsy but were present in genes overlapping syndromic epilepsies, EIEE, and a third group of all other types of epilepsies (Figure 2b). We also assessed 17 genes associated with disorders for which PMI evidence is emerging (Figures 1a and 2c and Table 1); at least 21% of individuals with a PosMD had positive molecular testing results specifically in these genes alone. Last, although not representing PMIs, we noted that another 95 and 282 individuals with PosMDs in this cohort were eligible for clinical treatment trials and condition‐specific clinical trials, respectively, based on molecular testing results (Table S4).
Figure 2.
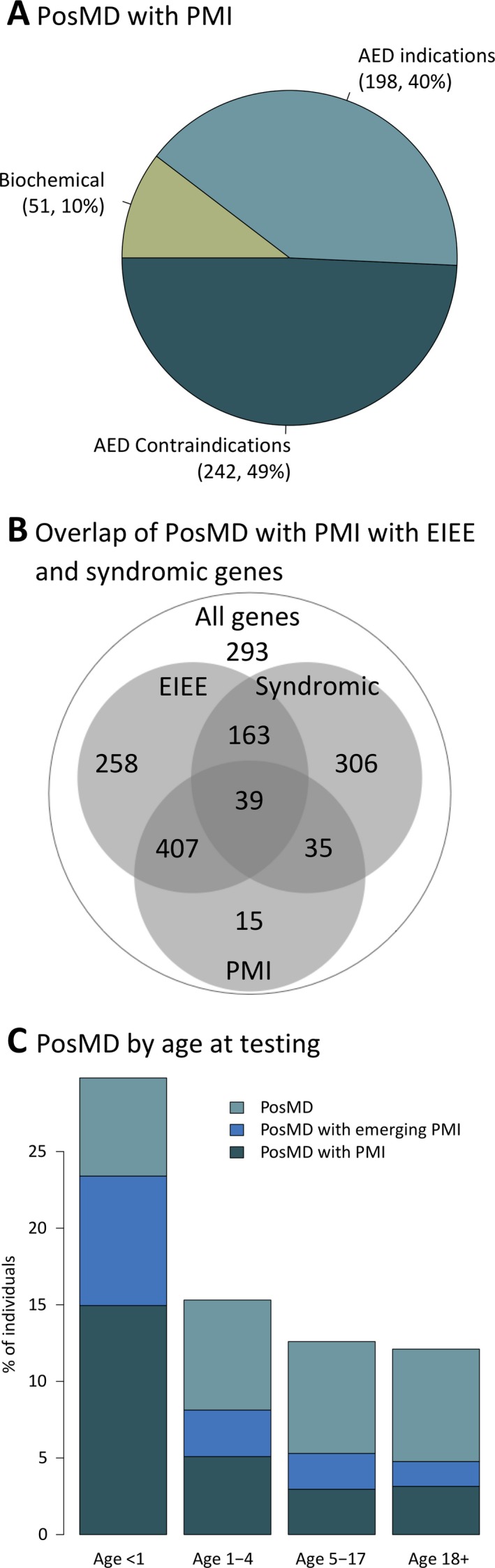
Positive molecular diagnoses (PosMDs) with possible precision medicine implications (PMIs) in epilepsy. Panel A shows the percentage of PosMDs related to various categories of precision medicine in epilepsy. Half of the PosMDs with possible PMI pointed to contraindications for certain anti‐epileptic drugs (AEDs). Approximately 10% of PosMDs with PMI were consistent with biochemical disorders that have established treatment options. Panel B shows the number of PosMDs with PMIs in genes in three overlapping categories of epilepsy disorders: early infantile epileptic encephalopathy (EIEE), Rett/Angelman spectrum of syndromic neurodevelopmental epilepsies, and a third group of all other forms of epilepsy. Panel C shows the positive diagnostic yield in various age groups separated by infancy (age <1 year), early childhood (age 1‐4 years), later childhood (age 5‐17 years), and adulthood (age ≥18 years). Colors indicate the proportion of individuals who received a PosMD with possible PMIs or those with emerging evidence of PMIs. A third group of PosMDs without PMIs is also shown at the top of each column
3.4. Age at diagnosis
Individuals with a PosMD ranged in age from newborns to 78 years (median, 4 years; average, 7 years); only 127 individuals (8%) with a PosMD were older than 18 years. A PosMD overall was much more frequent in children in their first year of life relative to the rest of the cohort (P < 0.001, chi‐squared; Figure 2c). Of the PosMDs specifically related to PMIs, 66% were in children younger than 5 years, 27% in children aged 5‐17, and the remaining 7% in adults. The age range for this subset was 2 days to 78 years (median, 2 years); only 33 individuals were adults. Children had PosMDs with PMIs related to biochemical disorders or epilepsies with indications or contraindications for AEDs, while adults had no PosMDs related to biochemical disorders and instead mostly had findings in SCN1A, and less frequently in KCNQ2, SCN2A, and TSC1.
3.5. Characteristics of reportable variants
The 9769 individuals in this study harbored 2101 (11%) variants classified as LP/P and another 16,373 as VUS (Tables S1 and S2). Nearly 80% of these were missense SNVs; the rest were divided among a group of truncating SNVs and ≤15 bp indels (17% together) and a key group of technically challenging variants (TCVs) that are often difficult to detect using a single traditional method such as Sanger sequencing, multiplex ligation‐dependent amplification, or chromosomal microarray. These TCVs included exonic CNVs, cytogenetic CNVs, large indels (>15 bp), mosaic SNVs, and polyalanine expansions in the ARX gene. We detected 402 intragenic and cytogenetic CNVs in 133 genes, mosaic pathogenic variants in 14 genes, mosaic VUS in 15 genes (Tables S2 and S3), and eight instances of polyalanine expansions in ARX. TCVs accounted for just 4% of reportable variants but constituted 16.5% of clinically significant LP/P variants overall. Approximately 45% of TCVs were classified as LP/P and contributed to at least 245 PosMDs (16.3% of all PosMDs; Figure 3c and Table S2). The LP/P TCVs were present in 81 genes, including several discovered relatively recently (eg, DEPDC5, PRRT2, NPRL3, TBC1D24, and GRIN2A) and ten related to disorders with PMIs (KCNQ2, SCN1A, SCN2A, TSC2, EPM2A, SLC6A8, SCN8A, ALDH7A, POLG, and TSC1). There were also seven instances of a clinically significant TCV that was compound heterozygous with a second pathogenic variant in a gene associated with AR or XL inheritance (two in CLN3 and one each in SLC13A5, PIGN, PPT1, WWOX, and KIAA2022; Table S3).
Figure 3.
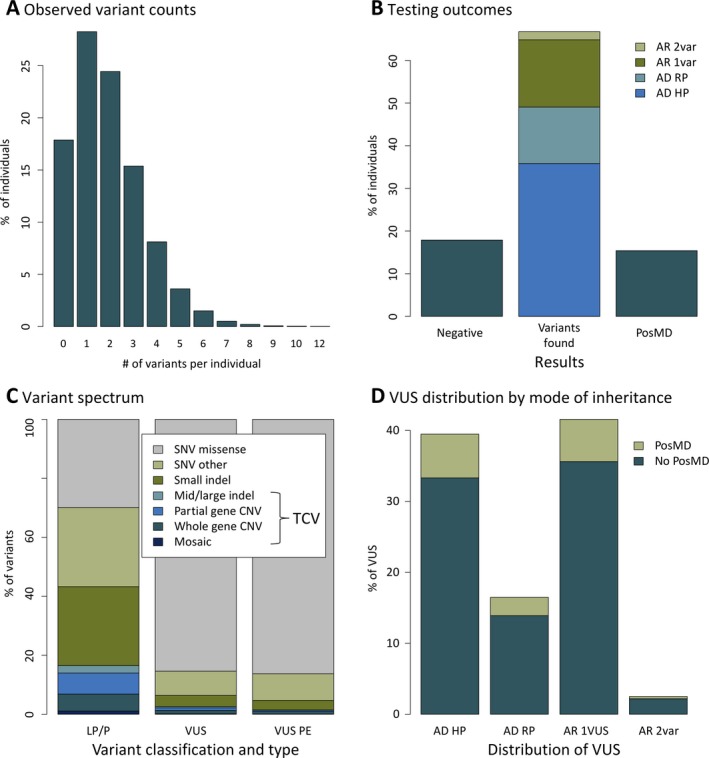
Distribution of clinically reportable variants. Panel A shows that variants identified in the 183 genes on the NGS panel were distributed in a range of one to 12 per individual. Panel B summarizes the proportion of individuals who received a negative report, a report describing a positive molecular diagnosis (PosMD), or an inconclusive report with a variant of uncertain significance or a single likely pathogenic/pathogenic variant, or both, that did not contribute to a PosMD. Panel C shows the wide range of variant types identified and the clinical classifications of each type. A significant number of clinically reportable variants that are technically challenging to identify with traditional methods were identified in this cohort. Panel D illustrates the distribution of variants of uncertain significance (VUS) identified in this study. The genes on the panel showed a 100‐fold range (0.02%‐9%) in the fraction of individuals who had at least one VUS in those genes. The VUS frequency was lower in the 57 genes associated with early‐onset and highly penetrant epilepsies compared with that in the remaining 103 solid evidence genes on the panel (P = 0.002, Wilcoxon rank sum). AD, autosomal dominant; AR, autosomal recessive; LP, likely pathogenic; P, pathogenic; PE, preliminary evidence; RP, reduced penetrance; var, variant; VUS, variant of uncertain significance
The 16 373 VUS were distributed in a range of one to 11 per person (Figure 3a). At least 15% of VUS were in individuals with a PosMD in another gene (Figure 3d). Conversely, among 8267 individuals without a PosMD, 75% had a VUS in at least one strong evidence gene (see Methods), 3% had a VUS in a preliminary evidence gene, and the remainder had only a single LP/P variant in a gene with AR inheritance or had no reportable variants (Figure 3b). The majority of VUS in strong evidence genes were single heterozygous alleles in genes associated with either AR inheritance (41% of VUS) or AD inheritance and reduced penetrance (17% of VUS; Figure 3d). Corroborating this observation, these two categories of genes had more rare sequence variation in healthy individuals in the gnomAD database compared with genes associated with AD inheritance and high penetrance (Figure S1). In that regard, 39% of individuals in this cohort still had a VUS in a gene associated with a highly penetrant disorder that therefore had the potential to reach clinical significance with parental testing or other studies.
We had initially classified 1612 variants as VUS but eventually reclassified them to LP/P (15%) or LB/B (85%). Results of parental testing for 846 probands led to reclassification in 54% of probands. Most reclassifications to LP/P occurred because parental testing demonstrated that the variant had arisen de novo. Reclassifications to LB/B were either because the variants were found to be inherited from an unaffected parent or due to other types of evidence, such as expansion of databases containing genomic information from healthy individuals or newly published literature. LP/P reclassifications contributed to 18.5% of all PosMDs overall, while the subset of LP/P reclassifications specifically related to disorders with PMIs contributed to 6.1% of all PosMDs. More than half of the PMI‐related LP/P reclassifications were in SCN1A, and a few were in SCN2A, KCNQ2, SCN8A, POLG, and TSC2. In seven individuals, the variant reclassification to LP/P occurred in a gene (TPP1, SLC2A1, ALDH7A1, or SLC6A8) associated with a treatable biochemical disorder.
4. DISCUSSION
Molecular testing is increasingly being used to identify genetic causes and confirm clinical diagnoses of epilepsy, but it remains far from standard practice.9 Our results demonstrate that multi‐gene panel testing frequently delivers important PosMDs that can guide precision medicine approaches to treating epilepsy. The observation of a high diagnostic yield in infants, and of all molecular diagnoses of treatable biochemical disorders in children younger than 5 years, complements studies that have shown positive clinical and health economic outcomes from early molecular testing in epilepsy.26, 27
The scope for precision medicine in epilepsy is expanding with the recognition of favorable treatment approaches in the presence of pathogenic variants in a growing list of genes. PMIs were immediately relevant to at least 33% of individuals with a PosMD in our study, which is considerably higher than the percentage observed in a smaller previously published analysis.22 That 51 individuals in our cohort received a diagnosis for a biochemical disorder, including several considered very rare, highlights the opportunity for precision medicine to help achieve a favorable clinical prognosis as early as possible using established treatments, even in rare disorders that might otherwise escape diagnosis. Aside from finding actionable results related to biochemical disorders, we identified a significant number of pathogenic variants in SCN1A in patients in whom sodium channel blockers would be contraindicated, as well as variants in a broad range of other genes that support the use of particular AEDs. We also identified pathogenic variants in genes associated with forms of epilepsy for which there is emerging evidence to support the use of specific AEDs, suggesting that the proportion of individuals who may possibly benefit from precision medicine in epilepsy could eventually increase considerably.
In addition to immediate PMIs, accessing ongoing clinical trials based on molecular testing results is another important opportunity for individuals with epilepsy. We estimate this to have an impact on another 25% of individuals with PosMDs in our cohort, demonstrating a good example of implementing the American College of Medical Genetics and Genomics policy advocating for patient access to emerging therapeutics.28, 29
Several genes discovered within only the last decade or initially thought to be rarely involved in epilepsy (eg, PCDH19, SYNGAP1, TPP1, PRRT2, DEPDC5) had high diagnostic yields in our study. Some of these are related to disorders for which new therapies are available or under development. As a compelling example of precision medicine, the discovery of pathogenic variants in TPP1 is critical because an enzyme replacement therapy has good efficacy when used early.10, 30 Similarly, there are ongoing clinical treatment trials aimed at ameliorating the phenotype in girls affected with epilepsy and at risk for neurodevelopmental deterioration due to pathogenic variants in PCDH19.31 Finally, mTOR inhibitors are being proposed for DEPDC5‐related epilepsy, further illuminating the growing opportunities for precision medicine in epilepsy.32, 33
Although genetic heterogeneity is extensive in epilepsy, half of the PosMDs in this study were explained by only eight genes and 80% by an additional 22 genes. This observation, corroborating results from another study,12 suggests that a core set of ~30 genes is essential in any molecular analysis of epilepsy. This would even be sufficient to address differential diagnoses in syndromic epilepsies. For instance, we found 16 PosMDs involving TCF4, associated with Pitt‐Hopkins syndrome, in individuals who had instead been diagnosed clinically with Rett, Angelman, or unspecified neurodevelopmental syndromes. WES can interrogate this core set of genes and a longer tail of genes rarely or newly implicated in epilepsy. However, because clinically important results from WES are often in genes that are available on panels and because WES has analytic limitations of coverage and relative insensitivity to TCVs,34, 35 an NGS panel is a useful first‐tier test that can interrogate most molecular etiologies in epilepsy and provide positive diagnoses rapidly and at relatively low cost.
Evidence‐based clinical interpretation of variants is key to identifying PosMDs and supporting precision medicine in epilepsy. However, because naturally occurring but latent disease‐causing variants exist alongside rare benign variants in the general healthy population, identifying the variants responsible for overt epilepsy in affected individuals is challenging.18, 19 Therefore, many rare variants are classified as VUS. After parental testing, VUS in genes with AD inheritance and high penetrance and homozygous or compound heterozygous VUS in genes with AR inheritance can be reclassified as LP/P and contribute to PosMDs. We identified de novo variants in many probands in this study, consistent with the expectation that such mutations cause several forms of epilepsy.36, 37, 38 Overall, we resolved the significance of 1612 VUS and provided additional PosMDs, including 91 with PMIs, emphasizing that a fair number of probands can benefit from VUS resolution studies.
5. LIMITATIONS
While this study describes the implications of genetic testing and high diagnostic yield in a large cohort, the absence of longitudinal clinical outcomes data for individuals who received positive testing results poses an obstacle to further understanding the effectiveness of precision medicine approaches in treating epilepsy. Furthermore, this study describes potential applications of precision medicine in epilepsy based on existing published literature on specific therapies, but further studies are still necessary to establish official professional guidelines for the use or avoidance of these therapies. Our group and others are currently conducting separate studies to evaluate the benefits of early molecular genetic testing and precision medicine in epilepsy. The second limitation of this study, common to diagnostic genetic laboratories,12 is that we could not define the study entry criteria at the outset since the cohort consisted of individuals referred for diagnostic genetic testing due to a variety of epilepsy presentations and may also be influenced by several factors, such as the expertise of referring clinicians in genetics, specialization in epilepsy at the referring institution, insurance coverage, economic factors, and others. In that respect, for example, adults with clinically recognizable genetic syndromes may have had previous genetic testing of only a small number of genes and not referred again for broader NGS panel testing. Lastly, the third limitation of this study is the absence of follow‐up WES analysis for individuals who received negative results on the NGS panel. This analysis would illuminate the additional diagnostic yield gained from considering very rare genetic causes of epilepsy. However, recent research studies have begun to address this question.15, 37
6. CLINICAL RELEVANCE
The various lines of evidence from this study support routine and early use of an NGS panel as an effective first‐tier test that offers a high diagnostic yield and substantial potential for precision medicine in epilepsy. Any multi‐gene testing approach should be sensitive enough to capture a broad spectrum of genetic variants, including small intragenic copy number variants and mosaic variants. The value of genetic testing will continue to increase as novel therapies are developed for different forms of epilepsy.
CONFLICTS OF INTEREST
Rebecca Truty, Nila Patil, Ali Entezam, Edward D. Esplin, Amy Fuller, Michelle Hogue, Britt Johnson, Amirah Khouzam, Yuya Kobayashi, Rachel Lewis, Keith Nykamp, Darlene Riethmaier, Jody Westbrook, Michelle Zeman, Robert L. Nussbaum, and Swaroop Aradhya are full‐time employees of Invitae, a genetic testing company. Dr. Raman Sankar and Dr. Joseph Sullivan are advisors to Invitae. The other authors have no conflicts of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Truty R, Patil N, Sankar R, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4:397–408. 10.1002/epi4.12348
REFERENCES
- 1. Thomas RH, Berkovic SF. The hidden genetics of epilepsy – a clinically important new paradigm. Nat Rev Neurol. 2014;10:283–92. [DOI] [PubMed] [Google Scholar]
- 2. Dunn P, Albury CL, Maksemous N, Benton MC, Sutherland HG, Smith RA, et al. Next generation sequencing methods for diagnosis of epilepsy syndromes. Front Genet. 2018;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poduri A. When should genetic testing be performed in epilepsy patients? Epilepsy Curr. 2017;17:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. EpiPM Consortium . A roadmap for precision medicine in the epilepsies. Lancet Neurol. 2015;14:1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Symonds JD, Zuberi SM, Johnson MR. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr Opin Neurol. 2017;30:193–99. [DOI] [PubMed] [Google Scholar]
- 6. Smith LA, Ullmann JFP, Olson HE, Achkar CME, Truglio G, Kelly M, et al. A model program for translational medicine in epilepsy genetics. J Child Neurol. 2017;32:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sands TT, Balestri M, Bellini G, Mulkey SB, Danhaive O, Bakken EH, et al. Rapid and safe response to low‐dose carbamazepine in neonatal epilepsy. Epilepsia. 2016;57:2019–30. [DOI] [PubMed] [Google Scholar]
- 8. Reif PS, Tsai MH, Helbig I, Rosenow F, Klein KM. Precision medicine in genetic epilepsies: break of dawn? Expert Rev Neurother. 2017;17:381–92. [DOI] [PubMed] [Google Scholar]
- 9. Berg AT, Coryell J, Saneto RP, Grinspan ZM, Alexander JJ, Kekis M, et al. Early‐life epilepsies and the emerging role of genetic testing. JAMA Pediatr. 2017;171:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson TB, Cain JT, White KA, Ramirez‐Montealegre D, Pearce DA, Weimer JM. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat Rev Neurol. 2019;15:161–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berg AT, Goldman S. Getting serious about the early‐life epilepsies: Lessons from the world of pediatric oncology. Neurology. 2018;90:842–48. [DOI] [PubMed] [Google Scholar]
- 12. Lindy AS, Stosser MB, Butler E, Downtain‐Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–71. [DOI] [PubMed] [Google Scholar]
- 13. Parrini E, Marini C, Mei D, Galuppi A, Cellini E, Pucatti D, et al. Diagnostic targeted resequencing in 349 patients with drug‐resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017;38:216–25. [DOI] [PubMed] [Google Scholar]
- 14. Gokben S, Onay H, Yilmaz S, Atik T, Serdaroglu G, Tekin H, et al. Targeted next generation sequencing: the diagnostic value in early‐onset epileptic encephalopathy. Acta Neurol Belg. 2017;117:131–38. [DOI] [PubMed] [Google Scholar]
- 15. Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early‐onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53:310–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18:898–905. [DOI] [PubMed] [Google Scholar]
- 17. Truty R, Paul J, Kennemer M, Lincoln SE, Olivares E, Nussbaum RL, et al. Prevalence and properties of intragenic copy‐number variation in Mendelian disease genes. Genet Med. 2018;21:114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cherepanova NS, Leslie E, Ferguson PJ, Bamshad MJ, Bassuk AG. Presence of epilepsy‐associated variants in large exome databases. J Neurogenet. 2013;27:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hildebrand MS, Myers CT, Carvill GL, Regan BM, Damiano JA, Mullen SA, et al. A targeted resequencing gene panel for focal epilepsy. Neurology. 2016;86:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Møller RS, Larsen LHG, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. 2016;7:210–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mercimek‐Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–16. [DOI] [PubMed] [Google Scholar]
- 23. Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53:1387–98. [DOI] [PubMed] [Google Scholar]
- 24. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, et al. Sherloc: a comprehensive refinement of the ACMG‐AMP variant classification criteria. Genet Med. 2017;19:1105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garcia J, Tahiliani J, Johnson NM, Aguilar S, Beltran D, Daly A, et al. Clinical genetic testing for the cardiomyopathies and arrhythmias: a systematic framework for establishing clinical validity and addressing genotypic and phenotypic heterogeneity. Front Cardiovasc Med. 2016;3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oates S, Tang S, Rosch R, Lear R, Hughes EF, Williams RE, et al. Incorporating epilepsy genetics into clinical practice: a 360° evaluation. NPJ Genom Med. 2018;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joshi C, Kolbe DL, Mansilla MA, Mason SO, Smith RJ, Campbell CA. Reducing the cost of the diagnostic odyssey in early onset epileptic encephalopathies. Biomed Res Int. 2016;2016:6421039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. ACMG Board of Directors . Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17:505–7. [DOI] [PubMed] [Google Scholar]
- 29. ACMG Board of Directors . Considerations in healthcare reform for patients and families with genetic diseases: a statement of the American College of Medical Genetics and Genomics. Genet Med. 2018;20:561. [DOI] [PubMed] [Google Scholar]
- 30. Markham A. Cerliponase alfa: first global approval. Drugs. 2017;77:1247–49. [DOI] [PubMed] [Google Scholar]
- 31. Zaccara G, Schmidt D. Do traditional anti‐seizure drugs have a future? A review of potential anti‐seizure drugs in clinical development. Pharmacol Res. 2016;104:38–48. [DOI] [PubMed] [Google Scholar]
- 32. Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Rotenberg A, et al. A mouse model of DEPDC5‐related epilepsy: neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis. 2018;111:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Myers KA, Scheffer IE. DEPDC5 as a potential therapeutic target for epilepsy. Expert Opin Ther Targets. 2017;21:591–600. [DOI] [PubMed] [Google Scholar]
- 34. Ostrander BEP, Butterfield RJ, Pedersen BS, Farrell AJ, Layer RM, Ward A, et al. Whole‐genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom Med. 2018;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. La Duca H, Farwell KD, Vuong H, Lu H‐M, Mu W, Shahmirzadi L, et al. Exome sequencing covers >98% of mutations identified on targeted next generation sequencing panels. PLoS ONE. 2017;12:e0170843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilfert AB, Sulovari A, Turner TN, Coe BP, Eichler EE. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li J, Cai T, Jiang Y, Chen H, He X, Chen C, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016;21:290–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50:1048–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials