


Abstract
Ribosome biogenesis is an essential process in all living cells, which entails countless highly sequential and dynamic structural reorganization events. These include formation of dozens RNA helices through Watson-Crick base-pairing within ribosomal RNAs (rRNAs) and between rRNAs and small nucleolar RNAs (snoRNAs), transient association of hundreds of proteinaceous assembly factors to nascent precursor (pre-)ribosomes, and stable assembly of ribosomal proteins. Unsurprisingly, the largest group of ribosome assembly factors are energy-consuming proteins (NTPases) including 25 RNA helicases in budding yeast. Among these, the DEAH-box Dhr1 is essential to displace the box C/D snoRNA U3 from the pre-rRNAs where it is bound in order to prevent premature formation of the central pseudoknot, a dramatic irreversible long-range interaction essential to the overall folding of the small ribosomal subunit. Here, we report the crystal structure of the Dhr1 helicase module, revealing the presence of a remarkable carboxyl-terminal domain essential for Dhr1 function in ribosome biogenesis in vivo and important for its interaction with its coactivator Utp14 in vitro. Furthermore, we report the functional consequences on ribosome biogenesis of DHX37 (human Dhr1) mutations found in patients suffering from microcephaly and other neurological diseases.
INTRODUCTION
In all living cells, ribosomes are essential nanomachines responsible for protein production. Ribosome biogenesis is a complicated pathway involving in budding yeast the synthesis of 84 core components: four ribosomal (r) RNAs and 80 ribosomal proteins, their modification, transport and faithful assembly (1). The process involves at least 200 protein trans-acting factors and 100 small nucleolar RNAs (snoRNAs), which interact transiently with maturing subunits aiding individual maturation steps to proceed productively, or preventing them to occur prematurely (2). Three of four mature rRNAs are produced from a single long polycistronic precursor synthesized by RNA polymerase I, thus requiring extensive processing including both endo- and exonucleolytic cleavages.
Ribosomal RNAs are highly structured molecules which undergo tremendous compaction during ribosomal subunit biogenesis through formation of dozens RNA helices and stem-loops. RNA helices may form between contiguous sequences, or involve sequences located far apart on the primary sequence of the rRNA thus bringing together otherwise distant domains of the molecule. Pre-rRNA folding is guided by numerous intervening factors of both proteinaceous (assembly factors) and ribonucleic (snoRNAs) nature.
Altogether, there is an important need for energy-consuming remodeling activities, including AAA ATPases, GTPases and helicases. Consistently, with twenty five members in budding yeast, the RNA helicases form the largest group of ribosomal assembly factors (3), of which Dhr1 is one member. While most helicases involved in ribosome biogenesis assembly belong to the DEAD-box class, Dhr1 is particular as it belongs to the DEAH-box family whose four most prominent members: Prp2, Prp16, Prp22 and Prp43 are involved in four consecutive steps of pre-mRNA splicing (4). Prp43 is also involved in pre-rRNA processing, and ribosomal assembly, being part of several maturing ribosomal subunits (5–9). Dhr1, in contrast, is not required for pre-mRNA splicing and is only known to be involved in early pre-rRNA processing steps leading to formation of 18S rRNA and small subunit (SSU) production (10).
The recent determination of high-resolution structures of ribosomal subunit precursors by cryo-EM has provided important insights into the function of assembly factors (reviewed in (11)). The structures of the early pre-ribosomal subunit(s) have notably provided evidences that the rRNA subdomains (body, head, etc.) of the nascent small subunit (SSU) are hold apart by the SSU-processome (the early acting assembly factors, (12–14)). Such structural organization suggests that the SSU-processome acts as an assembly platform which may provide a dedicated molecular environment aimed at i) protecting the nascent rRNA from degradation, ii) avoiding premature folding and the generation of ‘folding traps’, and iii) providing a ‘molecular barrier’ that, on one hand, favors r-proteins assembly and, on the other hand, avoid unproductive r-protein disassembly (early binding r-proteins are known to be biochemically weakly associated within early pre-ribosomes, (15)). Finally, various assembly events and structural remodeling steps still occur within and after SSU-processome action and release, until ribosomal subunits adopt their final structures.
An important step of small ribosomal subunit formation is the establishment of the central pseudo-knot (CPK, Figure 1), a long-range tertiary interaction that brings together through Watson-Crick base-pairing two 18S rRNA sequence motifs separated by more than 1 kb apart, and acting like a ‘zipper’ to juxtapose the 5′ and the 3′ major domains of the molecule (Figure 1). CPK formation is a dramatic irreversible structural transition essential to small ribosomal subunit biogenesis, and as such it is tightly controlled. In particular, it must not form prematurely. Timing of formation is regulated by the box C/D snoRNA U3, which interacts with, and sequesters, two sequence motifs important to establish the CPK. When Dhr1 was characterized, it was found to interact very tightly with U3, and the suggestion was made that it may act a ‘time keeper’ (similarly to what had been proposed for DEAH helicases in pre-mRNA splicing) providing the energy to resolve the U3–pre-RNA interactions, and then to dissociate U3 in order to allow CPK formation (10). This model was confirmed 15 years later, and the exact points of contact (nucleotide resolution) between Dhr1 and U3 revealed (16). In addition to binding to U3, Dhr1 interacts with multiple SSU-processome assembly factors, including the heterodimer Bud23-Trm112 (17,18), an m7G methyltransferase active on 18S rRNA (19,20), and Utp14, a protein activating Dhr1′ helicase function (21,22). Utp14 was proposed to act in a similar fashion as G-patch-containing proteins. However, in contrast to classical G-patch proteins such as Ntr1 and Paf1, which activate Prp43 respectively in pre-mRNA splicing and in ribosome biogenesis (8,23,24), Utp14 does not stimulate Dhr1 ATPase activity (21). Finally, it is interesting that the human homolog of Dhr1, the protein DHX37, which is also required for 40S biogenesis (25), has been linked to neurological diseases including microcephaly (26,27). To date, the effects of Dhr1 disease mutations on ribosome biogenesis have not been characterized molecularly.
Figure 1.

Schematic representation of the Dhr1-catalyzed structural transitions occurring between the 18S rRNA precursor associated with U3 snoRNA (left) and mature 18S rRNA (right). On the mature 18S rRNA (right), the central pseudo-knot (CPK) is highlighted by a red dashed circle. On the 18S precursor (left), the U3 snoRNA and its boxes A and A’ are colored in blue. The 18S rRNA sequences that base pair in the CPK are colored in green. Their interaction is prevented by the presence of U3 in the 18S precursor. The four rRNA domains (5′, central, 3′ major and 3′ minor) are represented using a color scheme also used in Figure 7D. Some specific Watson-Crick base pairing, i.e. those occurring between the 18S rRNA and U3 snoRNA or in the mature CPK, are depicted by lines. For the sake of clarity, the long 5′ ETS region located upstream of 18S rRNA, and a large portion of U3 snoRNA are not shown (depicted as dashed lines). The position of nucleotide G1575 is indicated on the 18S precursor (left) as an open circle; its modification to m7G carried out by the Bud23-Trm112 methyltransferase complex, is highlighted as a closed circle in the mature 18S (right). rRNA helices 11, 23, and 44 (h11, h23 and h44), which are contacted by Dhr1 are labelled (see Figure 7).
Despite tremendous progresses toward establishing the high-resolution structure of precursor ribosomes, numerous assembly factors remain to be mapped on the maturing ribosomes simply because they are not present on the purified precursor ribosomes analyzed by cryo-EM thus far. This is notably the case of many factors harboring catalytic activity, including most rRNA modification enzymes and remodeling activities, presumably because they only operate transiently on maturing subunits. Dhr1 is no exception, and, consequently, its exact location, interaction landscape, and, in fact, even its three-dimensional structure, are not available. U3 on the other hand was clearly located at the heart of the SSU-processome, literally holding apart the subdomains of the 18S rRNA (Figure 1 and (12–14)).
In this work, we have: (i) determined the first atomic resolution (2.3Å) structure of the helicase module of yeast Dhr1, revealing commonalities and differences to that of other DEAH-box helicases leading us to propose that our Dhr1 structure corresponds to an RNA substrate ‘pre-loading state’, a conformation not previously reported for this type of protein; (ii) identified and functionally characterized a remarkable carboxyl-terminal domain (CTD) demonstrating its importance to dissociate Dhr1 from pre-ribosomes, its requirement for pre-rRNA processing, and its involvement in the interaction between Dhr1 and its coactivator Utp14; (iii) analyzed the consequences on ribosome biogenesis in yeast cells of DHX37 disease mutations and (iv) tentatively proposed how Dhr1 progresses on precursor ribosomal subunits to unwind the U3 snoRNA during CPK formation.
MATERIALS AND METHODS
Protein over-expression and purification
Genes encoding for Saccharomyces cerevisiae Dhr1 and Utp14 proteins (intact proteins or domains) were cloned into pGEX-6P1 and pET28b expression vectors, respectively (Supplementary Table S1). Proteins were expressed in Escherichia coli and purified using a three-steps procedure consisting of affinity chromatography followed by an heparin column and a S200-16/60 size-exclusion column. When necessary, the GST tag fused to Dhr1 was removed by adding a 3C cleavage step after the affinity chromatography step. Detailed procedures can be found in Supplementary information.
Crystallization and structure determination
Diffracting crystals were obtained by mixing equal volume of Dhr1 helicase module (Dhr1-Hel) protein (9 mg/ml) with 0.2 M MgCl2; 0.1 M Tris pH 8.5 and 20% (w/v) PEG 8000 at 4°C following a 30 min incubation with trypsin (1:500 dilution w/w) at 20°C to chop the floppy regions. The structure was determined by molecular replacement using the structure of S. cerevisiae Prp43 DEAH RNA helicase as search model (28) and refined with BUSTER (29) to yield a final structure at 2.3 Å resolution with R = 20.5% and Rfree = 24.5%. Detailed procedures can be found in Supplementary information.
GST pull down experiments
Pull-down experiments were performed by mixing 130 pmol of GST-Dhr1 fusion proteins (GST-Dhr1-Hel GST-Dhr1-Hel-ΔCter and GST-Dhr1-Hel-ΔCTD) or GST alone with almost equimolar amount of His6-tagged Utp14. Buffer A (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 5 mM β-mercaptoethanol) was added to a final volume of 60 μl. The reaction mixtures were incubated on ice for 1 h. 10 μl were withdrawn as ‘input’ fraction for SDS-PAGE analysis. The remaining 50 μl were incubated with 500 μg of Glutathione Magnetic Agarose Beads (Thermo Scientific) equilibrated in buffer A to a final volume of 200 μl at 4°C for 1 h. Beads were washed three times with 500 μl of buffer A. Bound proteins were eluted with buffer A complemented with 25 mM reduced glutathione (pH 7.5). For a better resolution at higher molecular weight range (180–100 kDa), samples were resolved on 10% SDS-PAGE with an extended running time and visualized by SYPRO Ruby (BioRad; #S4942) staining.
Yeast strains and plasmids
To produce yeast strains expressing C-terminally truncated versions of DHR1, diploid cells (BY4743, available from Euroscarf) were transformed with suitable PCR cassettes (see Supplementary Table S1 for details). Clones selected on minimum medium lacking uracil were diagnosed by PCR on genomic DNA and by DNA sequencing of the junctions using primers LD2794 and LD268 (Supplementary Table S1).
For conditional expression of Dhr1 mutants, an haploid yeast strain expressing DHR1 under the control of a regulatable pMET promoter (pMET::HA-dhr1; (10)) was transformed with a low-copy plasmid expressing the corresponding DHR1 construct under its own promoter and terminator sequences. Detailed procedures can be found in Supplementary information.
Time course and yeast culturing
pMET-HA::dhr1 cells transformed with the desired plasmid were pre-grown to mid-log phase in minimal medium lacking leucine and methionine. To achieve depletion of endogenous Dhr1, methionine was added to a final concentration of 20 mM. Cells were maintained in mid-log phase during the entire duration of the experiment by dilution with pre-warmed medium. Total proteins and total RNAs were extracted after 0-, 6- and 12-h of transfer to methionine-containing medium.
Velocity gradient analysis
Cell extracts and sucrose gradient (10–50%) were prepared as described previously (30). Fractions were collected on a density gradient fractionation system (Teledyne ISCO).
Western blot analysis
Total protein fractions were separated on 10% SDS-polyacrylamide gels and transferred to a PVDF membrane. Western blotting signals were captured with a Chemidoc (Biorad). The following antibodies were used: anti-HTP (Sigma, 1:1000), anti-DHR1 (produced in this work, see Supplementary information for details, 1:2000), anti-GAPDH (Sigma, 1:5000), anti-HA antibodies (Sigma, 1:5000) and anti-rabbit peroxydase (1:10 000, Sigma).
RNA extraction and northern blotting
Total RNA extraction and northern blotting were performed essentially as described in (30). Oligonucleotides LD471 (anti-ITS1) and LD366 (anti-U3) used in the hybridizations are listed in Supplementary Table S1. For the analysis of high-molecular-weight RNA species, 10 μg of total RNA were separated on a 1.2% agarose–6% formaldehyde gel. For low-molecular-weight RNA species analysis, and for RNA extracted from sucrose gradient fractions, 8% urea-acrylamide gels were used. Phosphorimager quantification was performed using a Fuji FLA-7000 and the native Multi Gauge software (version 3.1). 25S/18S ratio were extracted from bioanalyzer electropherograms generated with the RNA 6000 nano kit (Agilent, #5067-1511). For U3 RNA analysis, the detected signal in each fraction was quantified by phosphorimager counting and represented graphically as a percentage of the total signal.
Additional Materials and Methods details are provided in Supplementary information.
RESULTS
Atomic resolution structure of the DEAH helicase module of Dhr1
The S. cerevisiae Dhr1 protein (hereafter named Dhr1) is a large essential protein mostly comprised of a DEAH helicase module (amino acids 315–1267) preceded by a poorly conserved N-terminal extension (NTD; amino acids 1–314; Figure 2A). This NTD interacts with the 18S rRNA m7G-methyltransferase Bud23–Trm112 complex via a region extending from residues 58 to 270 (17), which is not essential for Dhr1′ function (21). As the NTD is predicted to be unfolded, it was not included for structural studies. We expressed the Dhr1 helicase module (hereafter named Dhr1-Hel) in E. coli, purified it and initially obtained very thin crystals that suffered from serious diffraction anisotropy. Slightly thicker crystals were obtained by pre-incubating the purified protein with trypsin before setting the crystallization drops. This allowed us to collect a complete dataset at 2.3 Å resolution and to solve the structure of Dhr1 by molecular replacement by using the crystal structure of the related S. cerevisiae Prp43 as a search model ((28); Supplementary Table S2).
Figure 2.
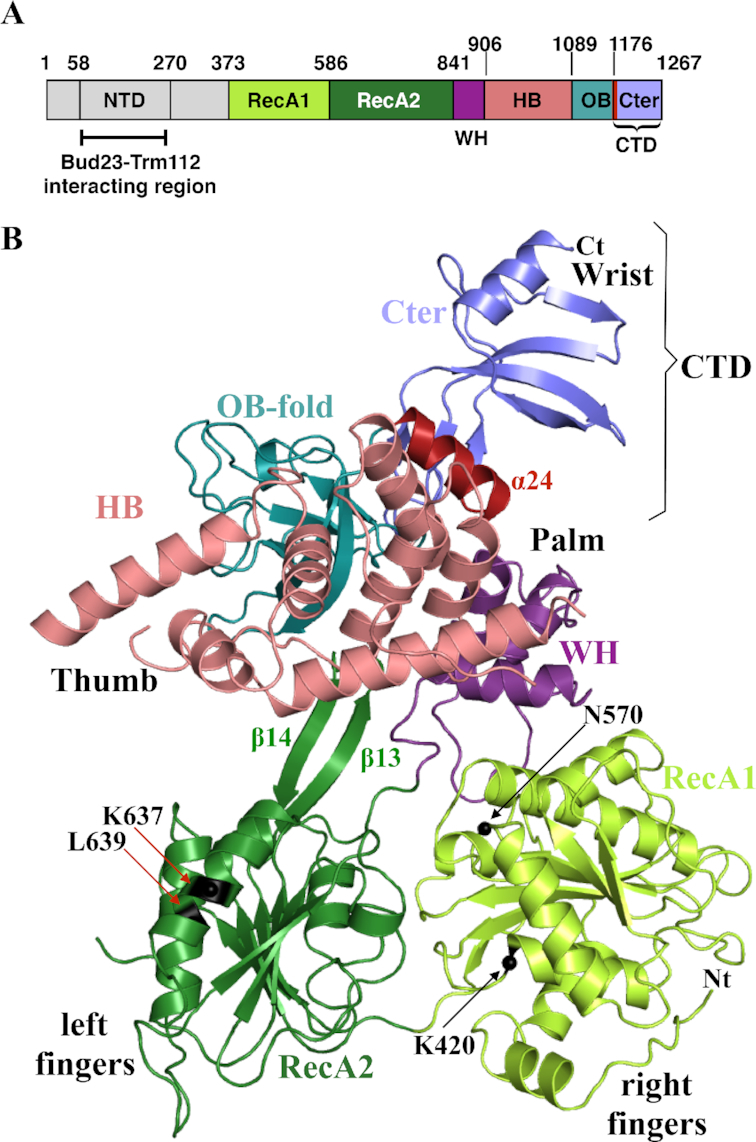
Protein domain organization and atomic structure of S. cerevisiae Dhr1 helicase module (Dhr1-Hel). (A) Schematic representation of Dhr1 organization with domain boundaries indicated. NTD, N-terminal domain; RecA1 and RecA2 are carrying the signatures responsible for ATPase and helicase activities of the protein; WH, Winged Helix; HB, Helical Bundle; OB, OB-fold domain; CTD, C-terminal domain. Helix α24 encompassing residues 1175–1187 from the CTD is colored in wine red. This color code is used in all figures unless otherwise specified. (B) Ribbon representation of Dhr1-Hel crystal structure. Protein domains named as in panel A. Nt and Ct: amino- and carboxyl-terminal end, respectively. The β13 and β14 β-strands of the RecA2 domain, proposed to act as a ‘bookend’, are indicated. The Cα atom of amino acids mutated in this study (K420, N570, K637 and L639, see Figure 5) are highlighted by black spheres and labelled.
Dhr1-Hel is composed of six domains: RecA1 and RecA2, which contain the residues responsible for ATP binding and hydrolysis, a Winged-helix (WH), an helical bundle (HB), an OB-fold, and an atypical carboxyl terminal domain (CTD, see Figure 2B). The six domains are arranged in a right-hand shape configuration ‘ready-to-grip’ an RNA substrate (Figure 2B). The RecA1 and RecA2 domains form respectively the right and left fingers of the hand while the WH, HB and OB-fold domains associate together to form the palm and the thumb. Finally, the CTD corresponds to the wrist. RecA domains are ubiquitously found in energy-consuming enzymes where they are involved in NTP binding and hydrolysis as well as in RNA binding. The RecA1 domain adopts a typical α/β domain fold made of a central seven-stranded parallel β-sheet surrounded by several α helices on both sides while the RecA2 domain central β-sheet contains only six β-strands (Supplementary Figure S1A and B). The Dhr1 RecA2 domain contains a β-hairpin structure (formed by the long β13 and β14 strands) that has been only observed so far in DEAH-box RNA helicases, such as Prp43 (Supplementary Figure S1B; (28,31–34)), Prp22 (35) and in DEXH-box DNA/RNA helicases (36–38). This β-hairpin structure was initially proposed to act as a ‘blade’ to separate DNA/RNA strands during unwinding, but more recently, it was suggested to act as a ‘bookend’ to hold the 5′ end of the single stranded nucleic acid fragment clamped between the RecA domains and the WH-HB domains (34). Most residues contacting directly ADP•BeF3− (ATP analog; (34)) in the structure of Chaetomium thermophilum Prp43 (hereafter named CtPrp43) in its RNA•ADP•BeF3− bound form are strongly conserved and belong to the RNA helicase motifs I, Ia, II, V and VI (Supplementary Figure S2). The WH domain is composed of three α helices and it tightly interacts with the HB domain, consisting of seven α helices. The OB-fold domain consisting of a β-barrel formed by five anti-parallel strands and one α helix, is packed against the HB domain. Altogether, the WH, HB and OB domains associate to form a single rigid auxiliary region, in compliance with what was observed in Prp43 ((28,32–34,39); Supplementary Figure S1C). Finally, the analysis of the 2Fo-Fc (contoured at 1σ) and Fo – Fc (contoured at 3σ) electron density maps revealed the presence of an additional domain at the C-terminal end of the protein (Supplementary Figure S3A and B), which could be modeled in these density maps by several iterative cycles of model building and refinement. This additional CTD (residues 1176–1262) is protruding from the OB-fold domain and folds as a concave four-stranded anti-parallel β-sheet surrounded by two α helices (helices α24 and α25), one at each end (Figure 2B). A CTD domain is also observed in the crystal structure of murine DHX37 (released while this paper was under revision (40)). For DHX37, the helix corresponding to helix α24 was considered to be part of the OB-fold domain on the basis of a former proposal on Prp43 (28,32). However, in the case of Prp43 this helix ends the protein which is not the case for Dhr1 or DHX37. For Dhr1, we consider that helix α24 truly belongs to the CTD because 40% of its surface is engaged in interaction with the remainder of the carboxyl terminal end of the protein while only 15% of its surface contacts the preceding OB-fold domain.
Dhr1 carries a remarkable carboxyl terminal domain homolog to single-stranded nucleic acid binding domains
Among the six domains observed in the crystal structure of Dhr1-Hel, five: the two RecA’s, the WH, the HB and the OB, are also found in Prp43 and Prp22 (28,32–35,39). In contrast, the last domain (CTD) is unique to Dhr1/DHX37 proteins. It has not been observed in the crystal structures of other members of the DEAH/DEXH RNA helicase family described so far, and it is not predicted by computational analysis to be present in any other DEAH-box RNA helicases. Typically, it is absent from yeast Dhr2, a protein highly homologous to Dhr1, albeit shorter and not involved in exactly the same pre-rRNA cleavage steps (10).
In order to identify protein domains with structural similarity to the CTD of Dhr1, we searched the Protein Data Bank with the DALI server (41). This led us to identify members of the DUF2128 (PF09901) family, which are commonly found in prokaryotes (for instance, L. lactis YdbC; rmsd of 2.9 Å over 58 Cα atoms; Figure 3A; (42)) but also in eukaryotes such as the human PC4 transcription cofactor (rmsd of 3–3.6 Å over 58 Cα atoms; (43)). Interestingly, in YdbC and PC4 proteins, this domain promotes the formation of homodimers raising the possibility that it may act similarly in the context of Dhr1 (42–44). However, a SEC-MALLS analysis performed on crystallized Dhr1-Hel showed that it is monomeric in solution in the experimental conditions tested (Supplementary Figure S3C) and hence that in the context of Dhr1, this CTD domain is not involved in homodimer formation.
Figure 3.

The CTD of Dhr1 is required for its recruitment to precursor ribosomes in vivo and for Utp14 binding in vitro. (A) Superimposition of YdbC (green) bound to ssDNA (black) onto Dhr1 CTD. (B) Mapping of the electrostatic potential (colored from –15 kBT/e− in red to 15 kBT/e− in blue) on the surface of the Dhr1 CTD. In panels B and C, the YdbC–ssDNA complex is represented with the same color code and in the same orientation as in A. The proposed path for the 3′ ssRNA extremity is shown as a dotted line. The electrostatic potential was calculated using the PBEQ Solver server (62). (C) Mapping of the sequence conservation at the surface of the Dhr1 CTD. Coloring is from grey (no conservation) to red (high conservation). The conservation score was calculated using the Consurf server from a sequence alignment of 45 fungal Dhr1 (63). (D) Velocity gradient analysis of strains expressing various Dhr1 constructs. Detection with anti-Dhr1 antibody in diploid BY4743 cells (first lane), and detection with anti-HTP in heterozygous DHR1-HTP/DHR1 (other lanes). The Dhr1 constructs used in this experiment are schematically depicted on the right of the sucrose gradient profiles. This experiment was repeated three times, a representative example is shown. (E) The CTD of Dhr1 has no significant influence on its RNA-dependent ATPase activity. Identical amounts of the indicated recombinant constructs were engaged in a classical green malachite ATPase assay ((64) and see Materials and Methods). The experiment was reproduced three times independently (the mean values and s.d. are shown). Note that poly-(A) RNA was used in these experiments. (F) The CTD of Dhr1 is enhancing the interaction with its coactivator Utp14 in vitro. The interaction between various Dhr1-Hel constructs and Utp14 was addressed by GST-pulldown assays. Input and eluate (GST-pulldown) fractions were analyzed on 10% SDS-PAGE and stained with Sypro Ruby. L: Molecular weight ladder.
YdbC and PC4 are also known to bind tightly to single-stranded DNA (ssDNA) as well as to ssRNA, albeit with lower affinity (42–44). The superimposition of YdbC–ssDNA complex onto the structure of Dhr1 CTD reveals that helix α24 clashes with the 3′ half of the ssDNA fragment bound to YdbC (see Figure 3A, helix α24 in wine red). This indicates that if ssRNA binds there it should be through an alternative mode of interaction (see below). In contrast, the region corresponding to the YdbC area interacting with the 5′ half of ssDNA fragment is fully accessible on Dhr1 (Figure 3A–C), and is well positioned to participate in ssRNA binding. However, no interaction could be detected in vitro between the isolated CTD and short ssRNAs (poly(U)15 or poly(A)15; data not shown). Furthermore, this superimposition reveals that a positively charged bent crevice at the interface between the OB-fold and the CTD domain of Dhr1 is ideally positioned to form an alternative binding route for the ssRNA 3′ half (shown as dotted lines in Figure 3B and C). The ‘walls’ of this crevice are formed by conserved residues typically known to be enriched in nucleic acid binding sites (Y1190, K1192, R1209, Q1238, K1240 and W1245; Figure 3C and Supplementary Figure S4). Hence, this specific carboxyl-terminal domain could influence Dhr1 recruitment to pre-90S ribosomes (see Figure 3), which are early precursor ribosomes containing the primary transcript (the 35S), or its immediate RNA processing byproducts prior to cleavage in internal transcribed spacer 1 that separate the precursors of the small (pre-40S) from those of the large (pre-60S) subunits. If this assumption is correct, the CTD may play important roles in early pre-rRNA processing mediated by Dhr1 (see Figure 4).
Figure 4.
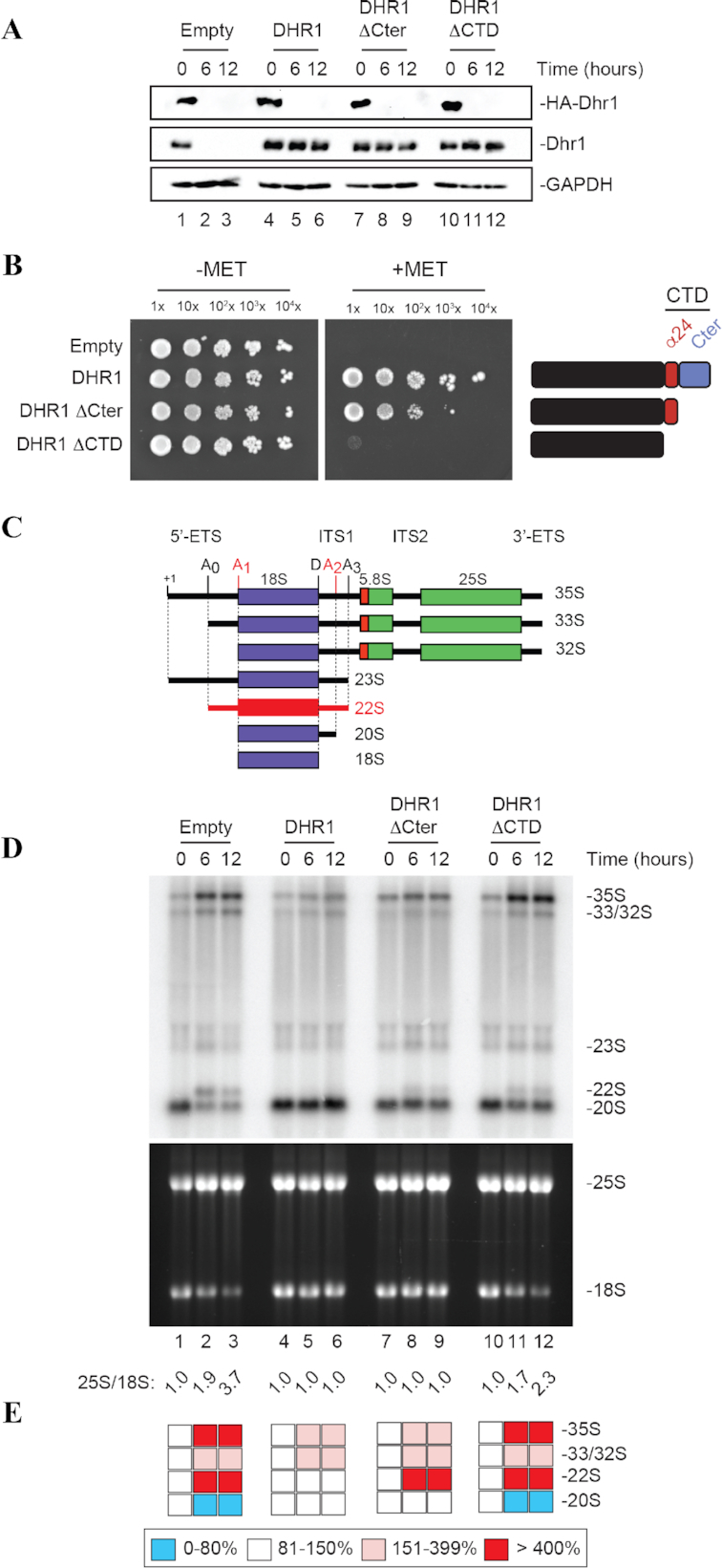
The CTD of Dhr1 is required for cell growth and small ribosomal subunit biogenesis. (A) The two Dhr1 CTD truncations (Dhr1ΔCTD and Dhr1ΔCter) are stably expressed. Western blot analysis showing the progressive extinction of expression of endogenous HA-Dhr1, specifically detected with an anti-HA antibody (top panels), and the expression of the complementing wild-type Dhr1 and the two CTD mutants (middle panels, detection with an anti-Dhr1 antibody). As loading control, blots were probed for GAPDH. The experiments in panels A and D are showing a time course of transfer from a medium lacking methionine (0-h time points) to a medium containing methionine (6- and 12-h time points). (B) The Dhr1ΔCTD and Dhr1ΔCter truncations impact cell growth differentially. Serial dilutions of the indicated yeast strains were incubated on agar plates for 3 days at 30°C in the absence (–MET) or presence (+MET) of methionine. (C) Structure of the yeast primary transcript (35S), of precursors (33S, 32S, 23S, 20S) in the pathway of synthesis of the 18S rRNA, and of the aberrant 22S RNA, typically accumulated in dhr1 mutants. Three out of four mature rRNAs are produced from a single polycistronic transcript by processing. Cleavage sites important for 18S rRNA production (A0, A1, A2, A3 and D) are shown. The 22S RNA extending from site A0 to A3 is produced in the absence of cleavages at sites A1 and A2 (highlighted in red). (D) The two Dhr1 CTD truncations inhibit pre-rRNA processing with distinct degree of severity. Top, Northern blot analysis detecting major pre-rRNA species (indicated to the right, and see panel C). The probe used was specific to internal transcribed spacer 1 (ITS1) sequence (see Materials and Methods). Bottom, ethidium bromide-stained denaturing agarose gel showing the amounts of 18S and 25S rRNAs. The 25S/18S ratio established from bioanalyzer electropherogramms is indicated below the gels. (E) Quantification of panel D. Pre-rRNA processing intermediates were quantified with a Phosphor imager. Results are shown as a heat-map.
The CTD of Dhr1 regulates its release from the SSU-processome in vivo
In the course of normal ribosomal subunit assembly, Dhr1 is recruited to precursor subunits where it unwinds duplexes formed between U3 and the pre-rRNA before being recycled to the pool of free proteins (10,16). Dhr1 is therefore associated with, and active on pre-ribosomes for a defined ‘time frame’. On the basis of its possible ssRNA-binding capacity, we were interested to learn if the carboxyl terminal domain of Dhr1 might be involved in regulating the dynamic association of the helicase with pre-ribosomes.
To test this hypothesis, we designed two C-terminally truncated Dhr1 constructs lacking either the entire CTD (residues 1–1175), hereafter referred as Dhr1ΔCTD, or only the C-terminal part located right after helix α24 (residues 1–1188), hereafter referred as Dhr1ΔCter. Initially, we aimed at constructing these two alleles directly on the chromosome of an haploid yeast strain. Despite repeated attempts, it was not possible, leading us to conclude that the CTD is essential, or at least very important, for cell growth (data not shown). This conclusion was later confirmed by use of a conditional expression system (see below Figure 4).
To circumvent this problem, we then manipulated the carboxyl terminal end of DHR1 on one of the two chromosomes of a diploid strain, leaving one chromosome, and thus one gene copy, intact. To become distinguishable, the manipulated gene copy was tagged with HTP (His6-TEV-ProtA). The distribution of Dhr1-HTP was found to be largely similar to that of untagged Dhr1, as established in a reference diploid wild-type yeast strain, indicating that the HTP-tagged construct is perfectly suitable to study the role of the carboxyl terminal end of Dhr1 (Figure 3D). Two yeast strains expressing either the Dhr1ΔCter or the Dhr1ΔCTD alleles fused to a C-terminal HTP-tag were then produced. Importantly, both these strains grew perfectly well indicating that neither truncations exert a dominant negative effect on cell growth (data not shown).
Analyses of the distribution of Dhr1 in these strains reveal that the two CTD truncations have a similar impact albeit with distinct severity (Figure 3D), namely there is an increase in the amounts of Dhr1 associated with large precursor ribosomes in the mutants (fractions 16–21) (stronger effect in Dhr1ΔCTD). This indicates that the CTD controls the release of Dhr1 from pre-ribosomes. Note that the amounts of Dhr1 associated with lighter fractions (fractions 4–5) is also increased in the mutants (stronger effect in Dhr1ΔCTD). This further suggests that the CTD truncated forms of Dhr1 may remain bound to 5′-ETS spacer fragments after A0-A2 cleavages due to inefficient release of the helicase from precursors. In this genetic background, we also attempted to introduce the ATPase deficient allele Dhr1-K420A, however this mutation was reported to be dominant negative (45), and, indeed, we were unable to produce a strain that could be successfully analyzed (data not shown).
The CTD of Dhr1 is required for efficient interaction with the coactivator Utp14 in vitro but not for RNA-dependent ATPase activity
To further deepen our understanding of the roles played by the CTD of Dhr1, we next tested in vitro whether it is mandatory for Dhr1 RNA-dependent ATPase activity (Figure 3E). As the degree of purity of Dhr1-Hel fragment is higher than that of entire Dhr1 protein, we performed these experiments using the Dhr1-Hel fragment deleted for the entire CTD (hereafter referred to as Dhr1-Hel-ΔCTD) or for the C-terminal region located downstream of helix α24 (hereafter referred to as Dhr1-Hel-ΔCter). We found that both constructs exhibit a similar ATPase activity as wild-type Dhr1-Hel (Figure 3E). As control, we used a protein harboring the K420A mutation, which is known to impact strongly the RNA-dependent ATPase activity of Dhr1 (16).
In parallel, and as it was formerly shown using a yeast two-hybrid assay that a large C-terminal region encompassing residues 839–1267 of Dhr1, i.e. a region including the WH, HB and OB domains as well as the newly-described CTD, is required for its interaction with Utp14 (21), we were interested to learn if this involves directly the CTD. To test this hypothesis, we have assessed the role of the CTD on the in vitro interaction between various recombinant GST-Dhr1 constructs (with or without the CTD) and full-length Utp14-His6 by GST-pulldown assays (Figure 3F). When the intact wild-type Dhr1-Hel was incubated with Utp14-His6, a band corresponding to Utp14 was clearly visible in the eluate in the gel stained with the sensitive Sypro Ruby dye (highlighted with a white arrow on Figure 3F; compare lanes 1, 4 and 9). This demonstrates that Dhr1 interacts weakly but specifically with full-length Utp14-His6. Interestingly, both Dhr1-Hel-ΔCTD and Dhr1-Hel-ΔCter constructs exhibit drastically reduced binding to Utp14-His6 by comparison to Dhr1-Hel (Figure 3F, compare lanes 9–11). This observation indicates that the CTD of Dhr1 contributes significantly to the interaction between Dhr1 and Utp14. The data also confirms that the interaction between Dhr1 and Utp14 is a direct one (21).
The CTD of Dhr1 is required for cell growth and small ribosomal subunit biogenesis
Having established that the CTD of Dhr1 is important to control the release of the helicase from pre-ribosomes in vivo and for its association with its coactivator Utp14 in vitro, we were interested to learn the exact impact of the CTD on yeast cell growth and small ribosomal subunit biogenesis.
Our work so far had informed us that the CTD is either important or essential for cell growth, thus we set up an experimental system to express CTD mutations conditionally (Figure 4 and materials and methods). Practically, the Dhr1ΔCTD and Dhr1ΔCter constructs were introduced on a plasmid in a strain in which the expression of the endogenous DHR1 gene can be progressively shut down by addition of methionine in the growth medium. To this end, a pMET promoter was inserted by homologous recombination on the chromosome upstream of the DHR1 gene in an haploid strain. To allow easy monitoring of protein levels, we also added a human influenza hemagglutinin A (HA)-tag at the amino-terminal end of Dhr1. Hence, when pMET::HA-dhr1 cells are grown in medium lacking methionine, HA-Dhr1 is readily detected (Figure 4A, 0-h time points), indicating that the protein is well expressed and metabolically stable. Upon methionine addition, the expression of endogenous HA-Dhr1 is shut down, and HA-Dhr1 level falls rapidly below western blot detection levels (as soon as after 6 h of transfer to non-permissive growth conditions, corresponding to ∼3 doubling times; Figure 4A, see 6- and 12-h time points).
The pMET::HA-dhr1 strain was transformed with a plasmid expressing either wild-type Dhr1 or one of the two carboxyl terminal truncations, namely: Dhr1ΔCTD or Dhr1ΔCter under the control of its own promoter. As control, pMET::HA-dhr1 cells were also transformed with an empty plasmid (Figure 4A). Upon addition of methionine to the medium, the endogenous HA-tagged Dhr1 was efficiently depleted in cells transformed by any of the plasmids (Figure 4A, see 6- and 12-h time points). In cells transformed with an empty control plasmid, no expression of Dhr1 was detected (Figure 4A, lanes 2–3, middle panel) and, as expected, these cells were unable to grow on medium containing methionine (Figure 4B and Supplementary Figure S5). In cells transformed with the plasmid encoding wild-type Dhr1, the protein was readily detected with an anti-Dhr1 antibody (Figure 4A, lanes 5–6) and cells grew well on methionine-containing agar plates (Figure 4B). Interestingly, the two Dhr1 CTD truncations impact cell growth differentially although they are stably expressed in cells (Figure 4A). The Dhr1ΔCter variant that retains the alpha helix α24 complements growth partially while the Dhr1ΔCTD variant lacking this helix does not restore growth at all (Figure 4B).
As Dhr1 is known to be required for pre-rRNA cleavage at sites A1 and A2 (10), we analyzed pre-rRNA processing in details by northern blotting in all the strains (Figure 4C–E). In the absence of cleavage at sites A1 and A2, the 33S pre-rRNA (corresponding to the primary transcript cleaved in the 5′-ETS at site A0) is processed at site A3 within ITS1, producing an aberrant 22S RNA, which is not further matured into 18S rRNA (Figure 4C and Supplementary Figure S8). The aberrant 22S RNA is formed at the expense of normal 20S pre-rRNA, which is the direct precursor of mature 18S rRNA. Hence, failure to cleave at sites A1 and A2 leads to 18S rRNA production impairment and cell death (10).
Total RNA was extracted from pMET::HA-dhr1 cells transformed with the above described plasmids and grown in the presence of wild-type Dhr1 (0-h time point) or in its absence (i.e. after addition of methionine, 6- and 12-h time points). Cells transformed with the control wild-type DHR1 plasmid showed no processing defect: (i) the levels of each precursor detected were largely normal (Figure 4D and E; compare lane 4 with lanes 5–6); (ii) there was no production of the aberrant 22S RNA and (iii) the ratio of 25S/18S rRNA was of 1.0, as in wild-type cells since these two rRNAs are produced from the same polycistronic transcript. In contrast, cells transformed with the empty control plasmid were strongly defective for pre-rRNA processing, as demonstrated by: (a) an accumulation of the long nucleolar precursors (35S and 33/32S pre-rRNAs), (b) the formation of the aberrant 22S RNA at the expense of 20S and (c) a severely reduced production of 18S rRNA and a 25S/18S rRNA ratio that increased from 1.0 to 3.7. Interestingly, we observed a graded pre-rRNA processing inhibition in the two CTD Dhr1 truncations with the Dhr1ΔCTD showing a more severe phenotype than the Dhr1ΔCter. Globally, the two CTD truncations affect pre-rRNA processing in a fashion highly similar to the one described in the absence of Dhr1 (empty plasmid control), i.e. by inhibiting cleavage at sites A1 and A2, but with varying degree of severity. The major difference between the two CTD truncations being that only in the Dhr1ΔCTD the processing inhibition is sufficiently important to affect the level of mature 18S rRNA production: the 25S/18S ratio reaches 2.3 with Dhr1ΔCTD while it remains at 1.0 with Dhr1ΔCter. This indicates that a certain processing inhibition threshold must be passed for detecting mature rRNA accumulation defects. Such threshold is reached with the Dhr1ΔCTD but not with the Dhr1ΔCter variant.
In conclusion, a thorough characterization of the CTD of Dhr1 has revealed that: (i) it is important for dissociating Dhr1 from the SSU-processome in cells, (ii) it is enhancing the interaction of Dhr1 with its coactivator Utp14 in vitro, (iii) it is indispensable for pre-rRNA processing reactions leading to production of mature 18S rRNA (cleavages A1 and A2), (iv) it is essential for cell growth, (v) it is not required for the RNA-dependent ATPase activity of Dhr1, (vi) it does not promote helicase dimer formation in the context of Dhr1, and, finally, (vii) it does not bind ssRNA (at least in the conditions tested in our assays).
We suggest that the strict requirement of the CTD of Dhr1 for its functions in ribosome biogenesis explains why Dhr2 over-expression (40% sequence identity and 59% sequence similarity with Dhr1), which lacks this CTD, cannot complement the deletion of DHR1 gene in S. cerevisiae (10). In most cases, the homology between related helicases is confined to the conserved motifs in the central area of the protein and rarely concerns the extensions whose functions remain largely undefined (46). For Dhr1, at least, we have now clearly established the importance of its CTD in small ribosomal subunit biogenesis.
Functional characterization of patient and animal mutations identified in vertebrate Dhr1 (DHX37)
Ribosomopathies are human syndromes often associated with cancer predisposition (47), resulting from ribosome assembly dysfunction caused by a mutation in a ribosomal protein or a ribosome assembly factor (48,49). Dhr1 is essential for growth in budding yeast (20), and mutations in DHX37, its metazoan ortholog, have been identified in patients suffering from microcephaly and other neurological disorders, as well as in animal models (fish) (26,27). Thus, in principle, Dhr1 should be added to the growing list of factors susceptible to cause ribosomopathies. However, a formal demonstration that the Dhr1 mutations identified in vertebrates cause ribosome biogenesis inhibitions was missing.
So far, two different homozygous mutations (N419K and R487H) have been identified in DHX37 (human Dhr1) in neurological patients (27), and zebrafish carrying the L489P mutation of Dhx37 exhibit abnormal motor response (26). The residue corresponding to human DHX37 N419 is N570 in S. cerevisiae Dhr1. It is located in a loop of the RecA1 domain and is at the interface with the WH domain (Figures 2B and 5A). Its side chain is engaged in an intricate hydrogen bonding network with main chain atoms from T572 and L573 from the RecA1 domain but also from F867 and P868 from the WH domain (Figure 5A). With the exception of T572, all of these residues are strictly conserved among Dhr1 proteins (Supplementary Figures S2 & S4) suggesting that this interaction network might be of functional importance. The substitution of this Asn by a Lys would prevent the formation of this dense interaction network. The two other described DHX37 mutants (R487H in human and L489P in zebrafish) are incidentally remarkably close to one another and they correspond to K637 and L639 in S. cerevisiae Dhr1, respectively. They are both located within helix α9 from the RecA2 domain (Figures 2B and 5B). L639 is a strictly conserved residue (Supplementary Figure S2), located in the center of helix α9 and pointing toward the hydrophobic core of the RecA2 domain (Figure 5B). Its substitution by a Pro residue, known to disrupt α helices, is strongly expected to affect the conformation of this region. In S. cerevisiae Dhr1, K637 is solvent exposed and has the same positively charged property as human R487. Its substitution by an histidine should not affect the overall fold of this region but could modulate interaction with a potential partner (protein or nucleic acid).
Figure 5.
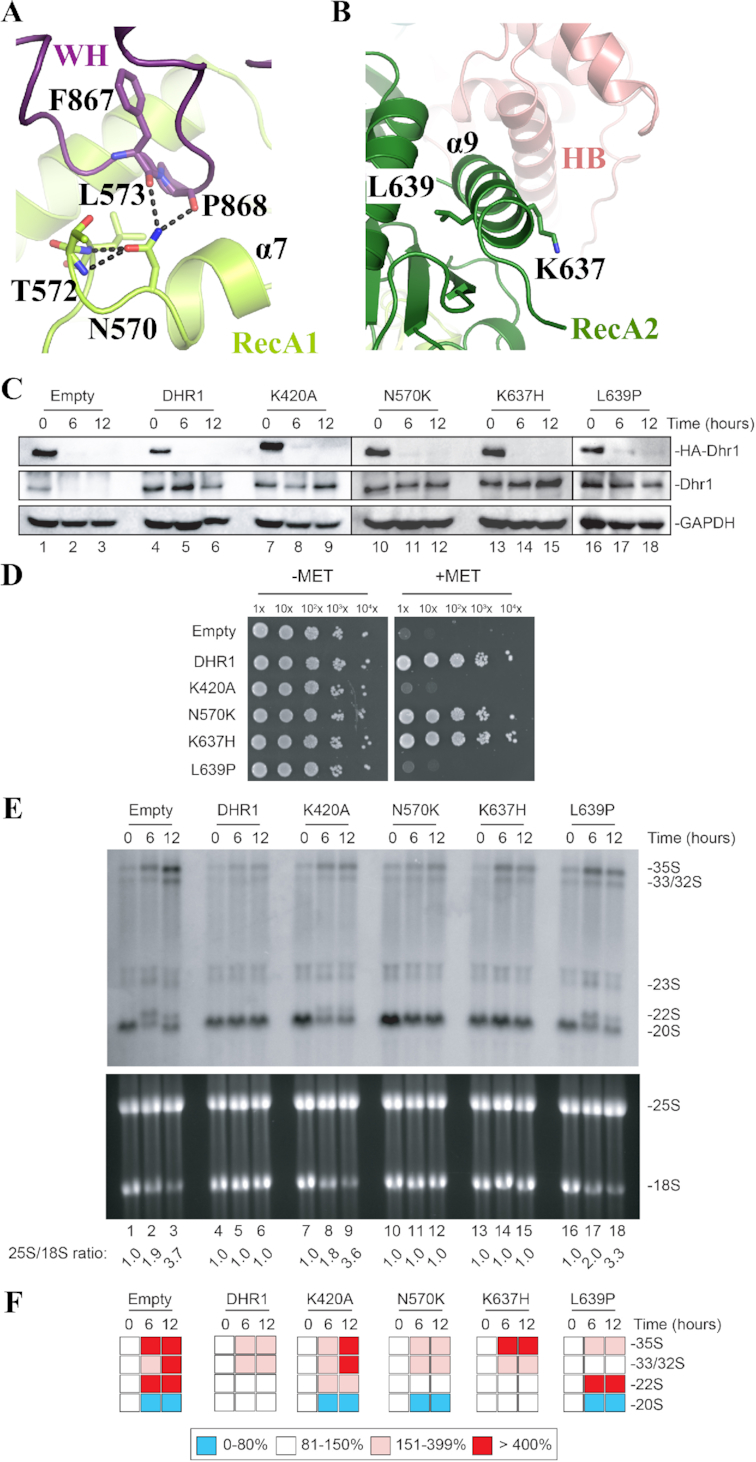
Functional analysis of DHX37 mutations associated with diseases. (A) The RecA1 mutation N570K (equivalent to N419 in human DHX37) is expected to disrupt the compact hydrogen bonding network (dashed black lines) normally formed between this residue and its neighbors. See also Figure 2B to localize the mutation in the global structure of the protein. (B) The RecA2 mutations K637H (R487 in human) and L639P (L489 in zebrafish) are located close to each other in helix α9. K367 is solvent exposed and positively charged, L639 is in the center of the helix and its substitution by a Proline is expected to disrupt the helix. (C) All four Dhr1 mutations are stably expressed. Western blot analysis showing the progressive extinction of expression of endogenous HA-Dhr1, specifically detected with an anti-HA antibody (top panels), and the expression of the complementing wild-type Dhr1 and four mutants (middle panels, detection with an anti-Dhr1 antibody). As loading control, blots were probed for GAPDH. The experiments in panels C and E are showing a time course of transfer from a medium lacking methionine (0-h time points) to a medium containing methionine (6- and 12-h time points). (D) Mutations K420A and L639P are deleterious for growth. Serial dilutions of the indicated yeast strains were incubated on agar plates for 3 days at 30°C in the absence (–MET) or presence (+MET) of methionine. (E) Mutations K420A and L639P are deleterious for ribosome biogenesis. Top, Northern blot analysis detecting major pre-rRNA species (indicated to the right). The probe used was specific to internal transcribed spacer 1 (ITS1) sequence (see materials and methods). Bottom, ethidium bromide-stained denaturing agarose gel showing the amounts of 18S and 25S rRNAs. The 25S/18S ratio established from bioanalyzer electropherogramms is indicated below the gels. (F) Quantification of panel E. Pre-rRNA processing intermediates were quantified with a Phosphor imager. Results are shown as a heat-map.
In order to assess the functional importance for cell growth and ribosome biogenesis of these three residues, we generated the N570K, K637H or L639P single point mutants at the equivalent positions in budding yeast Dhr1. In addition, we produced the Dhr1 K420A mutation known to inactivate the ATPase hydrolysis and unwinding function of the protein (16).
To assess the impact of these four substitutions, we used the same conditional system described above for the in vivo analysis of the CTD truncations (Figure 4). The pMET::HA-dhr1 strain was transformed with a plasmid expressing either wild-type Dhr1, or one of the four single point mutants (K420A, N570K, K637H or L639P), or an empty control plasmid. All four Dhr1 point mutants were stably expressed (Figure 5C, middle panel). The N570K and K637H Dhr1 mutants complemented growth while neither K420A nor L639P mutants did (Figure 5D). In parallel, we analyzed the doubling time of these different strains in liquid culture and observe that N570K and K637H Dhr1 mutants exhibit the same doubling time as the WT strain, while the K420A and L639P Dhr1 mutants have the same doubling time as the strain transformed with an empty vector (Supplementary Figure S5).
Interestingly, the K420A and L639P Dhr1 single point mutants impacted processing severely, showing the same phenotype as the one described with the empty control plasmid (Figure 5E and F). The two other Dhr1 mutations (N570K and K637H) have virtually no effect on processing, showing the same phenotype as the one described in cells transformed with control plasmid expressing wild-type DHR1. It is noteworthy that N570K leads to a marginal but reproducible reduction of 20S pre-rRNA but this has no consequences on mature rRNA formation (see Discussion).
Hence, among the three disease-related mutations identified in animal and human DHX37 and tested in yeast, only the L639P mutant exhibits growth and processing phenotypes as severe as those observed upon depletion of the WT Dhr1 protein. As anticipated from its location at the center of an α-helix in the RecA2 domain, the substitution of L639 by a Proline, known to break α-helices, is expected to destabilize this region of the protein. On the basis of sequence conservation, we therefore posit that the ribosomal processing defects we report here for the L639P mutation are responsible for the motor neurone defect and abnormal escape response observed in fishes expressing the Dhx37 L489P mutation (26). Future in vivo studies will be needed to characterize more deeply the impact of those human DHX37 and zebrafish Dhx37 mutants on ribosome biogenesis and eventually other processes in these respective organisms.
The Dhr1 helicase module adopts a substrate ‘pre-loading’ conformation compatible with an expansion RNA unwinding mechanism
So far, the only structural insights gained into the function of DEAH-box RNA helicases were obtained from studies on Prp43, an enzyme that contributes directly to both pre-mRNA splicing and pre-rRNA processing being physically associated with both the pre-mRNA spliceosome and the pre-rRNA processome (6,50) (see Introduction). Indeed, several Prp43 crystal structures from different organisms (S. cerevisiae (ScPrp43), Chaetomium thermophilum (CtPrp43) and human (HsDHX15)) were determined in the presence either of a diphosphate nucleotide (NDP forms; (28,32,39,51,52)), of an ATP analog (ATP form; (34)) or of both a poly(U) RNA fragment (ssRNA) and an ATP analog (RNA•ATP form; (33,34)). Each of these structures correspond to a structural ‘snapshot’ of the enzyme in motion.
To our knowledge, only three structures of DEAH-box RNA helicases (bovine DHX36 DNA/RNA helicase (38), C. thermophilum Prp22 splicing factor (35) and our structure of S. cerevisiae Dhr1) have been solved in the absence of any of its substrates (NTP or RNA). Those structures offer a unique opportunity to analyze conformational changes within domains, and domain movements, occurring upon NTP/RNA binding in this important family of enzymes. Note that the apo-form of bovine DHX36 DNA/RNA helicase adopts the same close conformation as its ADP-BeF3 or ADP-AlF4 bound forms (38) and hence will not be discussed in this analysis as it cannot be considered as ‘a true’ apo conformation.
First, the structural comparison of the different individual domains of Prp43 and Dhr1 reveals one major difference occurring within the RecA1 domain in a region, which has been previously proposed to act as a ‘bookend’ delineating the 3′ end of the Prp43 RNA channel (Figure 6A and B). In the structure of CtPrp43 RNA•ATP form (34), the loop connecting strands β3 and β4 (hereafter referred to as ‘3′ clamp’) together with residues from motif Ib located in helix α5, clamps the 3′ end of the ssRNA fragment via conserved residues (R180, F181 and R201; Figure 6B and C and Supplementary Figures S2 and S6). In Dhr1, the loop connecting strands β3 and β4 adopts a different conformation with F479 side chain occupying the position of the ssRNA 3′ end bound to Prp43, and R478 side chains matching with the position of CtPrp43 F181 (Figure 6A and B). In ScPrp43, the double mutant R177A/F178A (corresponding to R180A/F181A in CtPrp43) displays a strong slow growth phenotype while the R177A mutant is characterized by a cold-sensitive phenotype (33). As this region adopts a very similar conformation in the NDP, ATP or RNA•ATP forms of both CtPrp43 and ScPrp43, these different conformations between Dhr1 and Prp43 most likely reflect an intrinsic flexibility of this region. Interestingly, ScPrp43 and archaeal Hel308, a DEXH DNA helicase, have been crystallized with long nucleic acids and in these structures, the 3′ end of the single stranded nucleic acid wraps around this 3′ clamp (Supplementary Figure S6), further supporting that this region may have a conserved function in these DEXH helicases (31,33).
Figure 6.

RNA unwinding mechanism mediated by Dhr1. (A) Superimposition of CtPrp43 (grey) and Dhr1-Hel (light green) 3′ clamp and motif Ib (helix α5) regions from the RecA1 domains. (B) Ribbon representation of ssRNA (red) binding to the RecA1 domain of CtPrp43 (grey; (34)). The superimposed Dhr1 RecA1 domain is shown in light green. The side chains from the 3′ clamp (R180 and F181) as well as R201 from motif Ib that clamp the 3′ end of the ssRNA in CtPrp43 are shown as grey sticks. Equivalent residues from Dhr1 are shown as sticks in light green. (C) Surface representation of ssRNA (red) binding site on the RecA1 domain of CtPrp43 (grey). Same orientation as panel B. (D) Superimposition of Dhr1-Hel apo form onto CtPrp43 RNA•ATP form (gray). The two structures were superimposed using their RecA2 domain as anchor. The ssRNA bound to CtPrp43 is shown in red. (E) Focus on the superimposition of Dhr1-Hel apo form onto CtPrp43 RNA•ATP form (grey; the RecA2 domains were superimposed as in panel D). The side chains from R180 and F181 from CtPrp43 and R478 and F479 from Dhr1 3′ clamp motifs are shown as sticks. The ssRNA bound to CtPrp43 is shown in red. The distances between the 3′clamp (Cα atom of the arginine residue) and the long β-hairpin from RecA2 domain are shown by red dashed lines.
Second, the comparison of the different structures of Prp43, Prp22 and Dhr1 helicases reveals that the orientation of the various domains relative to each others differ significantly. In particular, the RecA1 is rotated by 26° (as calculated by the DynDom server; (53)) relative to the RecA2 domain in Dhr1 compared to the corresponding domains in both CtPrp43 ATP and RNA•ATP forms (Figure 6D). Such conformational changes between RecA domains in apo and nucleotide bound forms are typical for helicases as the amino acid residues directly interacting with the nucleotide are contributed by both RecA domains and hence nucleotide binding induces closure of the RecA domains (37). Similarly, the Dhr1 auxiliary domain rotates by 20° and 30° relative to the RecA2 domain compared to CtPrp43 ATP and CtPrp43 RNA•ATP forms, respectively (Figure 5D). The comparison of CtPrp22 and Dhr1 apo-structures also reveal significantly different conformations (rmsd value of 3.5–3.9 Å over 370–380 Cα atoms). Indeed, a rotation of Prp22 RecA1 domain (6° and 18° for Prp22 chains B and A, respectively) relative to RecA2 brings it closer to the later. Similarly, the Prp22 auxiliary domain rotates by 52° (chain A) or 38° (chain B) relative to the RecA2 domain compared to the conformation observed in Dhr1. Hence, the conformation of Dhr1 observed in our structure exhibits a more open conformation of both the nucleotide and the RNA binding sites by comparison to what is seen in the published structures of Prp43 and Prp22. We conclude that our Dhr1 structure likely corresponds to the conformation of the enzyme in a pre-RNA loading step (Supplementary Figure S7).
The differences highlighted above, namely: (i) distinct conformations of the 3′ clamp and (ii) rotation of RecA1 relative to RecA2, both result in an extended distance between the 3′ clamp of the RecA1 domain (residues R478 and F479 in Dhr1) and the long β-hairpin of the RecA2 domain (proposed to act as a bookend for the 5′ extremity of the ssRNA) in the Dhr1 apo form compared to the CtPrp43 RNA•ATP form (33) (Figure 6E). This distance increases from 27 Å in Prp43 to 33 Å in Dhr1, which is compatible with the accommodation of a 5-nucleotides stretch by Dhr1 in the absence of nucleotide (apo form) while only 4 nucleotides could be positioned between these two bookends in the ATP-bound form of DEAH helicases. This observation fits perfectly with the ‘expansion model’ recently proposed for several other DEAH-box RNA helicases, namely Prp43 (33), Prp22 (35) and DHX36 (38), to translocate along RNAs. This model posits that in its apo-form, the DEAH helicase accommodates 5 nucleotides between its 5′ bookend and 3′ clamp, while upon ATP binding, the RecA1 domain comes closer to the RecA2 domain to sandwich ATP, resulting in an enzyme that can now only ‘fit in’ 4 nucleotides between its 5′ bookend and 3′ clamp. Cycling between those two alternative conformations would allow DEAH RNA helicases to progress in a 3′ to the 5′ orientation in order to unwind RNA molecules as experimentally determined for Prp22 and Prp43 (33,54).
Role of Dhr1 in remodeling the CPK during small ribosomal subunit assembly
In order to test, directly in cells, if the inactivation of the Dhr1 ATPase and helicase activities impacts the association of U3 with pre-rRNAs, we established the distribution of U3 on velocity gradient using extracts from Dhr1-K420A catalytically-defective cells. The same amounts of total extracts from pMET::HA-dhr1 cells expressing the K420A mutation in the presence of wild-type Dhr1 (–MET), or in its absence (+MET), were separated by 5–50% sucrose gradients. After centrifugation, total RNA was extracted from the individual collected fractions, separated by denaturing acrylamide gel electrophoresis, and analyzed by Northern blotting with a probe specific to U3.
The expression of the Dhr1-K420A mutation leads to a major enrichment of U3 in fractions corresponding to 90S pre-ribosomes (from fractions 10 to 16; Figure 7A), indicating that U3 is more associated with large precursor subunits. This data confirms that the ATPase and helicase activities of Dhr1 are instrumental to the efficient release of U3 from pre-90S ribosomes in cells (16). A conclusion that correlates well with in vivo cross-linking analysis that revealed that Dhr1 preferentially interacts with the 5′ end region (residues 20–60) of U3 snoRNA and with pre-RNA fragments that form helices h11, h23 and h44 in the structure of mature 18S rRNA, and with in vitro unwinding assays (16).
Figure 7.

The involvement of Dhr1 in CPK formation and pre-90S assembly. (A) Velocity gradient analysis showing that in the absence of Dhr1′ catalytic activity, the box C/D snoRNA U3 remains bound to pre-90S ribosomes. Total cell extract from yeast cells expressing the K420A mutation in the presence of wild-type Dhr1 (–MET) or in its absence (+MET, see legend to Figure 4 for details) were separated on 5–50% sucrose gradients. RNA extracted from each collected fraction was resolved on denaturing polyacrylamide gels and analyzed by northern blotting with a probe specific to U3. The U3 signal detected in each lane was quantitated with a Phosphor imager and expressed as a percentage of total U3 signal (histogram in the upper part of the panel). For reference, positions of 40S, 60S, and 80S ribosomal peaks (as established by OD260) are indicated. TE, total extract control sample. It is not clear at this stage why U3 appears more abundant in the +MET panel (since the same amounts of total cell extracts were loaded on each -MET and +MET panels, but we note that an identical observation was made independently with the same K420A mutation (see Figure 2E in (16)). This experiment was repeated three times, a representative example is shown. (B) Surface representation of S. cerevisiae pre-90S particle as determined by cryo-EM at 3.8 Å overall resolution (58). Assembly factors modeled are colored in yellow and ribosomal proteins (RpS) in gray. For the sake of simplicity, the factors and RpS are not named on the figure, but their identify is known (see PDB 5WLC). The three rRNA helices with which Dhr1 interacts are shown: h11 (purple), h23 (pink) and h44 (red dashed line, this latter helix is largely undefined in the cryo-EM map likely due to high intrinsic flexibility). Nucleotides from pre-18S rRNA forming the CPK in mature 18S are colored in green in panels B to D (see also Figure 1); U3 is in blue. Selected assembly factors are highlighted (Utp14, Utp24-Krr1-Faf1, Bms1-Rcl1). The 18S rRNA is in salmon and the 5′ ETS in orange. (C) Simplified version of panel B (excluding factors not found in association with Dhr1-K420A in (16)). At this stage, Dhr1 has not been mapped on cryoEM structures because it was absent from the purified precursor ribosomes analyzed. Inset: surface representation of the Dhr1 helicase module at the same scale as pre-90S pointing toward its site of action: the U3 snoRNA (in blue) on the precursor small subunit. This is where we suggest Dhr1 binds after the release of the assembly factors Utp24-Krr1-Faf1 (see text for details). (D) ‘At the heart of the SSU-processome’: representation of RNA duplexes formed between the 5′ end of U3 and the 18S rRNA. The Dhr1-interacting protein Utp14 is colored in dark blue. U3 nucleotides identified as interacting directly with Dhr1 in in vivo cross linking studies, or as suppressing the growth defect of a cryo-sensitive dhr1 allele, are shown in cyan and black spheres, respectively (16). CPK formation is essential to small ribosomal subunit biogenesis because it brings together sequences from the four rRNA domains which are far apart in the primary structure. The four rRNA domains are colored as follows: 5′ domain (light grey), central domain (yellow), 3′ major domain (pink), and 3′ minor domain (light blue) according to the color scheme used in Figure 1 and in the inset. The duplexes formed between the U3 boxes A and A’ and the 18S rRNA are highlighted in black boxes. The G1575 nucleotide, which is N7-methylated by the Bud23-Trm112 complex is shown as blue spheres. The proposed path of the Dhr1 helicase onto the U3 snoRNA-pre-rRNA duplexes during unwinding is depicted as a dashed red arrow from 3′ to 5′ end (left to right). Inset: Cartoon representation of the 18S rRNA precursor bound to U3, using the same color code as in the 3D model representation.
DISCUSSION
RNA helicases are involved in all cellular processes and are particularly important to regulate the numerous structural transitions and remodeling events occurring during ribosomal subunit assembly (3,55). Twenty five RNA helicases have been involved in ribosome biogenesis thus far in budding yeast (56,57). Of these, the DEAH-box helicase Dhr1 is essential for small ribosomal subunit assembly as it regulates the timing of CPK formation by displacing the box C/D snoRNA U3 from pre-rRNAs, thus allowing essential Watson-Crick base-pairing to be formed at the heart of the mature 40S subunit ((16), see Figures 1 and 7D).
In this work, we have determined the crystal structure of the DEAH-box RNA helicase module (Dhr1-Hel) of S. cerevisiae Dhr1 revealing both commonalities and specificities with other members of this group. Globally, the structure of Dhr1 adopts the shape of ‘a hand’, with as most striking feature the presence of a CTD forming ‘a wrist’, which is absent in other proteins of the DEAH-box RNA helicase family (Figure 2). We provide important functional insights into the role of the CTD of Dhr1 in ribosome biogenesis. Notably, we show that the CTD of Dhr1 is required for: (i) cell growth, (ii) small ribosomal subunit pre-rRNA processing, (iii) efficient dissociation of Dhr1 from pre-ribosomes in vivo and (iv) interaction with the helicase coactivator Utp14 in vitro (Figures 3 and 4). As Utp14 is known to stimulate Dhr1 helicase activity in yeast (21), we further propose that in the absence of CTD, Utp14 cannot stimulate the helicase activity of Dhr1. This could explain the recycling defects observed in cells expressing the Dhr1ΔCTD and Dhr1ΔCter constructs.
Our conclusion that the CTD of Dhr1 is important for its association with Utp14 but not for its RNA-dependent ATPase activity is in agreement with a previous study that did not detect any influence of Utp14 on Dhr1′ ATPase activity (21). While this manuscript was under revision, the crystal structure of mouse DHX37 was determined, revealing the presence of a CTD which is structurally related to that of yeast Dhr1 but which, in addition, displays an extra seven α-helices-bundle (40). It is interesting that murine UTP14a (the ortholog of yeast Utp14) binds the CTD of DHX37 strongly stimulating its ATPase activity. We suggest that the species-specific differences between the mode of action of yeast and mammalian Utp14/UTP14a result from the presence of the extra helical bundle in murine DHX37.
By comparing our Dhr1 structure to conformational ‘snapshots’ of Prp43 bound to nucleotide and/or to RNA substrate, we observe that the RecA1 domain of Dhr1 is rotated by 26° relative to its RecA2 domain compared to Prp43 (Figure 6). On this basis, we propose that our Dhr1-Hel structure obtained without nucleotide or RNA, corresponds to a substrate ‘pre-loading’ state ready to accommodate a stretch of five nucleotides of RNA between its 5′ and 3′ bookends (Figure 6).
Finally, we have functionally characterized three mutations identified in an animal model or in human patients suffering from neurological disease and showed that one (L639P) impairs ribosome biogenesis severely (Figure 5). It is important to keep in mind that any mutations that would obliterate entirely the function of Dhr1 in ribosome biogenesis would be embryonic lethal, thus it is not surprising that mutations observed in vivo only inhibit Dhr1 functions partially. This is notably the case for mutation N570K, which leads to a marginal yet perfectly reproducible reduction of 20S pre-rRNA production (Figure 5E and F). A deeper characterisation of the impact of these mutations will require further work conducted directly in metazoan models where subtle effects can be monitored with dedicated assays (e.g. behavioral assays).
How does Dhr1 unwinds U3 from pre-rRNAs?
Despite recent advances in the structural characterization of precursor ribosomal subunits, it remains unclear how Dhr1 exactly interacts with precursor ribosomes because it was absent from the particles analyzed by cryo-EM thus far. In an attempt to understand how Dhr1 contacts and progresses onto maturing pre-ribosomes in order to release U3, we have combined our Dhr1-Hel structure with the published high-resolution cryo-EM structure of yeast pre-90S ribosomes and with nucleotide resolution information on the physical and functional contacts between Dhr1, the 18S rRNA and U3 (Figure 7B–D; (16,58)).
Using the published yeast pre-90S structures, which crucially lack Dhr1 (13,14), we produced a model in which we included the 18S rRNA, the U3 snoRNA and only the proteins (ribosomal proteins and assembly factors) which were affinity purified with the catalytically inactive allele of Dhr1 (the K420A mutant, see (16)), as we assumed that this is the best proxy for a Dhr1-containing particle (Figure 7C). In our model, the 18S rRNA helices h11 and h44, and to a lesser extent h23, which interact with Dhr1 according to UV cross-linking analyses (16), are localized at the surface of the pre-90S particle. However, they are too distant from one another to be bound simultaneously by Dhr1, suggesting that a large conformational change within pre-90S is either required prior to Dhr1 binding, or that such conformational reorganization is induced upon Dhr1 binding (Figure 7C).
Of particularly interest, is the striking absence in Dhr1 K420A-purified particles of the Utp24-Krr1-Faf1 complex (illustrated in white rendering in Figure 7B). Utp24 is an important SSU-processome component as it is the reported endonuclease responsible for the coupled pre-rRNA cleavages at sites A1/A2 (59). Indeed, Utp24 and Dhr1 binding sites on 18S rRNA, as determined by UV cross-linking (16,59), partially overlap, and the Utp24-Krr1-Faf1 complex ‘holds’ apart the helices h11 and h23 (in magenta and pink in Figure 7B and C, respectively), which are destined to unite in space with helix h44 after CPK formation (see also Figure 1). We propose that after A1/A2 processing, the Utp24-Krr1-Faf1 complex is dissociated from the SSU-processome, ‘opening up’ an area on the pre-ribosome to allow Dhr1 binding. Fully consistent with this idea, it was recently shown that 90S pre-ribosomes purified from cells depleted of Dim2 using Utp1 as bait contain Dhr1 but not Krr1 (60). In this model, the RNA duplex formed between the 5′ end of 18S rRNA (i.e. the region involved in CPK formation in mature 18S) and U3 snoRNA box A exhibits higher solvent accessibility and hence should be more accessible for Dhr1 binding (Figure 7D). The presence of Utp14 in the direct vicinity of U3 box A is also of particular interest as this protein was shown to bind and activate Dhr1 for U3 release, and we have now learned that the CTD of Dhr1 is required for optimal Dhr1-Utp14 interaction (Figures 3 and 7D; (21,22)).
Finally, on the basis of the experimentally demonstrated 3′-to-5′ overhang helicase activity of Dhr1 (16), we propose that the protein binds to the region of U3 formed by nucleotides 29 to 48 (Figure 7D), which were identified as Dhr1 direct contacts by UV cross-linking (16). It would first dissociate the duplex formed between the 5′ end of 18S rRNA (i.e. the region involved in CPK formation in mature 18S) and the U3 snoRNA box A, and then could unwind the duplex formed between U3 snoRNA box A’ (corresponding to U3 snoRNA 5′ end, see Figure 7D and inset) and 18S rRNA region encompassing nucleotides 1112–1122 (this sequence is located upstream of the 1139–1145 region, which base-pairs with the 5′ end of 18S rRNA to form the CPK). Other late assembly factors and some ribosomal proteins are likely to be recruited and to contribute to Dhr1 role in U3 snoRNA dissociation from pre-90S. Following U3 release, Dhr1 would be ideally positioned to ‘hand over’ faithfully folded pre-40S subunits (i.e. that have undergone CPK transition) to its well-established partners: the Bud23-Trm112 methyltransferase complex. The Bud23–Trm112 complex binds to late 40S subunit, on which CPK has been formed (61), and catalyzes the methylation of G1575 into m7G1575, a position located in the direct vicinity of the 5′ end of U3 snoRNA in pre-90S particles (58). This methylation is a late event occurring at the level of the 20S pre-rRNA, i.e. after U3-mediated A1 and A2 cleavages have occurred on pre-rRNAs (Figure 6D; (17,19–21)).
In summary, we propose that: (i) an important structural reorganization occurs within the SSU-processome such that rRNA helices h11, h23, and h44, which are spatially distant in the precursor, are brought together in order to be simultaneously bound by Dhr1, (ii) the Utp24 (the A1/A2 endoRNase)-Krr1-Faf1 subcomplex of assembly factors occupies the binding site of Dhr1, and must be displaced after RNA cleavage to allow Dhr1 to join pre-ribosomes and (iii) on the maturing ribosomes, Dhr1 unwinds first the helix formed between the 5′-end of 18S and the U3 box A, then releases the 18S rRNA-U3 box A’ duplex, to finally hand over faithfully folded pre-40S with CPK established to the m7G methylation machinery. These predictions will await the future structural characterization of Dhr1-bound pre-40S ribosomes to be validated and experimental support, in particular for the Dhr1 role in unwinding of box A’.
DATA AVAILABILITY
The coordinates of the final model and the reflection file used for refinement have been deposited to Protein Data Bank under accession code 6H57. The data that support the findings of this study are available upon request to the corresponding authors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank SOLEIL for provision of synchrotron radiation facilities.
Author contributions: A.R., C.J., G.B. and V.H.H. performed the experiments. D.L.J.L. and M.G. designed research; A.R., C.J., D.L.J.L. and M.G. analyzed the data. A.R., D.L.J.L. and M.G. wrote the paper. D.L.J.L. and M.G. contributed equally to this work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
M.G. acknowledges financial support from the CNRS ATIP-AVENIR program, the Agence Nationale pour la Recherche (ANR) [ANR-14-CE09-0016-02]; CNRS and Ecole Polytechnique. The collaboration between M.G. and D.L.J.L. teams was supported by the CNRS PICS program [PICS07484]. Research in the laboratory of D.L.J.L. is supported by the Université Libre de Bruxelles (ULB), the Fonds de la Recherche Scientifique [F.R.S./FNRS]; Région Wallonne (DGO6). D.L.J.L. is affiliated with the Center for Microscopy and Molecular Imaging (CMMI) and ULB Cancer Research Center (U-CRC). Work of VHH was supported by the CNRS, the Agence Nationale pour la Recherche (ANR) [ANR14-CE09-0016-01]; ‘Initiative d’Excellence’ program from the French State [‘DYNAMO’, ANR-11-LABX-0011]. Funding for open access charge: CNRS.
Conflict of interest statement. None declared.
REFERENCES
- 1. Pena C., Hurt E., Panse V.G.. Eukaryotic ribosome assembly, transport and quality control. Nat. Struct. Mol. Biol. 2017; 24:689–699. [DOI] [PubMed] [Google Scholar]
- 2. Lafontaine D.L. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat. Struct. Mol. Biol. 2015; 22:11–19. [DOI] [PubMed] [Google Scholar]
- 3. Martin R., Straub A.U., Doebele C., Bohnsack M.T.. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013; 10:4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cordin O., Beggs J.D.. RNA helicases in splicing. RNA Biol. 2013; 10:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lebaron S., Froment C., Fromont-Racine M., Rain J.C., Monsarrat B., Caizergues-Ferrer M., Henry Y.. The splicing ATPase prp43p is a component of multiple preribosomal particles. Mol. Cell Biol. 2005; 25:9269–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Combs D.J., Nagel R.J., Ares M. Jr, Stevens S.W.. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Mol. Cell Biol. 2006; 26:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leeds N.B., Small E.C., Hiley S.L., Hughes T.R., Staley J.P.. The splicing factor Prp43p, a DEAH box ATPase, functions in ribosome biogenesis. Mol. Cell Biol. 2006; 26:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lebaron S., Papin C., Capeyrou R., Chen Y.L., Froment C., Monsarrat B., Caizergues-Ferrer M., Grigoriev M., Henry Y.. The ATPase and helicase activities of Prp43p are stimulated by the G-patch protein Pfa1p during yeast ribosome biogenesis. EMBO J. 2009; 28:3808–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pertschy B., Schneider C., Gnadig M., Schafer T., Tollervey D., Hurt E.. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J. Biol. Chem. 2009; 284:35079–35091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Colley A., Beggs J.D., Tollervey D., Lafontaine D.L.. Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNP U3. Mol. Cell Biol. 2000; 20:7238–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klinge S., Woolford J.L. Jr. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019; 20:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kornprobst M., Turk M., Kellner N., Cheng J., Flemming D., Kos-Braun I., Kos M., Thoms M., Berninghausen O., Beckmann R. et al.. Architecture of the 90S pre-ribosome: a structural view on the birth of the eukaryotic ribosome. Cell. 2016; 166:380–393. [DOI] [PubMed] [Google Scholar]
- 13. Chaker-Margot M., Barandun J., Hunziker M., Klinge S.. Architecture of the yeast small subunit processome. Science. 2017; 355:eaal1880. [DOI] [PubMed] [Google Scholar]
- 14. Sun Q., Zhu X., Qi J., An W., Lan P., Tan D., Chen R., Wang B., Zheng S., Zhang C. et al.. Molecular architecture of the 90S small subunit pre-ribosome. eLife. 2017; 6:e22086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hector R.D., Burlacu E., Aitken S., Le Bihan T., Tuijtel M., Zaplatina A., Cook A.G., Granneman S.. Snapshots of pre-rRNA structural flexibility reveal eukaryotic 40S assembly dynamics at nucleotide resolution. Nucleic Acids Res. 2014; 42:12138–12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sardana R., Liu X., Granneman S., Zhu J., Gill M., Papoulas O., Marcotte E.M., Tollervey D., Correll C.C., Johnson A.W.. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol. 2015; 13:e1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Letoquart J., Huvelle E., Wacheul L., Bourgeois G., Zorbas C., Graille M., Heurgue-Hamard V., Lafontaine D.L.. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. PNAS. 2014; 111:E5518–E5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sardana R., Zhu J., Gill M., Johnson A.W.. Physical and functional interaction between the methyltransferase Bud23 and the essential DEAH-box RNA helicase Ecm16. Mol. Cell Biol. 2014; 34:2208–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. White J., Li Z., Sardana R., Bujnicki J.M., Marcotte E.M., Johnson A.W.. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol. Cell Biol. 2008; 28:3151–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Figaro S., Wacheul L., Schillewaert S., Graille M., Huvelle E., Mongeard R., Zorbas C., Lafontaine D.L., Heurgue-Hamard V.. Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575. Mol. Cell Biol. 2012; 32:2254–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu J., Liu X., Anjos M., Correll C.C., Johnson A.W.. Utp14 recruits and activates the RNA helicase Dhr1 To undock U3 snoRNA from the preribosome. Mol. Cell Biol. 2016; 36:965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Black J.J., Wang Z., Goering L.M., Johnson A.W.. Utp14 interaction with the small subunit processome. RNA. 2018; 24:1214–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka N., Aronova A., Schwer B.. Ntr1 activates the Prp43 helicase to trigger release of lariat-intron from the spliceosome. Genes Dev. 2007; 21:2312–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heininger A.U., Hackert P., Andreou A.Z., Boon K.L., Memet I., Prior M., Clancy A., Schmidt B., Urlaub H., Schleiff E. et al.. Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol. 2016; 13:320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choudhury P., Hackert P., Memet I., Sloan K.E., Bohnsack M.T.. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019; 16:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirata H., Ogino K., Yamada K., Leacock S., Harvey R.J.. Defective escape behavior in DEAH-box RNA helicase mutants improved by restoring glycine receptor expression. J. Neurosci. 2013; 33:14638–14644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M. et al.. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron. 2015; 88:499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walbott H., Mouffok S., Capeyrou R., Lebaron S., Humbert O., van Tilbeurgh H., Henry Y., Leulliot N.. Prp43p contains a processive helicase structural architecture with a specific regulatory domain. EMBO J. 2010; 29:2194–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O.S., Vonrhein C. et al.. 2016; Cambridge: Global Phasing Ltd. [Google Scholar]
- 30. Sharma S., Langhendries J.L., Watzinger P., Kotter P., Entian K.D., Lafontaine D.L.. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015; 43:2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buttner K., Nehring S., Hopfner K.P.. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007; 14:647–652. [DOI] [PubMed] [Google Scholar]
- 32. He Y., Andersen G.R., Nielsen K.H.. Structural basis for the function of DEAH helicases. EMBO Rep. 2010; 11:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He Y., Staley J.P., Andersen G.R., Nielsen K.H.. Structure of the DEAH/RHA ATPase Prp43p bound to RNA implicates a pair of hairpins and motif Va in translocation along RNA. RNA. 2017; 23:1110–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tauchert M.J., Fourmann J.B., Luhrmann R., Ficner R.. Structural insights into the mechanism of the DEAH-box RNA helicase Prp43. eLife. 2017; 6:e21510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamann F., Enders M., Ficner R.. Structural basis for RNA translocation by DEAH-box ATPases. Nucleic Acids Res. 2019; 47:4349–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He Y., Andersen G.R., Nielsen K.H.. The function and architecture of DEAH/RHA helicases. Biomol. Concepts. 2011; 2:315–326. [DOI] [PubMed] [Google Scholar]
- 37. Ozgur S., Buchwald G., Falk S., Chakrabarti S., Prabu J.R., Conti E.. The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J. 2015; 282:850–863. [DOI] [PubMed] [Google Scholar]
- 38. Chen M.C., Tippana R., Demeshkina N.A., Murat P., Balasubramanian S., Myong S., Ferre-D’Amare A.R.. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature. 2018; 558:465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murakami K., Nakano K., Shimizu T., Ohto U.. The crystal structure of human DEAH-box RNA helicase 15 reveals a domain organization of the mammalian DEAH/RHA family. Acta Crystallogr. F, Struct. Biol. Commun. 2017; 73:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boneberg F., Brandmann T., Kobel L., van den Heuvel J., Bargsten K., Bammert L., Kutay U., Jinek M.. Molecular mechanism of the RNA helicase DHX37 and its activation by UTP14 in ribosome biogenesis. RNA. 2019; 25:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Holm L., Laakso L.M.. Dali server update. Nucleic Acids Res. 2016; 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rossi P., Barbieri C.M., Aramini J.M., Bini E., Lee H.W., Janjua H., Xiao R., Acton T.B., Montelione G.T.. Structures of apo- and ssDNA-bound YdbC from Lactococcus lactis uncover the function of protein domain family DUF2128 and expand the single-stranded DNA-binding domain proteome. Nucleic Acids Res. 2013; 41:2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brandsen J., Werten S., van der Vliet P.C., Meisterernst M., Kroon J., Gros P.. C-terminal domain of transcription cofactor PC4 reveals dimeric ssDNA binding site. Nat. Struct. Biol. 1997; 4:900–903. [DOI] [PubMed] [Google Scholar]
- 44. Werten S., Moras D.. A global transcription cofactor bound to juxtaposed strands of unwound DNA. Nat. Struct. Mol. Biol. 2006; 13:181–182. [DOI] [PubMed] [Google Scholar]
- 45. Granneman S., Bernstein K.A., Bleichert F., Baserga S.J.. Comprehensive mutational analysis of yeast DEXD/H box RNA helicases required for small ribosomal subunit synthesis. Mol. Cell Biol. 2006; 26:1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Linder P., Jankowsky E.. From unwinding to clamping - the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011; 12:505–516. [DOI] [PubMed] [Google Scholar]
- 47. Sulima S.O., Hofman I.J.F., De Keersmaecker K., Dinman J.D.. How Ribosomes Translate Cancer. Cancer Discov. 2017; 7:1069–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narla A., Ebert B.L.. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010; 115:3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Farley K.I., Baserga S.J.. Probing the mechanisms underlying human diseases in making ribosomes. Biochem. Soc. Trans. 2016; 44:1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arenas J.E., Abelson J.N.. Prp43: An RNA helicase-like factor involved in spliceosome disassembly. PNAS. 1997; 94:11798–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tauchert M.J., Fourmann J.B., Christian H., Luhrmann R., Ficner R.. Structural and functional analysis of the RNA helicase Prp43 from the thermophilic eukaryote Chaetomium thermophilum. Acta Crystallogr. F, Struct. Biol. Commun. 2016; 72:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robert-Paganin J., Halladjian M., Blaud M., Lebaron S., Delbos L., Chardon F., Capeyrou R., Humbert O., Henry Y., Henras A.K. et al.. Functional link between DEAH/RHA helicase Prp43 activation and ATP base binding. Nucleic Acids Res. 2017; 45:1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayward S., Berendsen H.J.. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998; 30:144–154. [PubMed] [Google Scholar]
- 54. Tanaka N., Schwer B.. Characterization of the NTPase, RNA-binding, and RNA helicase activities of the DEAH-box splicing factor Prp22. Biochemistry. 2005; 44:9795–9803. [DOI] [PubMed] [Google Scholar]
- 55. Sloan K.E., Bohnsack M.T.. Unravelling the Mechanisms of RNA Helicase Regulation. Trends Biochem. Sci. 2018; 43:237–250. [DOI] [PubMed] [Google Scholar]
- 56. de la Cruz J., Kressler D., Linder P.. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 1999; 24:192–198. [DOI] [PubMed] [Google Scholar]
- 57. Rodriguez-Galan O., Garcia-Gomez J.J., de la Cruz J.. Yeast and human RNA helicases involved in ribosome biogenesis: current status and perspectives. Biochim. Biophys. Acta. 2013; 1829:775–790. [DOI] [PubMed] [Google Scholar]
- 58. Barandun J., Chaker-Margot M., Hunziker M., Molloy K.R., Chait B.T., Klinge S.. The complete structure of the small-subunit processome. Nat. Struct. Mol. Biol. 2017; 24:944–953. [DOI] [PubMed] [Google Scholar]
- 59. Wells G.R., Weichmann F., Colvin D., Sloan K.E., Kudla G., Tollervey D., Watkins N.J., Schneider C.. The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 2016; 44:5399–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sturm M., Cheng J., Bassler J., Beckmann R., Hurt E.. Interdependent action of KH domain proteins Krr1 and Dim2 drive the 40S platform assembly. Nat. Commun. 2017; 8:2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ameismeier M., Cheng J., Berninghausen O., Beckmann R.. Visualizing late states of human 40S ribosomal subunit maturation. Nature. 2018; 558:249–253. [DOI] [PubMed] [Google Scholar]
- 62. Jo S., Vargyas M., Vasko-Szedlar J., Roux B.Æ, Im W.. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008; 36:W270–W275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N.. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016; 44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pugh G.E., Nicol S.M., Fuller-Pace F.V.. Interaction of the Escherichia coli DEAD box protein DbpA with 23 S ribosomal RNA. J. Mol. Biol. 1999; 292:771–778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates of the final model and the reflection file used for refinement have been deposited to Protein Data Bank under accession code 6H57. The data that support the findings of this study are available upon request to the corresponding authors.