


Abstract
Bacterial toxin–antitoxin pairs play important roles in bacterial multidrug tolerance. Gcn5-related N-acetyltransferase (GNAT) toxins inhibit translation by acetylation of aminoacyl-tRNAs and are counteracted by direct contacts with cognate ribbon–helix–helix (RHH) antitoxins. Our previous analysis showed that the GNAT toxin KacT and RHH antitoxin KacA of Klebsiella pneumoniae form a heterohexamer in solution and that the complex interacts with the cognate promoter DNA, resulting in negative autoregulation of kacAT transcription. Here, we present the crystal structure of DNA-bound KacAT complex at 2.2 Å resolution. The crystal structure revealed the formation of a unique heterohexamer, KacT–KacA2–KacA2–KacT. The direct interaction of KacA and KacT involves a unique W-shaped structure with the two KacT molecules at opposite ends. Inhibition of KacT is achieved by the binding of four KacA proteins that preclude the formation of an active KacT dimer. The kacAT operon is auto-regulated and we present an experimentally supported molecular model proposing that the KacT:KacA ratio controls kacAT transcription by conditional cooperativity. These results yield a profound understanding of how transcription GNAT–RHH pairs are regulated.
INTRODUCTION
Prokaryotic toxin–antitoxin (TA) modules were initially discovered due to their ability to stabilize plasmids by post-segregational killing (1,2). However, TA modules are also present in the chromosomes of many free-living prokaryotes, often in multiple numbers (3,4). Chromosome-encoded TA modules have several biological functions, including bacteriophage defence by abortive infection, multidrug tolerance (persistence) and gene stabilization (5). Based on the nature of the antitoxin and the mechanism by which it counteracts the toxin, TA modules have been divided in six types (type I–VI). Usually, type II TAs encode a metabolically unstable antitoxin and a stable toxin that form a tight, non-toxic complex. Moreover, the metabolic instability of the antitoxin is essential to toxin activation. Based on toxin sequence similarities, Type II TA modules have been divided into gene families, many of which are abundant in both Bacteria and Archaea (6). Most type II toxins are RNases that inhibit bacterial cell growth by degrading essential RNAs. For example, RelE family of toxins cleave mRNAs positioned at the ribosomal A-site (7,8).
Recently, it was shown that type II toxins containing the Gcn5-related N-acetyltransferase (GNAT) fold inhibit translation by inactivation of tRNA. GNAT toxins transfer an acetyl group from acetyl coenzyme A (AcCoA) to the amine group of charged tRNAs, thereby preventing the tRNA from engaging productively in the peptidyltransferase reaction at the ribosome (9). Six examples of GNAT toxins have been analysed, namely, TacT, TacT2 and TacT3 of Salmonella enterica Typhimurium (10,11), AtaT of enterohemorrhagic Escherichia coli O157:H7 (12), GmvT of Shigella flexneri pINV plasmids (13) and KacT of Klebsiella pneumoniae HS11286 (14) (Supplementary Figure S1). The crystal structures of GNAT toxin proteins, including TacT, KacT and AtaT, have been recently reported. Our structure analysis suggested that two GNAT toxin KacT monomers form a dimer and that each monomer binds one AcCoA molecule (14).
The GNAT toxins are inactivated by direct interaction with cognate antitoxins that contain ribbon–helix–helix (RHH) DNA-binding domains. Putative GNAT–RHH TA pairs are frequent within bacterial chromosomes. For example, 123 Salmonella enterica strains encode 661 GNAT–RHH loci, 365 Escherichia coli strains 261 loci, 129 Klebsiella pneumoniae strains 204 loci and finally 56 Mycobacterium tuberculosis strains 58 loci (14).
Type II toxins are inactivated by cognate antitoxins through direct protein–protein contact that involves a number of different interaction mechanisms (15). In many cases, the C-terminal region of the antitoxin wraps around the toxin like a pincer and masks the active site or the target-binding site (5). For example, RelB antitoxin interacts with the active site of RelE to block RelE’s enzymatic activity (16), whereas CcdA antitoxin binds to the surface of CcdB that interacts with DNA gyrase (17). In these cases, the C-terminal domain of free antitoxin is intrinsically disordered and becomes partially folded upon binding to cognate toxin (15). Usually, the N-terminal domain of type II antitoxins contains a DNA-binding motif that also dimerizes the proteins. In GNAT TA pairs, the antitoxins contain RHH or helix-turn-helix motifs in their N-termini (3). In K. pneumoniae, the RHH antitoxin KacA and the GNAT toxin KacT also form a heterohexamer, KacA4KacT2 (14). Escherichia coli AtaRT protein complex is arranged as a heterohexameric AtaT-AtaR4-AtaT architecture (18).
Bacterial type II TA operons are autoregulated by the antitoxin that binds, often cooperatively, to operators in the promoter region. Often, but not always, the TA complex binds stronger and with higher cooperativity to the operators, i.e. the toxin acts as a corepressor of transcription (15). Interestingly, some of the type II TA operons are found to be regulated by a principle called ‘conditional cooperativity’ (19). If [Toxin] < [Antitoxin], then toxin increases cooperative binding of antitoxin to operator sequences and thereby helps in repressing transcription. However, if [Toxin] > [Antitoxin], then the opposite occurs: the toxin destabilizes the operator•TA complex. The underlying molecular mechanisms of conditional cooperativity have been elucidated in a few cases only, such as in phd/doc of P1 and relBE of E. coli (19,20). The overall effect of transcriptional regulation by conditional cooperativity is an increased TA operon transcription under conditions where the free toxin level is high (21).
The TacAT complex of S. enterica binds to the promoter region of the TacAT operon and autoregulates transcription; notably, TacT-mediated acetylation of TacA alters the promoter binding pattern of TacAT but seemingly does not influence the basal tacAT transcription level (22). Recently, the crystal structure of the AtaRT–operator DNA complex suggested that the heterohexameric arrangement is also crucial for transcription autoregulation (23). The N-terminal RHH domain of KacA of K. pneumoniae recognizes a palindromic sequence in the promoter region (14). KacA and KacT copurify as KacA4KacT2 heterohexameric complex that binds operator DNA with high affinity (14). However, the molecular mechanisms underlying the transcriptional autoregulation of the kacAT operon is not known.
Here, we report the crystal structure of K. pneumoniae acetyltransferase-type TA module KacAT bound to a 27 base pair operator DNA fragment at a resolution of 2.2 Å. Four KacA antitoxin and two KacT toxin molecules form a unique heterohexamer, denoted KacT-KacA2-KacA2-KacT to reflect its spatial organization. This remarkable structure provides a framework for understanding the mechanism of toxin inactivation in the family of acetyltransferase-type TA systems.
MATERIALS AND METHODS
Expression and purification of a selenomethionine-substituted KacAT complex
To express KacT and KacA as a complex, a 588 base pair kacT gene PCR product was obtained by using the primers 5′-CAGGATCCGTCTTCTGGTGAGAACCTGTACTTCCAATCCAATATTGGAATGGAGCAGCAACTGACGATTGAG-3′ (BamH I site in italics) and 5′-CGAAGCTTTTATGACTCATCGTCAGTAAAGAGC-3′ (Hind III site in italics). A 279 base pair kacA gene PCR product was obtained by using the primers 5′-CGCATATGATGCCCGCACTTAAAAAGCAGC-3′ (Nde I site in italics) and 5′-GCGGTACCTTACCTGGTTTGTAGACGCTTCGC-3′ (Xho I site in italics) and then cloned into the BamH I/Hind III site and Nde I/Xho I site of pACYCDuet to yield the pACYC::kacAT plasmid. To produce the selenomethionine (Se-Met)-substituted KacAT complex, 0.5 L of M9 minimal broth with 25 μg/ml chloramphenicol was inoculated overnight with 5 ml of BL21(DE3) cells containing the KacAT coexpression construct. Cells were grown at 37°C with shaking. When an OD600 of 0.5 was reached, 25 mg of leucine, 25 mg of isoleucine, 25 mg of valine, 50 mg of phenylalanine, 50 mg of lysine and 50 mg of threonine were added to the culture. After 30 min, 30 mg of selenomethionine was added, and protein expression was then induced overnight at 16°C by the addition of 0.2 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). The cells were harvested and lysed by sonication in 25 ml of lysis buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 0.5 mM phenylmethylsulfonyl fluoride (PMSF)). After centrifugation, the cleared lysate was applied to a column with 2 ml of NiNTA resin (Qiagen), washed with 50 ml of wash buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 25 mM imidazole) and finally eluted with 10 ml of elution buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 500 mM imidazole). TEV protease was added, and the sample was dialyzed overnight in TEV buffer (25 mM Tris, pH 8.0, 500 mM NaCl). The sample was loaded onto a Ni-NTA resin column again, and the flow through was collected. The sample was concentrated and subjected to size exclusion column chromatography using the Superdex 200 Increase 10/300 column (GE Healthcare) equilibrated with SEC buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl). The fractions from gel filtration were analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and fractions containing pure KacAT protein complex were pooled and concentrated to ∼25 mg/ml and stored in aliquots at –80°C for further experiments.
Crystallization, data collection and structure determination
To prepare the double-stranded DNA duplex used for crystallization, the Fw 5′-AAATGTACGGTTATTAACCGTACATGA-3′ and Rv 5′-TCATGTACGGTTAATAACCGTACATTT-3′ oligos were resuspended in SEC buffer, mixed with 500 μM duplex, heated at 96°C for 5 min and slowly cooled to 25°C. The DNA duplex and protein were mixed in a 1.5:1 molar ratio with 10 mg/ml protein. The initial crystallization screening was carried out using the sitting drop, vapour-diffusion technique in 96-well microplates. Trays were set using the Crystal Gryphon LCP platform (Art Robbins Instruments, CA) and commercial crystallization kits (Hampton Research, Valencia, CA). The drops were set up by mixing equal volumes (0.3 μl) of the protein and the precipitant solutions equilibrated against 50 μl of the precipitant solution. Subsequent optimizations were manually performed in a 24-well plate using the sitting drop vapour diffusion method. Crystals of selenomethionine-derivatized KacAT bound to DNA were grown by the sitting drop vapour diffusion method in 0.05 M ammonium sulfate, 0.05 M BIS-TRIS, pH 6.5, and 30% pentaerythritol ethoxylate (15/4 EO/OH) for ∼2 days at 18°C. Ammonium sulfate was used as a cryoprotectant and added in two increments to a final concentration of 2 M. The crystals were flash-frozen in liquid nitrogen prior to data collection.
The complete selenium single-wavelength anomalous dispersion dataset was collected at the BL19U beamline of the Shanghai Synchrotron Radiation Facility. All diffraction data processing was carried out using the HKL3000 program (24). A total of 12 heavy atom sites were identified and used to determine the initial phases with the software Autosol in the PHENIX suite (25); model building was then automatically started in Autobuild in the PHENIX suite. The model was manually built in Coot (26), and iterative refinement was performed with refmac5 (27). All crystallographic figures were drawn in PyMOL (Schroedinger, LLC, New York).
Escherichia coli growth assay
To examine the effect of KacT and different KacA mutants on the growth of E. coli, variants of the pACYC::kacAT plasmid were constructed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Overnight cultures of strain BL21(DE3) carrying different plasmids were diluted 1:100 into fresh LB broth with chloramphenicol. After 90 min of growth, IPTG was added to induce toxin transcription. Growth was monitored by determining OD600 values at 30-min intervals.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed on the MicroCal iTC200 system (GE Healthcare). Titrations were carried out at 15°C, and in a typical experiment, 10 μM DNA, protein, or the mutants were loaded into the sample cell and titrated against 100 μM KacAT in the injection syringe. Data were analysed using the Origin-based software provided by the manufacturer.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) is carried out by mixing 1 pmol PkacAT promoter DNA probe with a 6′-FAM modification at the 5′-end with 4 pmol purified wild-type and mutated KacA protein and KacTY145F proteins at a range of concentrations in EMSA buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5% glycerol, 0.8 μg/μl Salmon Sperm DNA) at room temperature for 20 min. The mixtures were then subjected to electrophoresis in native 8% polyacrylamide gel at 100 mA for 70 min. The images of gels were obtained by using Bio-Rad Molecular Imager Gel Doc XR+ System.
Size exclusion chromatography with multi-angle static light scattering (SEC-MALS)
SEC was using a Superdex 200 increase 10/30 column (GE Healthcare) at room temperature equilibrated in buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl. A 0.1-ml aliquot of protein (1 mg/ml) was loaded onto the column and eluted at a flow rate of 0.5 ml/min. The molar mass of KacA, KacTY145F, KacA-KacTY145F complex, and the mixture of KacTY145F and KacA with the ratio of KacTY145F:KacA = 1:2 were determined by analytical SEC performed in-line with a MALS DAWN HELEOS II instrument (Wyatt Technologies, Inc.) coupled to refractive index instrument (Optilab T-rEX, Wyatt Technologies, Inc.). The molar mass of chromatographed protein was calculated from the observed light scattering intensity and differential refractive index using ASTRA software (Wyatt Technologies, Inc.) based on a Zimm plot analysis using a refractive index increment, dn/dc = 0.185 ml/g (28).
Promoter analysis by lacZ fusions
To generate the reporter plasmid, a 114-bp kacATpromoter DNA was inserted upstream of the lacZgene of promoterless plasmid pLACZ, resulting in the formation of the fusion plasmid pLACZ-PkacAT. E. coli BL21(DE3) was transformed first with pLACZ-PkacAT, and then the plasmid pCDFDuetkacTkacA in which the coding region of kacT and kacA was independently fused to a T7 promoter, followed by the introduction of pBAD33kacA or pBAD33kacT with respective kacA or kacT under the Para promoter (Supplementary Figure S7). The transformants were grown in LB supplemented with 0.5 mM IPTG and 0.2% arabinose for 5 h. β-Galactosidase activity was then determined according to the standard Miller method, using SDS and chloroform to permeabilize the cells (29).
Data access
The atomic coordinates and structure factors for the reported crystal structure of DNA-bound KacAT have been deposited in the Protein Data Bank (PDB) under accession number 5ZGN.
RESULTS
Overall structure of the KacAT–DNA complex
The KacA and KacT proteins were coexpressed in E. coli and copurified as a stable complex (14). Our previous analysis showed that the KacA4KacT2 complex binds to its operator by recognizing the palindromic sequence that spans the transcription start site of kacAT (14). We performed crystallization trials for KacA4KacT2 in the presence or absence of various DNAs, which were all adapted from the palindromic sequence. The apo-KacAT sample did not crystalize, but we successfully crystallized and solved the structure of the KacA4KacT2 complex bound to a 27 base pair DNA duplex, referred to as KacAT–DNA hereafter.
Crystals of the KacAT–DNA complex were grown under the conditions composed of 0.05 M ammonium sulfate, 0.05 M BIS-TRIS (pH 6.5) and 30% pentaerythritol ethoxylate (15/4 EO/OH). The structure was solved by the single anomalous dispersion (SAD) method using a selenomethionine derivative of the KacAT protein and refined to 2.24 Å resolution with Rwork and Rfree values of 0.214 and 0.255, respectively (Supplementary Table S1). The crystal belongs to the P1 space group; each asymmetric unit contains two toxin molecules (KacT1 and KacT2), four antitoxin molecules (KacA1, KacA1′, KacA2 and KacA2′) and one 27 base pair DNA duplex. In the structure, two KacT and four KacA molecules pack together and form a stable W-shaped KacA4KacT2 complex, which has a dyad axis perpendicular to the DNA double helix (Figure 1). The KacA4KacT2 complex forms a unique KacT–KacA2–KacA2–KacT arrangement; the overall architecture of the KacAT complex is consistent with the oligomerization state observed in analytical size exclusion chromatography (14), consistent with the proposal that the structure represents the fundamental biological entity of the KacAT protein.
Figure 1.
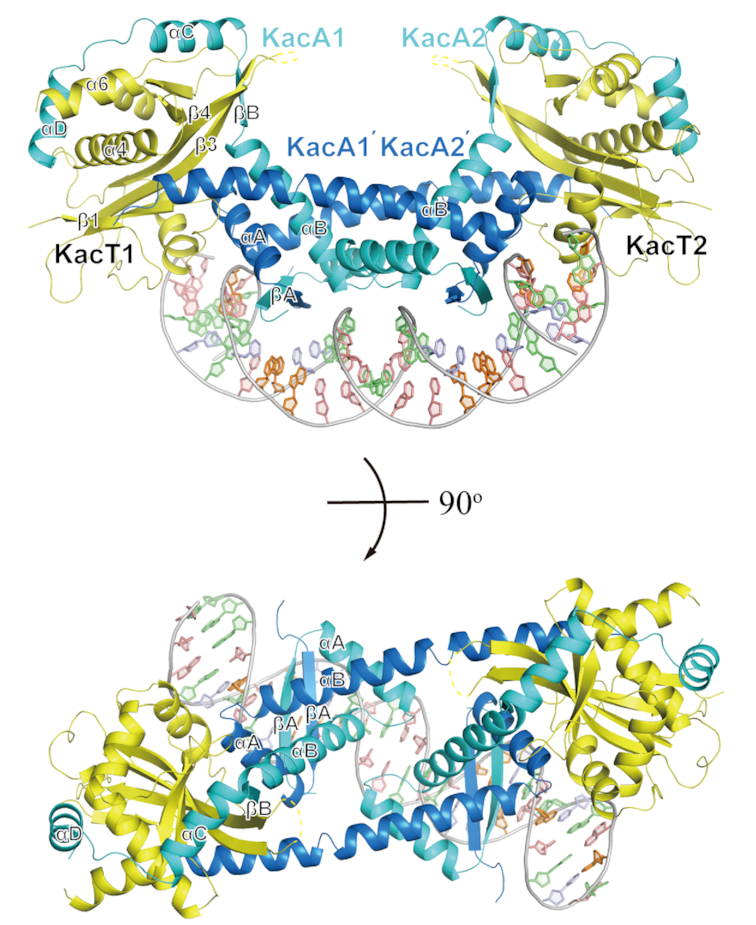
Overall structure of the K. pneumoniae W-shaped heterohexameric complex KacT-KacA2-KacA2-KacT bound to operator DNA shown in two orthogonal views with KacT in yellow, one KacA in blue and the other KacA in cyan. Secondary structure elements are indicated. The disordered part of the KacA-bound KacT is shown by a yellow dashed line.
Organization of KacA molecules in the KacA4KacT2 complex
In the KacAT–DNA complex structure, four KacA molecules form two homodimers, KacA1–KacA1′ and KacA2–KacA2′ (Figure 2A). The RHH domain in the N-terminus of KacA (88 aa) consists of one β–strand (βA, aa 5–13) and two helices (αA and αB composed of aa 14–28 and 31–51, respectively). As revealed by structural superposition (Figure 2B), the overall folds of the RHH domains are very similar; the rmsd values between the four RHH domains are all within the range of 0.1–0.3 Å. Interestingly, however, the C-terminal regions of the KacA molecules adopt two completely different conformations. In KacA1 and KacA2, the C-terminal regions form one flexible hook-like conformation, composed of one β–strand (βB, aa 52–56) and two helices (αC and αD); αC and αD consist of aa 57–70 and aa 75–86 and are connected by a short linker (71PPAP74). Unlike KacA1 and KacA2, KacA1′ and KacA2′ form one long extended αB helix, including both aa 31–51 of the RHH domain and aa 52–68 of the C-terminal region. In addition, the 17 C-terminal residues (aa 72–88) of KacA1′ and KacA2′ are not observed in the structure.
Figure 2.
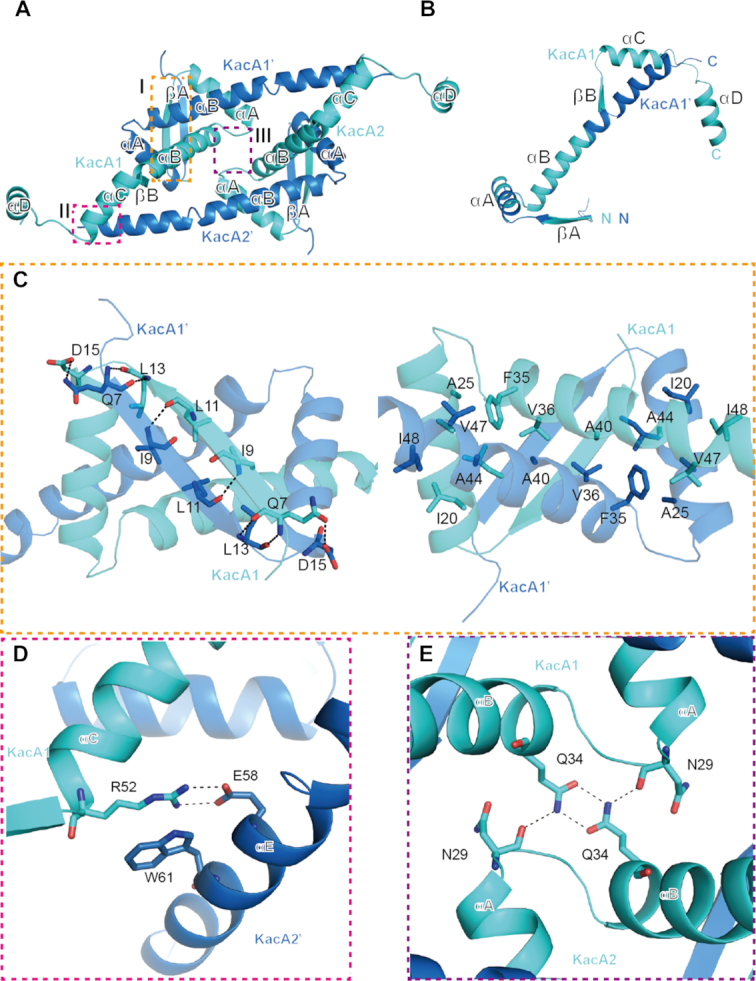
Interactions within and between two heterodimers of the RHH antitoxin KacA. (A) Overall structure of two KacA heterodimers viewed from the top. The interactions within and between the two KacA heterodimers identified by the dashed line boxes (I, II and III) are detailed in panels (B–E). Residues participating in the interface are drawn in stick representation and labelled in all figures. Hydrogen bonds are indicated by black dashed lines in all figures. (B) Superposition of two antitoxins in alternate conformations. (C) Details of the interactions at site I of the RHH domain shown in two views with 180 rotation along the horizontal axis. (D) Details of the interactions at site II, showing the interactions between KacA1 and KacA2′. (E) Details of the interactions at site III, showing the interactions between KacA1 and KacA2.
Dimerization of KacA1–KacA1′ or KacA2–KacA2′ is mediated by the interactions among the N-terminal RHH domains. As shown in KacA1–KacA1′, the βA strands of the two KacA molecules form one antiparallel β–sheet (Figure 2C, left panel) stabilized by the backbone hydrogen bonds between the paired residues (N7-L13 and I9-L11). Helices αA and αB of the RHH domains are also involved in the dimerization of KacA (Figure 2C, right panel). Residues I20, I21, A24, A25, A26 and I27 of αA from KacA1 form hydrophobic contacts with residues A44, A45, V47 and I48 of αB from KacA1′. In addition to the αA helices, the αB helices of KacA1 and KacA1′ also form hydrophobic interactions with each other via the side chains of F35, V36, V37, A38 and A40 (Figure 2C, right panel).
The two KacA homodimers are further assembled into a tetramer conformation in the structure. As depicted in Figure 2D, the side chains of R52 of KacA1 and E58 of KacA2′ form a salt bridge; the distance is 3.0 Å between the R52 NH2 atom and the E58 OE2 atom and is even shorter (2.7 Å) between the NH1 and OE1 atoms. R52 of KacA1 also forms a stable hydrophobic stacking interaction with the side chain of W61 of KacA2′; the average distance between the two side chains is ∼3.5 Å. Around the pseudo-dyad axis, KacA1 and KacA2 form a H-bond network (Figure 2E), via N29 and Q34. The two Q34 residues form two H-bonds with each other; the distances between the OE1 and the interacting NE2 atoms are all ∼2.8 Å. The conformations of the Q34 side chains are further stabilized by their H-bond interactions with the main chain O atoms of N29.
KacT and KacA interactions
The KacAT heterohexamer adopts a KacT–KacA2–KacA2–KacT arrangement; as depicted in Figure 3A, each KacT interacts with three KacA molecules in the structure: KacA1, KacA1′ and KacA2′ for KacT1, and KacA2, KacA2′ and KacA1′ for KacT2. The interaction modes of KacT1 and KacT2 are very similar; for simplicity, only the detailed interactions of KacT1 are described below.
Figure 3.

Details of KacA–KacT interactions. (A) Overview of the KacT–KacA interactions within the KacA4T2 heterohexamer. The interactions between the KacT and KacA identified by dashed line boxes (I, II, III and IV) are detailed in panels (B–F). The residues forming the interfaces are drawn in stick representation and labelled in all figures. Colouring as in Figure 1. (B) Details of the interactions at binding site I, showing the interactions between KacT and KacA1 around the βB strand. (C) Details of the interactions at binding site II, showing the interactions between KacT and KacA1 around the αC helix. (D) Details of the interactions at binding site III, showing the AcCoA-binding site identified in the KacT Y145F structure. (E and F) Details of the interactions at binding site IV, showing the KacT dimer interface site identified in the KacT Y145F structure. Panel (E) is the close-up view of the interactions between KacT and KacA2′. Panel (F) is the close-up view of the interactions between KacT and KacA1′. Hydrogen bonds are indicated by black dashed lines.
The C-terminal region (βB-αC-αD) of KacA1 attaches to the surface and forms extensive interactions with KacT1 (Figure 3B–D). The βB strand of KacA1 forms a parallel β-sheet with the β3 strand of KacT1; in addition to the backbone H-bond interactions (R52-C69 and M54-C69), the β-sheet conformation is also stabilized by the H-bond interaction between the side chains of R53 of KacA1 and E71 of KacT (Figure 3B). The three N-terminal residues (N57, E58 and S60) of helix αC of KacA1 form several H-bonds with E156, V154 and R72 of KacT1, whereas the middle and C-terminal residues (including W61, V64, A67 and I68) of KacA1 αC mainly form hydrophobic interactions with F70, F133 and F162 of KacT1. The αC-αD connecting linker of KacA1 packs along α6 of KacT1; the backbone O atom of P72 of KacA1 forms one very stable H-bond (2.7 Å) with the side chain OG atom of S167 of KacT1 (Figure 3C). Like αC, helix αD of KacA1 also forms H-bond interactions at its N-terminus and hydrophobic interactions at its middle and C-terminus with KacT1. The H-bond interactions are mediated by N75 and R77 of KacA1 and E110 and R147 of KacT1. The hydrophobic interactions are mediated by the side chains of L78 and L85 of KacA1 and M111, H115, M117 and Y163 of KacT1 (Figure 3D).
Unlike KacA1, KacA1′ mainly uses the residues of its αA helix to interact with KacT1 (Figure 3E). The side chain of E23 of KacA1′ αA forms three H-bonds: one between its OE1 atom and the N atom of the KacT1 Q43 and the other two between its OE2 atom and the N atoms of I44 and L45 of KacT1; the H-bond distances are all ∼2.8 Å, suggesting that this H-bond network is very stable. Via the backbone O atom of I27 and the side chain ND2 atom of N29, KacA1′ forms two additional H-bond interactions, which are with the side chain NH2 atom of R96 and the backbone O atom of A27 of KacT1, respectively. The relative orientations of KacA1′ and KacT1 are further stabilized by the hydrophobic interactions that are mediated by E22, E23 and A26 of KacA1′ and H35, H40 and D41 of KacT1.
Via M65, which resides near the C-terminus of the long extended helix αB, KacA2′ forms strong hydrophobic stacking interactions with KacT1. As depicted in Figure 3F, the side chain of KacA2′ M65 points towards the 42GQIL45 region of KacT1, and the distances between the CE atom of KacA2′ M65 and the backbone C atoms of the four KacT1 residues are all within the range of 3.4–3.6 Å. In addition to hydrophobic interactions, KacA2′ and KacT1 also form two H-bond interactions: one (2.9 Å) is between the backbone O atom of KacT1 V119 and the side chain NE2 atom of KacA2′ Q70, and the other (2.8 Å) is between the backbone O atom of KacA2′ I68 and the OH group of KacT Y48.
KacA affects AcCoA binding and dimerization of KacT
As indicated by the superposition of KacT in the KacAT–DNA complex and the apo-KacT structure (Figure 4A), KacA binding does not cause a significant change in the overall fold of KacT. However, there are minor subtle conformational changes in the AcCoA-binding region (Figure 4B). KacT has very high affinity to AcCoA as KacT can form a natural complex with AcCoA when overexpressed and purified. The KacAT complex was overexpressed using the same E. coli system as we used to produce apo-KacT, but AcCoA was not bound in the protein–DNA complex. Instead, one SO42− ion is observed at the KacAT–DNA complex structure and mimicked the α-phosphate group of AcCoA. In the apo-KacT structure, the side chains of E110 and R147 forms a H-bond (3.0 Å) interaction; via its NH2 atom, R147 also forms a H-bond (2.9 Å) interaction with the O9A atom of AcCoA. Interestingly, in the KacAT–DNA complex structure, the side chain of R147 is disordered to some degree, indicated by its very high B-factor (∼90 Å2). Compared to the apo-KacT structure, the side chain of E110 is rotated ∼180° around the CA-CB bond and forms one H-bond (3.1 Å) with the side chain of R77 of KacA in the KacAT–DNA complex structure. As revealed by structural superposition, formation of the E110-R77 bond conflicts with the ribose group and the 3′-phosphate group of the AMP motif of AcCoA. Taken together, these observations clearly suggest that KacA can impair the binding between AcCoA and KacT, thereby inhibiting the tRNA acetylation activity of KacT.
Figure 4.

Comparison of the KacTY145F structure and the KacAT–DNA structure. The structure of the apo-KacTY145F molecule (PDB ID: 5XUN) is coloured grey, whereas the structure of the KacT molecule in the KacAT–DNA structure (PDB ID: 5ZGN) is coloured yellow. The KacA molecule is coloured cyan, and the KacA′ molecule is coloured blue. (A) Superposition of KacA-bound KacT and isolated KacTY145F yielding an rmsd of 0.732 Å. (B) Enlarged view of the comparison of the KacTY145F structure and the KacAT–DNA structure at the AcCoA binding site. (C) Enlarged view of the comparison of the KacTY145F structure and the KacAT–DNA structure at the KacT dimer interface. (D) Growth curves of E. coli BL21(DE3) cells expressing KacT paired with the wild-type or KacA mutants from pACYCDuet. Expression was induced by the addition of IPTG. Data show mean and standard deviation of n = 3 independent experiments.
As revealed by the apo-KacT structure (14), KacT forms a dimer. Residues G67, Y86 and G128 are directly involved in the KacT–KacT dimerization (Supplementary Figure S2). The point mutant toxins, KacTY86F and KacTG67WG128W, are not toxic to K. pneumoniae HS11286-RR2ΔkacAT cells (Supplementary Figure S2C). An approximately 2218 Å2 area is buried in the dimer interface, which consists of extensive hydrogen-bond and hydrophobic interactions. The surfaces of the dimerization regions are highly positively charged, which may play a role in the binding of substrate tRNA, which is highly negative in charge. However, as revealed by structural superposition (Figure 4C), the dimerization interface of KacT is occupied by the three interacting KacA molecules. As a consequence, the KacT subunits exist as two monomers, which are ∼54 Å apart from each other in the KacAT–DNA complex structure (Figure 1). The amino acids that are involved in the KacT dimer interface form stable interaction with KacA. The hydrophobic surface of KacT forms strong interaction with M65 of KacA, and the OH group of Y48 in KacT forms a H-bond with I68 of KacA.
Taken together, the above observations indicate that KacA can impair the AcCoA binding and dimerization of KacT, thus preventing tRNA acetylation by KacT. To support these structural interpretations, we constructed two KacA mutants. As depicted in Figure 4D, replacing R77 with alanine (for the mutant KacAR77A) abolished the protein’s ability to counteract the toxicity of KacT. M65, I68 and Q70 of KacA are directly involved in the interaction with the dimerization interface of KacT. Like KacAR77A, co-expression of the KacAM65AI68AQ70A mutant could not neutralize the toxicity of KacT.
DNA recognition by KacA
In the structure of the KacTA•DNA complex (Figure 1), the 27 base pair DNA substrate adopts a B-form-like conformation in which the DNA is strongly bent towards the ends. The DNA is compressed at the minor groove of the central AT-rich region, whereas it is expanded at the major groove of the KacA-interacting area. The operator DNA is a pseudo palindrome sequence and is recognized by the RHH domains of KacA. The β-sheet formed by the β1 strands of KacA1 and KacA1′ inserts into the major groove and forms several sequence-specific interactions with the DNA (Figure 5A). As depicted in Figure 5B, the side chain of R12 of KacA1 forms one H-bond (3.0 Å) with the nucleobase of G5. D10 forms one H-bond (3.2 Å) with the nucleobase of C8, and the C8-pairing G20 forms two H-bonds with the side chain of R8 of KacA1′, and the distances are all ∼2.8 Å. Similar to G20, the nucleobase of G9 also forms two H-bonds (2.7 and 2.9 Å) with the side chain of R8 of KacA1; interestingly, the relative orientation of the KacA1 R8 is more similar to that of the KacA1′ R8 than to that of the KacA1 R12. The side chain of D10 of KacA1′ forms one weak H-bond (3.2 Å) with the nucleobase of the G10-pairing C18.
Figure 5.

Interaction between KacA and DNA. (A) Schematic overview of the interactions between KacA and DNA. Dashed lines indicate hydrogen bonds or electrostatic interactions with phosphate-sugar backbone and bases. The residues in KacA are shown in colours matching those in Figure 1. (B) Details of the sequence-specific interactions of the antitoxin KacA heterodimer with bases in the operator DNA. (C) Measurement of the binding affinity between the palindromic sequence and the KacAT complex containing KacA mutants using ITC. Individual peaks from titrations were integrated and presented in a Wiseman plot.
Isothermal titration calorimetry (ITC) analysis (Figure 5C), which measures the binding affinity between the palindromic sequence and the KacAT complex containing KacA mutants, showed that the R8 or R12 mutations both abolished DNA binding ability of the complex. In addition, the electrophoretic mobility shift assay (EMSA) of the KacA antitoxins using the PkacAT promoter as target DNA also revealed that the wild-type KacA is able to bind to the operator DNA (Supplementary Figure S3). However, two antitoxins variants, KacAR8A and KacAR12A, lose DNA binding ability while the variant, KacAD10A, binds much weaker to the operator than wild-type KacA. In agreement with the structural position, KacAR8A or KacAR12A does not alter its oligomeric state in the solution when compared with that of the wild-type KacA, a dimer with the molecular weight of ∼20 kDa (Supplementary Figure S5). Deficiency in operator binding of KacA mutant does not essentially affect its interaction with KacT as the N-terminus 10 residues truncation of KacA can neutralize the KacT toxic effect in coupled expression (14). Together, these results suggested that the R8 and R12 residues of KacA are critical for operator DNA recognition.
Interaction between KacA, KacT and the operator DNA in vitro
We analysed the effect of varying the TA ratio on binding of KacA antitoxin to operator DNA using EMSA (Figure 6A). A constant, low concentration of KacA was used to monitor the effect of increasing the concentration of KacT on KacA binding. KacA alone binds to operator DNA (Figure 6A, band IV of lane 3), whereas KacT alone does not (lane 2). Increasing the TA ratio changed the gel-shift signal to the shifted DNA operator bands (band I–IV, lanes 4–6, 8, Figure 6A), indicating that KacA and KacT form the distinct complexes when bound to operator DNA. The strongest shifted band (band II, Figure 6A) in lane 8 with the molar ratio of [KacTY145F]:[KacA] = 1:2 corresponds to that for purified KacAT heterohexamer, KacT2KacA4. Interestingly, a further increase of the TA ratio prevented DNA binding (lanes 9–11, Figure 6A) as evidenced by the release of DNA substrates. Thus, we propose that an excess of KacT destabilizes the KacA•KacT•operator complex in vitro. By inference, this result indicates that kacAT transcription is regulated by conditional cooperativity in vivo, involving transcriptional repression at low KacT:KacA ratios and derepression at high KacT:KacA ratios.
Figure 6.

Interplay between KacA, KacT and operator DNA. (A) EMSA test of PkacAT promoter DNA with fixed KacA and increasing concentration of KacTY145F. The four shifted DNA bands are marked with band I, II, III and IV, respectively. (B) Analytical gel filtration chromatography for the mixture of KacTY145F and KacA ([T]:[A] = 1/2) by using a Superdex Increase 200 10/300 size exclusion column. (C) SDS-PAGE of the component elutes from peak α, β and γ in the retention window indicated in panel (B). (D) SEC-MALS chromatogram of KacA, KacTY145F and KacTA complex. The chromatogram shows the refractive index signal of KacA (gry), KacTY145F (red) and TA-complex (blue) with the derived molar masses indicated by the horizontal lines. (E) SEC-MALS chromatogram of the mixture of KacTY145F and KacA ([T]:[A] = 1/2). The chromatogram shows the refractive index signal with three peaks, corresponding to peak α, β and γ in panel (B). (F) EMSA test of PkacAT promoter DNA with the component elutes from peak α, β and γ. The mixture of KacT145F and KacA ([T]:[A] = 1/2) was used as a control.
To provide stoichiometric evidence for KacA and KacT on the shifted bands in lane 8, they were subjected to SEC and SEC-MALS (Figure 6). Peak α, β and γ were clearly detected for the KacTY145F and KacA mixture ([T]/[A] = 1/2) in SEC analysis (Figure 6B), and contain KacTY145F and KacA of various molar ratios (Figure 6C). Their molecular weights were calculated to be ∼225, ∼85 and ∼20 kDa by using SEC-MALS (Figure 6E), respectively. Notably, peak β with a molecular weight of ∼85 kDa (Figure 6E) is very close to that for KacA4T2 heterohexmer (∼80 kDa). In the EMSA with the PkacAT promoter as target DNA, the proteins of peak β can form a shifted band with the same position as that for KacAT heterohexamer (Figure 6F). Peak α with the molecular weight of ∼225 kDa might correspond to a KacA8–KacT8 complex (predicted to be 240 kDa). The average calculated MW for Peak γ is ∼20 kDa, and the subsequent SDS-PAGE analysis of its sub-eluates revealed decreasing content of KacT with increasing elution volume (Supplementary Figure S5). In addition, the KacTY145Fand KacA mixtures of [T]/[A] = 1/4 (Figure 6A, lane 6) and [T]/[A] = 1/1 (Figure 6A, lane 9) also give the three and two peaks, respectively, with variable relative height or area in analytical gel filtration chromatography (Supplementary Figure S5c and e). Thus, we conclude that the KacT and KacA could form the diverse complexes with different stoichiometries; however, the KacAT heterohexamer with the arrangement of KacA4–KacT2 is stable and exhibits the strongest binding to operator DNA.
To further test the effect of the interplay between KacA and KacT on the binding pattern of KacAT complex to the operator DNA, we performed EMSA binding assays using either wild-type or mutated KacA (Supplementary Figure S6). When the KacAR8A and KacAR12A mutant proteins were added to the EMSA mixture, no band shift was observed, further supporting the critical roles of R8 and R12 in operator binding. When the KacAΔ61-88 mutant was added to the EMSA mixture, the super-shift band that represents the operator binding with the KacTA complex disappeared, due to the deletion of the KacT-interacting region in KacA.
Transcription regulation by KacT–KacA in vivo
To assess the functional relevance of the KacAT complex binding to its own promoter and if excess KacT toxin would de-repress transcription, we transcriptionally fused the kacAT promoter (PkacAT) to lacZ. Plasmid pLACZ-PkacAT was co-transformed into E. coli BL21(DE3) with pCDFDuet::kacT::kacA and pBAD33::kacA or pBAD33::kacT, resulting in different [KacT]:[KacA] ratios in vivo [corresponding to >1, = ∼1 and <1, respectively (Supplementary Figure S7)]. According to the LacZ activity, PkacAT alone was active, whereas the KacAT complex repressed the PkacAT promoter (Figure 7). Notably, repression at [KacT]:[KacA] = ∼1 was stronger than that of [KacT]:[KacA] < 1 as well as that of [KacT]:[KacA] > 1. Indeed, PkacAT was de-repressed by excess levels of KacT toxin and was thus regulated by conditional cooperativity.
Figure 7.
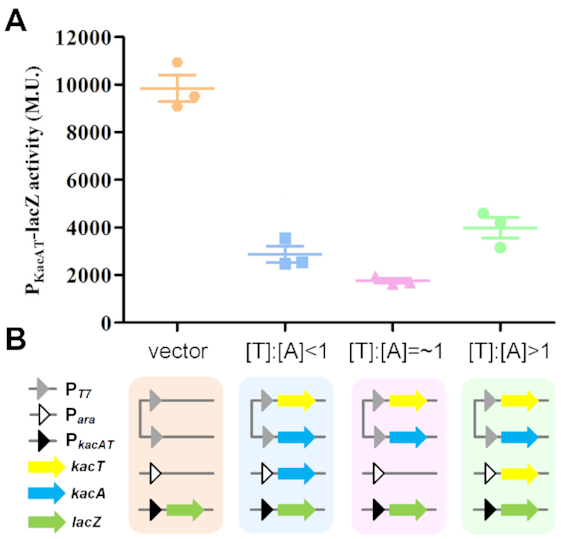
Transcription regulation analysis by lacZ fusions. (A) LacZ activities of strains harbouring PkacAT promoter-lacZ fusions in the presence of varied ratio of [KacT]:[KacA]. Enzymatic activities were determined 5 h after the addition of IPTG (0.5 mM) to induce kacA and arabinose (0.2%) to induce kacT. Data are presented as a scatterplot showing mean and standard error of three independent experiments. (B) Schematic representation of the expression of transcriptional reporter lacZ fusion subjected to regulation by the KacTA complex at varied ratios of [KacT:KacA]. Each coloured rectangle represents an E. coli BL21(DE3) cell harbouring three plasmids (more details available in Supplementary Figure S6), each of which is shown as a continuous line. Plasmid pLACZ-PkacAT (with the PkacAT promoter denoted by a black triangle) in BL21(DE3) together with the KacA and KacT expression plasmid pCDFDuet::kacT::kacA (both with PT7 promoters denoted by grey triangles) and pBAD33::kacA (with Para denoted by white triangle) or pBAD33::kacT (with Para), establishing the different [KacT]:[KacA] ratios in vivo.
DISCUSSION
In this study, we elucidated the direct interaction of the GNAT toxin with the RHH antitoxin via the crystal structure of the K. pneumoniae KacAT complex bound to its cognate operator DNA. Both the structural analysis presented here and our previous biochemical data (14) demonstrate that the KacAT complex forms a compact heterohexameric assembly that we denote as KacT–KacA2–KacA2–KacT to reflect its spatial organization. In this structure, the two KacA-bound KacT molecules are at opposite ends of the W-shaped conformation. This positioning represents the molecular basis for how the binding of the antitoxin KacA inhibits the acetyltransferase activity of KacT. In general, toxins of well-characterized TA complexes have been found to be inactivated by two different mechanisms: (i) the active site of the toxin is buried when the toxin is bound to its cognate antitoxin, such as in YefM-YoeB (30), RelBE (19), MazEF (31) and DinJ-YafQ (32) and (ii) the target binding site of the toxin is obstructed by its cognate antitoxin, such as CcdBA (17) and HigBA (33). However, the mechanism underlying the inhibition of the GNAT toxin KacT by the RHH antitoxin KacA utilizes a unique mechanism; KacA counteracts KacT by preventing both the dimerization of KacT and the binding site of AcCoA (Figure 4). Structural analysis of KacT (14), as well as other GNAT toxins, TacT (10) and AtaT (23), showed that the dimeric state of the toxin is essential for acetyltransferase activity. Comparison of the topology of KacT in the KacTY145F dimer and the KacAT–DNA (Figure 4A) complex reveals that there are only minor structural rearrangements in KacT; however, the four KacA molecules prevent the contact between the two KacT molecules necessary to form a dimer (Figure 4C).
In the W-shaped conformation of the KacAT heterohexamer, KacA blocks the dimerization of KacT by using two different folding states of the C-terminal region of KacA. The C-terminal region of KacA1 exhibits a βB-αC-αD fold and wraps around the KacT monomer, whereas the folding of a portion of the C-terminal region of KacA1′ into a complete α helix can only be attributed to the presence of Pro70 and interact with the hydrophobic interface of KacT.
RHH superfamily antitoxin proteins require dimerization to form a single DNA-binding domain (34). Our KacAT–DNA complex structure revealed that the four KacA molecules in the KacAT hexamer form two DNA-binding motifs and interact with the two arms of one palindrome sequence in the PkacAT promoter region. Most recently, a closely related homologue of the KacAT TA system, the AtaRT protein complex from E. coli O157:H7 EDL933 (AtaT is the GNAT toxin with 67% BLASTp identities to KacT while AtaA is the RHH antitoxin with 73% BLASTp identities to KacA) was also shown to form a heterohexameric protein complex binding a palindromic sequence at the ataAT operator with the AtaAT–DNA complex structure determined at 3.36 Å resolution (23), similar to our KacAT–DNA complex structure determined at 2.24 Å resolution (Supplementary Figure S8). Both the GNAT–RHH–DNA complex structures obtained by the independent observations in two different organisms support the unique DNA binding organization of RHH molecules in the GNAT–RHH hexamer. In addition, the structure of the KacAT complex of K. pneumoniae exhibits some resemblance to that of the structure of VapBC (FitAB) of Neisseria gonorrhoeae (PDB ID: 2H1O) (35). Antitoxin FitA possesses an N-terminal RHH DNA-binding domain, and the four FitA molecules in a FitAB octamer unit also form two DNA-binding motifs that interact with the palindrome sequence in the fitAB promoter region. Interestingly, despite the resemblance of the dimerization of the RHH domains in FitAB–DNA and our KacAT–DNA complex, the detailed organizations of the four RHH-containing antitoxin molecules are entirely different because the two FitA homodimers are widely separated in the FitAB–DNA complex structure.
Conditional cooperativity is the transcriptional autoregulation mode among some type II TA modules, for examples ccdAB, phd/doc and relBE. In the proposed model of a mechanism for conditional cooperativity regulation based on E. coli RelBE, RelBE forms a RelB4RelE2 complex that binds operator DNA (36). The structure of KacAT–DNA is consistent with this speculation and we, therefore, investigated the binding of KacA to the operator in the presence of varying amounts of KacT (Figure 6). Indeed both our in vitro EMSA test (Figure 6) and in vivo lacZ reporter system (Figure 7) support that the kacAT promoter is regulated by conditional cooperativity. At increasing [KacT]:[KacA] ratios between 0 and 1, a series of distinct shifted bands appear, similar to the observation of the Doc modulates the affinity of Phd for the promoter DNA (20). When the [KacT]:[KacA] ratio exceeds 1, the KacAT–DNA complexes apparently resolve and the band corresponding to unbound DNA re-appears. The subsequent analyses of the SEC-MALS suggested that the KacAT–DNA complex formed distinct compositions. The strongest band in lane 8 in Figure 6A might correspond to the stable DNA-bound KacAT heterohexamer (KacA4–KacT2–DNA), which was also observed by the co-expression of the kacT and kacA genes in our previous in vivo study (14). The weak band below and above the one corresponding to the DNA-bound heterohexamer might reflect the unstable diverse compositions. These in vitro EMSAs indicated that KacA and KacT form a series of complexes when bound to operator DNA in vivo. Further experimental work is needed to investigate the KacAT complex arrangements with the presence/absence of the promoter DNA inside a living organism.
We propose a molecular model that explains the transcriptional regulation in kacAT operon (Figure 8). The levels of toxin and antitoxin proteins vary according to the growth state of the bacterial cell. During unstressed (i.e. rapid) growth conditions, KacA is expressed in excess of KacT; thus, the KacA antitoxin molecules bind to the KacT toxin molecules, resulting in a stable heterohexamer KacA4KacT2 that prevents KacT from forming a dimer and binds strongly to the kacAT operator to repress transcription. Under stress conditions that yield slow growth, KacA is degraded, resulting in excess, monomeric KacT. The free KacT binds the unoccupied C-terminal tails of KacA inside the heterohexamer via a high-affinity interaction at the same place as the DNA, thus abolishing transcriptional repression, and eventually lead to the formation of the active KacT dimer. The molecular basis of the conditional cooperativity includes the high-affinity interaction and the low-affinity interaction between different toxin and antitoxin molecules, which is supported by the structural analysis of Phd-Doc (20). By examining the interaction between KacT and KacA in the KacAT–DNA structure, we found that the interaction between KacT and KacA may also involve two different modes: the KacA C-terminal region folding into βB-αC-αD and wrapping around KacT (Figure 3B–D) can be considered the high-affinity interaction, while the interaction between KacA′ and KacT can be considered the low-affinity interaction (Figure 3E and F).
Figure 8.
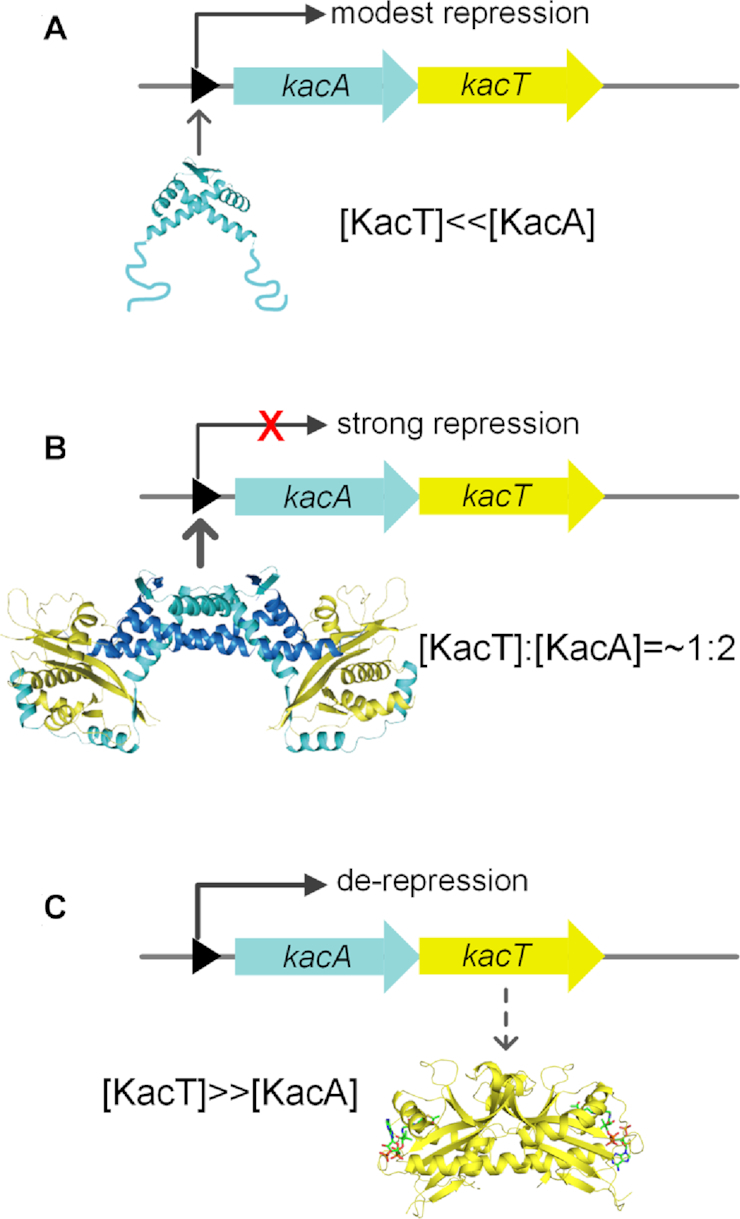
Model explain transcriptional regulation of the kacAT operon by conditional cooperativity. (A) At [A] > [T], antitoxin KacA binds weakly to kacAT operator DNA, resulting in modest transcriptional repression. (B) At an [A]:[T] ratio close to 1:2, KacT and KacA form a stable heterohexameric complex that binds strongly to operator DNA, resulting in efficient repression. (C) At [T] > [A], the excess of KacT destabilizes operator binding, resulting in de-repression of transcription, replenishment of KacA antitoxin. This complicated mode of transcriptional regulation by the T:A ratio secures replenishment of antitoxin during states of high toxin levels and thus, slow growth. Thus, this mechanism may reduce fortuitous activation of toxin and may also allow for the resuscitation of persister cells as well as promote restart of cell growth after the termination of stressful conditions (21).
In summary, we presented the crystal structure of the KacAT•operator complex, revealing direct interactions between GNAT toxin, RHH antitoxin and operator DNA. Inactivation of KacT by KacA occurs via a unique molecular mechanism that entails blocking of AcCoA binding and dimerization of KacT. The mechanism was illustrated by the unusual KacAT W-shaped conformation of KacT–KacA2–KacA2–KacT complex. The heterohexameric structure of KacAT also yields a mechanistic explanation underlying transcriptional regulation by conditional cooperativity. We propose that other GNAT–RHH TA loci may be regulated by a similar principle. Due to the important role of acetyltransferase-type toxins in persister cell formation, these results from the KacAT module might facilitate investigations of the transcription regulatory role of the recently characterized GNAT–RHH TA loci in antibiotic stress or other stresses in bacterial pathogens.
DATA AVAILABILITY
The atomic coordinates and structure factors for the reported crystal structure of DNA-bound KacAT have been deposited in the Protein Data Bank (PDB) under accession number 5ZGN.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to BL19U beamline staff at the Nation Center for Protein Science Shanghai (NCPSS) for their expert assistance during X-ray diffraction data collection. We are also very grateful to Jun Zhang, Jie Li and other staffs at Large-scale Protein Production System of NCPSS for their expert assistance to SEC-MALS.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The 973 program, Ministry of Science and Technology, China [2015CB554202]; National Key R&D Program of China [2018YFE0102400]; National Natural Science Foundation of China [31670074, 31870721, 31871250 and 21661140002]; Key Research and Development Project of China [2016YFA0500600]; Danish Natural Research Foundation [DNRF120]; Novo Nordisk Foundation. Funding for open access charge: the 973 program, Ministry of Science and Technology, China [2015CB554202].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jaffé A., Ogura T., Hiraga S.. Effects of the ccd function of the F plasmid on bacterial growth. J. Bacteriol. 1985; 163:841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerdes K., Rasmussen P.B., Molin S.. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:3116–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xie Y., Wei Y., Shen Y., Li X., Zhou H., Tai C., Deng Z., Ou H.Y.. TADB 2.0: an updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2017; 46:D749–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shao Y., Harrison E.M., Bi D., Tai C., He X., Ou H.-Y., Rajakumar K., Deng Z.. TADB: a web-based resource for Type 2 toxin–antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011; 39:D606–D611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harms A., Brodersen D.E., Mitarai N., Gerdes K.. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell. 2018; 70:1–17. [DOI] [PubMed] [Google Scholar]
- 6. Pandey D.P., Gerdes K.. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005; 33:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pedersen K., Zavialov A.V., Pavlov M.Y., Elf J., Gerdes K., Ehrenberg M.. The bacterial toxin RelE displays Codon-Specific cleavage of mRNAs in the ribosomal A site. Cell. 2003; 112:131–140. [DOI] [PubMed] [Google Scholar]
- 8. Neubauer C., Gao Y.-G., Andersen K.R., Dunham C.M., Kelley A.C., Hentschel J., Gerdes K., Ramakrishnan V., Brodersen D.E.. The structural basis for mRNA recognition and cleavage by the Ribosome-Dependent endonuclease RelE. Cell. 2009; 139:1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jurėnas D., Garcia-Pino A., Van Melderen L.. Novel toxins from type II toxin-antitoxin systems with acetyltransferase activity. Plasmid. 2017; 93:30–35. [DOI] [PubMed] [Google Scholar]
- 10. Cheverton A.M., Gollan B., Przydacz M., Wong C.T., Mylona A., Hare S.A., Helaine S.. A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell. 2016; 63:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rycroft J.A., Gollan B., Grabe G.J., Hall A., Cheverton A.M., Larrouy-Maumus G., Hare S.A., Helaine S.. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat. Commun. 2018; 9:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jurėnas D., Chatterjee S., Konijnenberg A., Sobott F., Droogmans L., Garcia-Pino A., Van Melderen L.. AtaT blocks translation initiation by N-acetylation of the initiator tRNAfMet. Nat. Chem. Biol. 2017; 13:640–646. [DOI] [PubMed] [Google Scholar]
- 13. McVicker G., Tang C.M.. Deletion of toxin–antitoxin systems in the evolution of Shigella sonnei as a host-adapted pathogen. Nat. Microbiol. 2016; 2:16204. [DOI] [PubMed] [Google Scholar]
- 14. Qian H., Yao Q., Tai C., Deng Z., Gan J., Ou H.-Y.. Identification and characterization of acetyltransferase-type toxin-antitoxin loci in Klebsiella pneumoniae. Mol. Microbiol. 2018; 108:336–349. [DOI] [PubMed] [Google Scholar]
- 15. Page R., Peti W.. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016; 12:208–214. [DOI] [PubMed] [Google Scholar]
- 16. Overgaard M., Borch J., Gerdes K.. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the Ribbon–Helix–Helix motif in RelB. J. Mol. Biol. 2009; 394:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Jonge N., Garcia-Pino A., Buts L., Haesaerts S., Charlier D., Zangger K., Wyns L., De Greve H., Loris R.. Rejuvenation of CcdB-Poisoned gyrase by an intrinsically disordered protein domain. Mol. Cell. 2009; 35:154–163. [DOI] [PubMed] [Google Scholar]
- 18. Yashiro Y., Yamashita S., Tomita K.. Crystal structure of the enterohemorrhagic Escherichia coli AtaT-AtaR Toxin-Antitoxin complex. Structure. 2019; 27:476–484. [DOI] [PubMed] [Google Scholar]
- 19. Overgaard M., Borch J., Jorgensen M.G., Gerdes K.. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008; 69:841–857. [DOI] [PubMed] [Google Scholar]
- 20. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R.D., Charlier D., van Nuland N.A.J., Loris R.. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010; 142:101–111. [DOI] [PubMed] [Google Scholar]
- 21. Cataudella I., Trusina A., Sneppen K., Gerdes K., Mitarai N.. Conditional cooperativity in toxin–antitoxin regulation prevents random toxin activation and promotes fast translational recovery. Nucleic Acids Res. 2012; 40:6424–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. VanDrisse C.M., Parks A.R., Escalante-Semerena J.C.. A toxin involved in Salmonella persistence regulates its activity by acetylating its cognate antitoxin, a modification reversed by CobB sirtuin deacetylase. MBio. 2017; 8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jurenas D., Van Melderen L., Garcia-Pino A.. Mechanism of regulation and neutralization of the AtaR-AtaT toxin-antitoxin system. Nat. Chem. Biol. 2019; 15:285–294. [DOI] [PubMed] [Google Scholar]
- 24. Cymborowski M., Otwinowski Z., Chruszcz M., Minor W.. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 25. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meyer M., Morgenstern B.. Characterization of gelatine and acid soluble collagen by size exclusion chromatography coupled with multi angle light scattering (SEC-MALS). Biomacromolecules. 2003; 4:1727–1732. [DOI] [PubMed] [Google Scholar]
- 29. Griffith K.L., Wolf R.E.. Measuring β-Galactosidase activity in Bacteria: Cell growth, permeabilization, and enzyme assays in 96-Well arrays. Biochem. Biophys. Res. Commun. 2002; 290:397–402. [DOI] [PubMed] [Google Scholar]
- 30. Kamada K., Hanaoka F.. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell. 2005; 19:497–509. [DOI] [PubMed] [Google Scholar]
- 31. Kamada K., Hanaoka F., Burley S.K.. Crystal structure of the MazE/MazF Complex: Molecular bases of Antidote-Toxin recognition. Mol. Cell. 2003; 11:875–884. [DOI] [PubMed] [Google Scholar]
- 32. Maehigashi T., Ruangprasert A., Miles S.J., Dunham C.M.. Molecular basis of ribosome recognition and mRNA hydrolysis by the E. coli YafQ toxin. Nucleic Acids Res. 2015; 43:8002–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schureck M.A., Maehigashi T., Miles S.J., Marquez J., Cho S.E., Erdman R., Dunham C.M.. Structure of the Proteus vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J. Biol. Chem. 2014; 289:1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan W.T., Espinosa M., Yeo C.C.. Keeping the wolves at Bay: antitoxins of prokaryotic type II Toxin-Antitoxin systems. Front. Mol. Biosci. 2016; 3:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mattison K., Wilbur J.S., So M., Brennan R.G.. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of Toxin-Antitoxin heterodimers containing pin domains and Ribbon-Helix-Helix motifs. J. Biol. Chem. 2006; 281:37942–37951. [DOI] [PubMed] [Google Scholar]
- 36. Boggild A., Sofos N., Andersen K.R., Feddersen A., Easter A.D., Passmore L.A., Brodersen D.E.. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure. 2012; 20:1641–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors for the reported crystal structure of DNA-bound KacAT have been deposited in the Protein Data Bank (PDB) under accession number 5ZGN.