


Abstract
The potent antiretroviral protein APOBEC3G (A3G) specifically targets and deaminates deoxycytidine nucleotides, generating deoxyuridine, in single stranded DNA (ssDNA) intermediates produced during HIV replication. A non-catalytic domain in A3G binds strongly to RNA, an interaction crucial for recruitment of A3G to the virion; yet, A3G displays no deamination activity for cytidines in viral RNA. Here, we report NMR and molecular dynamics (MD) simulation analysis for interactions between A3Gctd and multiple substrate or non-substrate DNA and RNA, in combination with deamination assays. NMR ssDNA-binding experiments revealed that the interaction with residues in helix1 and loop1 (T201-L220) distinguishes the binding mode of substrate ssDNA from non-substrate. Using 2′-deoxy-2′-fluorine substituted cytidines, we show that a 2′-endo sugar conformation of the target deoxycytidine is favored for substrate binding and deamination. Trajectories of the MD simulation indicate that a ribose 2′-hydroxyl group destabilizes the π-π stacking of the target cytosine and H257, resulting in dislocation of the target cytosine base from the catalytic position. Interestingly, APOBEC3A, which can deaminate ribocytidines, retains the ribocytidine in the catalytic position throughout the MD simulation. Our results indicate that A3Gctd catalytic selectivity against RNA is dictated by both the sugar conformation and 2′-hydroxyl group.
INTRODUCTION
Cytidine deaminases perform a variety of functions ranging from diversification of antibodies to defense against viral infection. Four members of the APOBEC3 (A3) family of cytidine deaminases (A3D, A3F, A3G and A3H) have varying degrees of effectiveness in restricting HIV-1 infection (1–6). Restrictive A3 proteins are encapsidated during viral replication by associating with viral and cellular RNAs, and transported in the budding virion to the target cell. During the course of viral reverse transcription, a transient single-stranded DNA (ssDNA) intermediate is formed. Restrictive A3 proteins bind to the ssDNA intermediate and deaminate cytosine bases to uracil in preferred polynucleotide contexts (5′-TC for A3D, A3F and A3H, and 5′-CC for A3G) (7). Upon copying of the ssDNA intermediate to form the dsDNA required for successful integration of the HIV-1 genome into the host DNA, mutated uracils base pair with adenines resulting in G to A hypermutation and loss of coding integrity (8). Interestingly, even though these restrictive A3 proteins bind tightly to RNA in the cell (9,10), they do not catalyze cytosine deamination in the context of RNA (11,12). The mechanism by which these A3 proteins distinguish between relatively rare single stranded DNAs and the abundant single stranded RNA present in the cellular milieu has been a perplexing question. Without the ability to selectively exclude ribocytidines from deamination, mRNA would acquire lethal amounts of nonsense and missense mutations[e.g., (13)], and without the ability to interact with RNA, A3 proteins would not be able to exert restrictive pressure during HIV infection since encapsidation is essential for deamination of the HIV-1 genome [e.g. (6)]. Sharma and co-workers observed the deamination of RNA by A3G in natural killer cells, lymphoma cell lines and CD8-positive T cells under specific conditions, such as cellular crowding and hypoxia, but not in cells under normal conditions (14). Since A3G strongly disfavors ribocytidine as a substrate in vitro (11,12), the physiological function of RNA deamination by A3G remains elusive.
Structures of the catalytically active subunits of A3A, A3B, A3C, A3F, A3G and A3H have been determined in the absence of ssDNA by us (15–19) and others (20–29). We (30) and another group (31) also determined structures of A3A bound to ssDNA, which provided insights of static interactions between substrate ssDNA and protein at the catalytic site. In the A3A–ssDNA co-crystal structures, the ssDNA exists in a tightly curved conformation with three nucleotides (the target deoxycytidine and flanking nucleotides) forming hydrogen bonds and π–π stacking interactions with A3A, and the sugar of the target deoxycytidine adopting the C2′-endo conformation typically found in DNA (30,31). Most recently, we determined the structure of the ssDNA-bound A3G catalytic domain using a variant of A3Gctd (A3G-CTD2) that has strong affinity for ssDNA containing a hotspot sequence, 5′-TCCCA (32). In comparison to the A3A–ssDNA co-crystal structure, the ssDNA has a more extended conformation and larger contact surface with A3G-CTD2, by interacting with five nucleotides instead of only three. Although this co-crystal structure provided atomic details of static interactions between the hotspot nucleotides and the protein, the mechanism by which A3G strongly disfavored ribocytidine as a substrate was not revealed. In particular, a 2′-OH could fit within the spatial position of the 2′-H without significant steric hindrance (32). Previously, Nabel et al. reported that the C2′-endo sugar conformation was important for the efficiency of deoxycytidine deamination catalyzed by human activation induced deaminase (AID) and mouse APOBEC1 (33). This finding may or may not be applicable for A3G because AID and mouse APOBEC1 are substantially different from A3G in regard to physiological targets; AID deaminates deoxycytidines in particular 5′-A/T-2A/G-1C0 hotspots of the immunoglobulin genes undergoing transcription (34), whereas APOBEC1 deaminates a specific cytidine in the apolipoprotein B (Apob) pre-mRNA (35,36). Importantly, APOBEC1 but not A3G, requires an additional factor for deamination site selection and activity in cells; an RNA-binding protein, namely the APOBEC1 complementation factor or A1CF (37,38). For AID, different studies have found that various proteins interact with AID (39,40). In this study, we interrogate the differences in the interaction modes of A3Gctd for substrate or non-substrate ssDNAs, and the exclusion mechanisms for ribocytidine from deamination. We show that the mode of interaction including extent, intensity and time-scale, determined by NMR titration experiments, clearly distinguish the catalytically productive binding mode for substrate ssDNA from the inactive mode for non-substrate. In addition, we reveal the importance of 2′-endo sugar conformation for catalytically productive binding using 2′-deoxy-2′-fluorine substituted cytidines as substrates. Furthermore, molecular dynamics (MD) simulations indicate that 2′-OH causes the target ribocytidine to dislocate from the catalytic position for A3Gctd but not for A3A, consistent with A3A’s ability to deaminate ribocytidine.
MATERIALS AND METHODS
Plasmid generation and protein purification
The pGEX-6P-1 vector (GE Healthcare Life Science) containing the C-terminal catalytic domain of A3G (A3Gctd), residues 191–384, with the previously reported 2K3A mutations (L234K, C243A, F310K, C321A, C356A) (15) was used as the template for Quikchange mutagenesis (Stratagene/Agilent Technologies) to introduce E259A substitution. Escherichia coli were transformed with the plasmid, grown to OD 0.5 at 37°C followed by a reduction in temperature to 17°C for 30 min, and protein expression was induced using a 0.1 mM final concentration of isopropyl β-d-1-thiogalactopyranoside (IPTG). The cells were lysed using sonication into buffer containing 50 mM sodium phosphate pH 7.3, 100 mM NaCl, 2 mM DTT, 0.002% Tween 20. Following high-speed centrifugation, the supernatant was bound to glutathione sepharose resin (GenScript) and washed under high salt and high detergent conditions, 400 mM NaCl and 0.06% Tween 20, followed by two washes in low salt and low detergent conditions, 30 mM NaCl and 0.002% Tween 20. The GST-tag was removed using PreScission protease (GE Healthcare Life Science) in 50 mM sodium phosphate buffer at pH 7.3 with 100 mM NaCl, 2 mM DTT and 0.002% Tween 20. Following cleavage, the protein was dialyzed into sample buffer containing 50 mM sodium phosphate pH 6.0, 100 mM NaCl, 2 mM DTT, 0.002% Tween 20 and 50 μM ZnCl2.
NMR spectroscopy
All multi-dimensional NMR spectra were acquired on an 850 MHz Bruker Ascend spectrometer equipped with a 5 mm Z-gradient TCI cryoprobe. Samples contained a final volume of 300μL (97% H2O/3% D2O, v/v), and spectra were taken at 293 K. Backbone resonance assignments for the A3Gctd-2K3A-E259A mutant were derived using TROSY versions of a standard set of triple resonance spectra (HNCA, HN(CO)CA, HNCACB, HN(CO)CACB, HNCO, HN(CA)CO) on uniformly 15N/13C labeled protein with 85% random deuteration at pH 7.3. Assignments were transferred to pH 6.0 HSQC spectrum by titrating pH to identify relevant peak shifts. 15N-HSQC with ssDNA titrations were collected on 0.2 mM 15N-labeled A3Gctd-2K3A-E259A samples at pH 6.0 with unlabeled ssDNA at ratios of 1:1, 1:2 and 1:5 (A3Gctd-2K3A-E259A:ssDNA). Each titration point was collected with 128 transients and 100 real data points in the indirect 15N dimension. Chemical shift and intensity changes were monitored through a series of spectra at varying relative concentration ratios. Chemical shift changes were calculated using the equation:
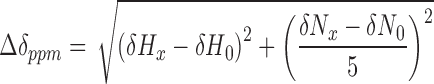 |
Intensity changes were calculated using the difference in peak height at the center of the 15N-HSQC peak between the unbound and bound spectra divided by the unbound peak height. Real-time 1D 1H NMR deaminase assays were performed on Bruker Avance III 600 MHz NMR spectrometer at 20°C in buffer containing 50mM sodium phosphate pH 6.0, 100 mM NaCl, 1 mM DTT, 10 μM ZnCl2 and 0.002% Tween-20. Oligonucleotide substrate concentrations of 150 μM were used in the assays with enzyme concentrations ranging from 1.5 to 50 μM. Spectra were analyzed using Topspin 3.5 software package (Bruker Corporation, Billerica, MA, USA).
DNA oligomers
Oligonucleotides containing standard DNA and RNA bases were synthesized by Integrated DNA Technologies (IDT). Oligonucleotides containing 2′-deoxy-2′-fluororibonucleic acid (2′-F-RNA) and 2′-deoxy-2′-fluoroarabonucleic acid (2′-F-ANA) at the underlined cytosine position in the 5′-ATTCCCAATT oligonucleotide were synthesized by Boston Open Labs.
Microscale Thermophoresis assay (MST)
The binding affinity of purified A3Gctd-2K3A-E259A with 9nt ssDNAs (IDT), including 5′-AATCCCAAA, 5′-AATCCdeoxyUAAA, 5′-AATCdeoxyUdeoxyUAAA, 5′-AATCCriboseCAAA, 5′-ATTCC(2′-F-ANA)CAATT and 5′-ATTCC(2′-F-RNA)CAATT, were measured using Monolith NT.115 (Nano Temper Technologies) (41). RED-tris-NTA fluorescent dye solution was prepared at 100 nM in the MST buffer (50 mM phosphate pH 6.0, 100 mM NaCl, 1 mM DTT, 0.002% Tween-20, 20 μM ZnCl2). A3Gctd-2K3A-E259A was mixed with dye at final concentration of 100 nM and incubated for 30 min at room temperature followed by centrifugation at 15 000 g for 10 min. The ssDNAs were prepared to stock concentration of 64 mM for AATCCCAAA, 5′-AATCCdeoxyUAAA, 5′-AATCdeoxyUdeoxyUAAA, 5′-AATCCriboseCAAA, or 32 mM for 5′-ATTCC(2′-F-ANA)CAATT and 5′-ATTCC(2′-F-RNA)CAATT) in the MST buffer. To determine the binding affinity, 10 μl of ssDNA solution at 16 different concentrations, ranging from 32 mM to 0.24 μM, or 16 mM to 0.12 μM for 5′-ATTCC(2′-F-ANA)CAATT and 5′-ATTCC(2′-F-RNA)CAATT, were prepared in LoBind centrifuge tubes (Fisher Scientific), then 10 μl of fluorescent labelled A3Gctd-2K3A-E259A solution (100 nM) was added to each tube. The mixtures were incubated at 4°C to reach equilibrium. Each incubated solution was loaded into a Nano Temper MST premium coated capillary. The measurement was performed at room temperature using 40% LED power and 20% MST power. The experiment was repeated three times using freshly purified protein at each time, and data analysis was carried out using Nano Temper analysis software (MO affinity).
Molecular dynamics simulations
The structures of wild type A3Gctd with 5′-TCCCAA or 5′-TCCrCAA were modeled starting from ssDNA-bound A3Gctd crystal structure (PDB ID: 6BUX) through program Modeller 9.15 using basic modeling. The structures of wild type A3A with 5′-AATCGAA or 5′-AATrCGAA were modelled based on A3A DNA-bound crystal structure (PDB ID: 5KEG) using the same method. The phosphate groups of 5′ T base in all structures were removed to prevent strong electronegative environment. All molecular dynamics simulations were performed using Desmond (42) from Schrödinger. The models were first optimized using Protein Preparation Wizard at pH 6.5. The simulation systems were then built through Desmond System Setup using OPLS3 force field (43). Simple point charge (SPC) water model was used for solvation with cubic boundary conditions and 12 Å buffer box size. The final system was neutral and had 0.15 M sodium chloride. The simulation system was first energy minimized with gradually reduced restraints (1000, 5, 0 force constant) on backbone and solute heavy atoms. A multi-stage MD simulation protocol was used. Briefly, the system was simulated using NPT ensemble with gradually increased simulation time (24, 50 and 500 ps) and decreased restraints on the solute heavy atoms to no restraints. The final production stage was performed at 300 K and 1 bar with no restraints using NPT ensemble. 100 ns MD simulations were performed for all DNA-bound structures. The analysis of MD simulations was performed separately for each trajectory. The RMSD and RMSF of protein and DNA molecule were calculated using Simulation Interactions Diagram from Schrödinger. Hydrogen bond occupancies over the trajectories and the side chain dihedral angles were calculated using program VMD. A hydrogen bond was defined as having a donor-acceptor distance of <3.6 Å and involving polar atoms nitrogen, oxygen, sulfur and fluorine. The donor-hydrogen-acceptor angle was defined as being less than 30 degrees. The trajectories from MD simulations for RMSD, distance and dihedral analysis were aligned based on whole molecules.
RESULTS
Assigning NMR signals of A3Gctd-2K3A-E259A at pH 6.0
Wild-type A3Gctd has weak affinity for ssDNA at neutral pH, making detection difficult of significant NMR chemical shift perturbations upon ssDNA binding (15,21). Enzymatic kinetics analysis of A3Gctd at pH 6 suggested that A3Gctd bound ssDNA with a higher affinity (44), but wild-type A3Gctd was not stable enough to conduct lengthy NMR experiments at that pH with high protein concentration. Therefore, we used a variant A3Gctd, termed A3Gctd-2K3A that contained five amino acid substitutions (L234K, C243A, F310K, C321A and C356A) which enhance the solubility and stability of protein, without altering catalytic activity, structure, or HIV-1 restriction (15,17,45). To observe interaction and compare differences between substrate and non-substrate ssDNAs without ongoing catalytic reaction, we produced a catalytically inactive variant of A3Gctd-2K3A by introducing a single alanine point mutation at the catalytic glutamate (E259A), termed A3Gctd-2K3A-E259A. We completed the assignment of backbone NMR signals of A3Gctd-2K3A-E259A by using standard triple resonance NMR experiments at pH 7.3, then transferred the assignments to the spectrum recorded at pH 6.0 by following peak shifts throughout pH titration from pH 7.3 to pH 6.0. We were able to assign most of the resolved NMR signals in the 15N-HSQC spectrum at pH 6.0 (Figure 1).
Figure 1.
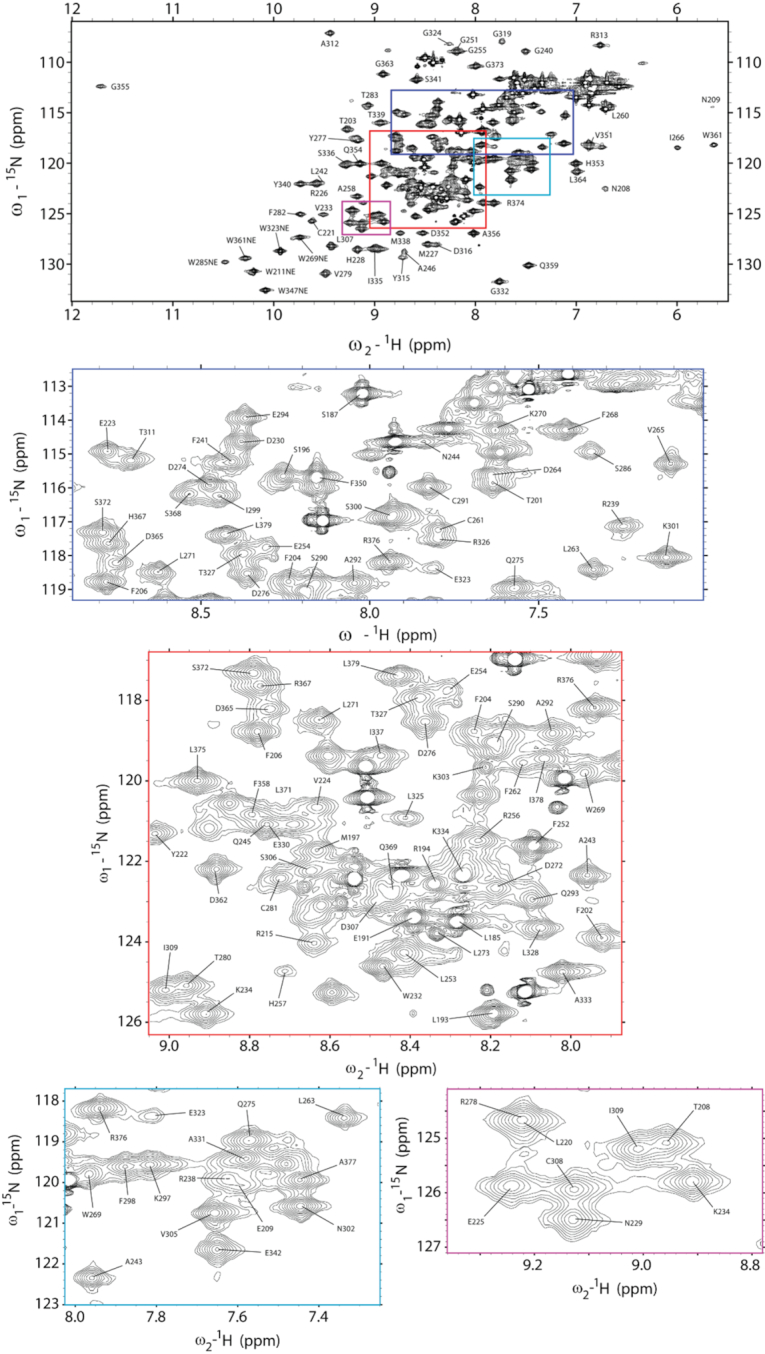
NMR signal assignments of A3Gctd-2K3A-E259A at pH 6.0. 1H–15N HSQC spectrum of A3Gctd-2K3A-E259A. Inset locations indicated with colored boxes correspond to expanded inset spectrum borders.
Identification of ssDNA-binding surfaces of A3Gctd
A3Gctd deaminates the 3′ C in the 5′-CCC motif 45 times more efficiently than the middle C, and does not deaminate the 5′ C in vitro (44,46–48). In order to determine interactions that are responsible for this disparity, we mixed ssDNA to a sample containing catalytically inactive A3Gctd-2K3A-E259A and observed interaction between the protein and ssDNA. We mixed the substrate ssDNA (5′-AATCCCAAA), the intermediate product (5′-AATCCdeoxyUAAA), the final product (5′-AATCdeoxyUdeoxyUAAA), or a ribocytidine substituted ssDNA (5′-AATCCrCAAA) to A3Gctd-2K3A-E259A, and compared chemical shift perturbations (CSP) and signal intensity changes of their 15N-HSQC spectra.
The 15N-HSQC spectrum of A3Gctd-2K3A-E259A showed substantial perturbations upon adding 5′-AATCCCAAA (Figure 2A). This data was quantified as described in Methods and plotted as CSP (red with right axis) and signal intensity changes (gray with left axis) in Figure 2C. Both analyses revealed three primary regions perturbed upon 5′-AATCCCAAA binding. These three regions, binding regions 1, 2 and 3 or BR1, BR2 and BR3, form a continuous surface in the 3D structure of ssDNA-free A3Gctd (PDB ID: 4ROV) (25) (Figure 2D). BR1 spans residues T201-L220, which includes residues located in helix1 (T201-N207) and loop1 (N208-T218). Especially, W211 and R215, both located in loop1, lost >70% of their signal intensity suggesting direct interactions with DNA. BR2 spans residues R238-K270, and includes β-sheet2′, loop3 and helix2. Residues sequentially close to N244 and H257, both located in loop3, showed substantial CSP and intensity changes (Figure 2C). These changes are likely caused by the direct interaction of N244 and H257 with the target deoxycytidine, as observed in the co-crystal structure of A3Gctd-ssDNA (32). It is noteworthy that R238, located in the short loop between β-sheet2 and β-sheet2′, showed substantial perturbation, although it is not located at the catalytic site. Furthermore, F262, L263, V265 and W269, all located in helix2 with their side chains directed toward the inside of the protein and forming a hydrophobic core, displayed DNA-bound as well as DNA-unbound NMR signals following substrate addition (Figure 2B), indicating slow exchange dynamics between bound and unbound states. BR3 included W285 and T311-E330, which contains loop7 (T311-G319), previously suggested to be important for recognition of the hotspot sequence (20,49,50). Especially, W285 located at the catalytic pocket (15,20) as well as loop7 residues, A312, R313, Y315 and D316, displayed substantial CSP with slow exchange dynamics (Figure 2B). These perturbations were consistent with the co-crystal structure (32) as R313, Y315 and D316 had direct interactions with ssDNA. The exchange of bound and unbound states of loop7 residues likely destabilized helix4 (E323–E330), since residues located in helix4 showed >60% reductions in signal intensity.
Figure 2.
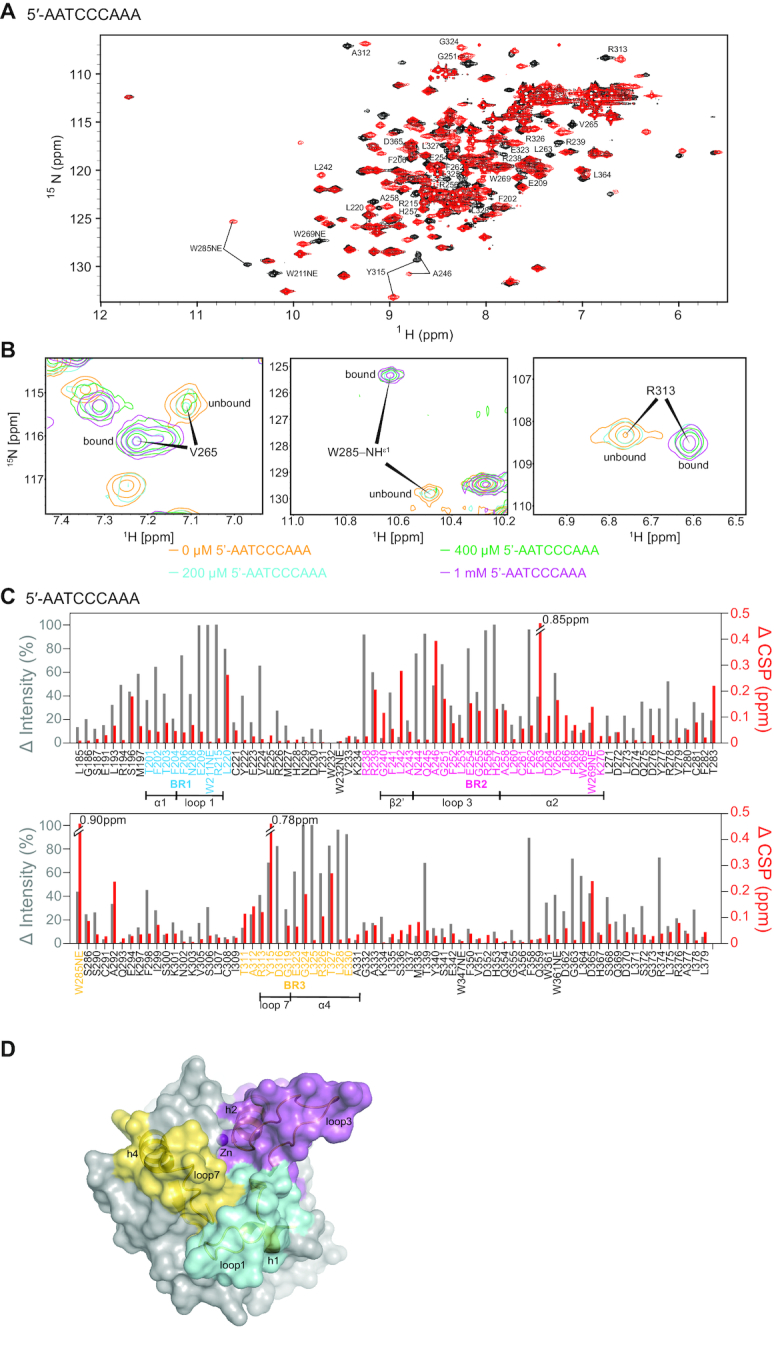
Chemical shift perturbation and signal intensity changes upon binding 5′-AATCCCAAA. (A) 1H–15N HSQC spectrum of 0.2 mM A3Gctd-2K3A-E259A mixed with 1 mM 5′-AATCCCAAA (red) overlaid onto 0.2 mM A3Gctd-2K3A-E259A (black). Significantly shifted peaks are labeled. (B) NMR signals of residues in slow exchange regime upon addition of 5′-AATCCCAAA. DNA-unbound signals are labeled unbound, whereas DNA-bound signals are labeled bound. Intensities of unbound signals decrease, while intensities of bound signals increase, upon increment of the ssDNA concentration. (C) Quantification of peak intensity changes (gray bars, left axis) and chemical shifts changes (red bars, right axis). Residues in BR1, BR2 and BR3 are colored blue, magenta and yellow, respectively. Secondary structures within the binding regions are shown under the residues. (D) Three ssDNA binding regions are shown on the surface of the structure of ssDNA-free wild type A3Gctd (PDB ID# 4ROV). Binding region 1 (BR1, cyan) spans residues 201–220, binding region 2 (BR2, magenta) spans residues 238–270, and binding region 3 (BR3, yellow) spans the non-consecutive residues 285, 311–330. Secondary structures of binding regions are shown in cartoon models.
We next compared CSP and intensity changes for 5′-AATCCCAAA with the intermediate product, 5′-AATCCdeoxyUAAA, by subtracting the changes for 5′-AATCCCAAA from the changes for 5′-AATCCdeoxyUAAA (‘delta – delta’ plot, Figure 3A). Raw data including NMR spectra, quantified CSP and intensity changes upon the addition of 5′-AATCCdeoxyUAAA are provided in Supplementary Figures S1A and S2A, respectively. We found that 5′-AATCCdeoxyUAAA engaged all three binding regions described above for 5′-AATCCCAAA, however, the key difference was that BR1 residues displayed reduced chemical shift changes and signal intensity changes (appearing as negative red and gray bars in Figure 3A), indicating lesser interaction of BR1 with 5′-AATCCdeoxyUAAA. In addition, Figure 3A revealed that the exchange rate between bound and unbound states became faster with 5′-AATCCdeoxyUAAA than 5′-AATCCCAAA, as residues in BR2 and BR3 indicated reduced chemical shift changes (negative red bars) but increased intensity reduction (positive gray bars) caused by line-broadening due to exchange between bound and unbound states. The faster exchange rate with 5′-AATCCdeoxyUAAA was also evident in the spectrum (Supplementary Figure S1A) since there was no residue showing two distinct bound and unbound signals, as had been displayed upon binding 5′-AATCCCAAA (Figure 2B). 5′-AATCCdeoxyUAAA contained a 5′-CC deamination motif, and the underlined C was presumably positioned at the catalytic site. The lesser interaction with 5′-CC compared to 5′-CCC was consistent with deamination efficiency since A3Gctd deaminates 5′-CCC 45-times more efficiently than 5′-CCdeoxyU (44).
Figure 3.

Comparison of chemical shift perturbations and intensity changes upon binding substrate and non-substrate ssDNAs. ssDNA oligomers were mixed with A3Gctd-2K3A-E259A at 1 mM: 0.2 mM concentration ratio, and 1H–15N HSQC spectra were acquired (spectra are provided in Supplementary Figure S1). Peak intensity changes and chemical shifts changes upon addition of ssDNA were quantified for each ssDNA (provided in Supplementary Figure S2), then ‘delta—delta’ plots were made by subtracting the changes for 5′-AATCCCAAA from the changes for (A) 5′-AATCCdeoxyUAAA, (B) 5′-AATCdeoxyUdeoxyUAAA and (C) 5′-AATCCriboseCAAA. (D) shows the ‘delta – delta’ plot where the changes for 5′-AATCC(2′-F-ANA)CAAA have been subtracted from the changes for 5′-AATCC(2′-F-RNA)CAAA. The differences of chemical shift changes and signal intensity changes are shown by red bars (right axis) and gray bars (left axis), respectively.
Next, we compared a non-substrate ssDNA with the substrate by using ‘delta – delta’ plots, subtracting the changes for 5′-AATCCCAAA from the changes for 5′-AATCdeoxyUdeoxyUAAA (Figure 3B). NMR spectrum, quantified CSP and intensity changes upon mixing with 5′-AATCdeoxyUdeoxyUAAA are provided in Supplementary Figures S1b and S2b, respectively. 5′-AATCdeoxyUdeoxyUAAA is the final product of the deamination of 5′-CCC as A3Gctd does not deaminate the 5′-TC motif in vitro (46,47,51). All three binding regions, BR1, BR2 and BR3, demonstrated greatly reduced chemical shift changes and signal intensity changes compared with 5′-AATCCCAAA (appearing as negative red and gray bars in Figure 3B), indicating interactions were lost. Although interactions with BR1 and BR3 were almost completely lost, W211NE (BR1), R215 (BR1) and D316 (BR3) retained significant reduction of signal intensities, suggesting that these residues still engage the DNA (Supplementary Figure S2b). We tested another non-substrate ssDNA containing a ribocytidine at the target position, 5′-AATCCriboseCAAA, by using ‘delta–delta’ plots (Figure 3C). NMR spectrum and CSP, intensity changes are provided in Supplementary Figures S1c and S2c, respectively. Figure 3C displayed very similar profile to Figure 3B as all three binding regions substantially reduced both chemical shift changes and signal intensity changes compared with 5′-AATCCCAAA. Especially, BR1 residues lost interaction with the exception of W211NE and R215. BR3 was slightly more involved in the interaction with 5′-AATCCriboseCAAA than 5′-AATCdeoxyUdeoxyUAAA as BR3 residues showed smaller loss of signal intensity changes (shorter negative gray bars in Figure 3C).
The affinities of A3Gctd-2K3A-E259A for above substrate and non-substrate ssDNAs were assayed directly by using microscale thermophoresis (MST) (41). The apparent dissociation constant, Kd, was determined for 5′-AATCCCAAA, 5′-AATCCdeoxyUAAA, 5′-AATCdeoxyUdeoxyUAAA and 5′-AATCCriboseCAAA to be 1.57 ± 0.16, 2.17 ± 0.25, 2.76 ± 0.28 and 6.65 ± 0.86 mM, respectively (Table 1; binding curves are provided in Supplementary Figure S3). Although the differences of Kd values among the ssDNAs were small, the direction of changes of Kd values supported deamination activity of A3Gctd as it showed stronger affinity for the substrate (5′-AATCCCAAA) and weaker affinity for the product (5′-AATCdeoxyUdeoxyUAAA), and the intermediate product (5′-AATCCdeoxyUAAA) showed a Kd value between the substrate and the product. 5′-AATCCriboseCAAA displayed an affinity weaker than that of the product 5′-AATCdeoxyUdeoxyUAAA, indicating that a ribocytidine was disfavored more than deoxy-uridine for binding by A3Gctd.
Table 1.
Apparent Kd values of A3Gctd-2K3A-E259A for binding substrate and non-substrate ssDNAs.
| ssDNA sequence | K d [mM] |
|---|---|
| 5′-AATCCCAAA | 1.57 ± 0.16 |
| 5′-AATCCdUAAA | 2.17 ± 0.25 |
| 5′-AATCdUdUAAA | 2.76 ± 0.28 |
| 5′-AATCCrCAAA | 6.65 ± 0.86 |
| 5′-AATCC(2′-F-RNA)CAAA | 3.76 ± 0.30 |
| 5′-AATCC(2′-F-ANA)CAAA | 1.74 ± 0.49 |
Collectively, NMR and MST experiments showed that A3Gctd has multiple substrate and non-substrate ssDNA binding modes with similar affinities, but one conformation involved interaction with BR1, slightly enhancing binding, and presumably positioned the target cytosine base into the active site for the deamination to occur.
Effects of sugar conformation on ssDNA binding and deamination
Two potential mechanisms could exclude ribocytidines from catalysis by A3Gctd: the presence of the hydroxyl moiety at the sugar C2′ position of the ribocytidine or the conformation of the sugar; ribose prefers the C3′-endo conformation whereas deoxyribose prefers the C2′-endo conformation (Figure 4). To discriminate between these two possible mechanisms, we tested two fluorinated cytidine substrates, the first containing a fluorine substituted for the C2′ hydroxyl of the ribose (2′-deoxy-2′-fluororibonucleic acid, 2′-F-RNA) and the second containing an arabinose sugar with the C2′ hydroxyl substituted for fluorine (2′-deoxy-2′-fluoroarabonucleic acid, 2′-F-ANA) (Figure 4). The 2′-F-RNA cytidine presumably had the C3′-endo conformation of the un-substituted ribose base, while the 2′-F-ANA cytidine presumably preferred the C2′-endo conformation typically seen in DNA (52). Fluorine substitution retains an electronegative atom at the C2′ position to mimic the presence of an oxygen atom with significantly weaker capability to form a hydrogen bond.
Figure 4.

Comparison of nucleotide sugar conformation. Varying functional group and stereochemistry at the C2′ position of the ribose ring impacts the ribose ring conformation. (A) Stereochemistry at the C2′ position of the nucleotides used in this study. (B) Preferred conformations of the ribose ring containing specified substitutions in polynucleotide contexts. Exo-conformations indicated by vertical lines connecting functional groups at the indicated stereo-centers. Endo-conformations indicated by horizontal lines connecting functional groups.
We added 5′-ATTCC(2′-F-ANA)CAATT or 5′-ATTCC(2′-F-RNA)CAATT to a sample of A3Gctd-2K3A-E259A (NMR spectra and CSP/intensity changes are provided in Supplementary Figures S1S and E and S2D and E). Addition of 5′-ATTCC(2′-F-ANA)CAATT resulted in moderate CSP and substantial intensity changes in all BR1, BR2 and BR3 regions (Supplementary Figure S2d). On the contrary, addition of 5′-ATTCC(2′-F-RNA)CAATT showed limited CSP and moderate reduction of NMR signal intensities across the protein, but did not display the intensive BR1 interaction (Supplementary Figure S2e), suggesting that 5′-ATTCC(2′-F-RNA)CAATT is not a substrate. Figure 3d shows the ‘delta-delta’ plot where the CSP and intensity changes for 5′-ATTCC(2′-F-ANA)CAATT are subtracted from the changes for 5′-ATTCC(2′-F-RNA)CAATT. 5′-ATTCC(2′-F-ANA)CAATT caused substantially increased reduction of NMR signal intensities compared with 5′-ATTCC(2′-F-RNA)CAATT (negative gray bars in Figure 3D) in BR1 and BR2, but less extent in BR3.
To compare differences in affinity, apparent dissociation constant, Kd, values were determined using MST. Kd values were 1.73 ± 0.48 mM and 3.76 ± 0.30 mM for 5′-ATTCC(2′-F-ANA)CAATT and 5′-ATTCC(2′-F-RNA)CAATT, respectively (Table 1, binding curves are provided in Supplementary Figure S3). The Kd value of 5′-ATTCC(2′-F-ANA)CAATT was similar to that of substrate ssDNAs, including 5′-AATCCCAAA (Kd = 1.57 ± 0.15 mM) and 5′-AATCCdeoxyUAAA (Kd = 2.17 ± 0.25 mM), whereas the Kd value of 5′-ATTCC(2′-F-RNA)CAATT was between the Kd values of two non-substrate ssDNAs, 5′-AATCdeoxyUdeoxyUAAA and 5′-AATCCriboseCAAA.
Since both NMR signal intensity changes and Kd values suggested that 5′-ATTCC(2′-F-ANA)CAATT might be a substrate for the deamination catalyzed by A3Gctd, we conducted 1D 1H NMR deamination assays. Over the course of 8 hours, we observed the appearance of the H5 signal from the deaminated C2′-F-arabinose uracil product 5′-ATTCC(C2′-F-ANA)UAATT at 5.58 ppm, followed by the appearance of another H5 signal at 5.68 ppm from the uracil from the product of deamination of the middle deoxycytidine, 5′-ATTCdeoxyU(C2′-F-ANA)UAATT (Figure 5C). The deamination speed for the 2′-F-ANA cytidine was 0.06 ± 0.01 reactions/min (Supplementary Figure S4). We also tested whether 5′-ATTCC(2′-F-RNA)CAATT could be deaminated by A3Gctd-2K3A, but over the course of 8 h, no uracil signal was observed, confirming that the 2′-F-RNA cytidine was not a substrate (Figure 5D).
Figure 5.

Real-time NMR deamination assays. (A) 1D 1H spectra series of 150 μM 5′-ATTCCCAATT mixed with 1.5 μM A3Gctd-2K3A. The H5 signal of uracil from the deamination product 5′-ATTCCdUAATT appears at 5.64 ppm, another H5 signal of uracil from 5′-ATTCdUdUAATT product appears at 5.73 ppm with concurrent shifting of the 3′ dU signal to 5.66 ppm. (B) 1D 1H spectral series of 150 μM 5′-ATTCCrCAATT mixed with 50 μM A3Gctd-2K3A. No deamination product was observed. (C) 1D 1H spectra series of 150 μM 5′-ATTCC(2′-F-ANA)CAATT mixed with 30 μM A3Gctd-2K3A. A doublet signal of the (2′-F-ANA)U, which is the deamination product of (2′-F-ANA)C, was observed at 5.58 ppm. A doublet signal of the uracil resulted from deamination of the middle C, 5′-CU(2′-F-ANA)U, later appears at 5.68 ppm with concurrent shifting of the 3′ (2′-F-ANA)U to 5.60 ppm. (D) 1D 1H spectral series of 150 μM 5′-ATTCC(2′-F-RNA)CAATT mixed with 30 μM A3Gctd-2K3A. No deamination product was observed.
Molecular dynamics simulations of A3Gctd–ssDNA and A3A–ssDNA complexes
To reveal the atomic-level mechanism for how A3Gctd strongly disfavors ribocytidine (rC) as a substrate, we investigated the stability of ssDNA in the active site through molecular modeling and molecular dynamics (MD) simulations. We modeled 5′-TCCCAA and 5′-TCCrCAA with wild type A3Gctd based on the ssDNA-bound A3Gctd crystal structure (PDB ID: 6BUX) and performed MD simulations. Both MD simulations converged during the 100 ns simulation time. The deoxycytidine (dC) remained in the crystal structure conformation at the catalytic site during the simulations with 5′-TCCCAA (Figures 6A–C, 7A, blue and B). However, in the simulations with 5′-TCCrCAA, ssDNA still bound, but rC shifted ∼3 Å away relative to the starting position within 10 ns of the MD simulation (Figures 6D–F, 7A, red and C). The relocation of rC was due to conformational rearrangements induced by the hydroxyl group attached to 2′C (2′-OH) in rC. H257, which is in close proximity to rC, can form a hydrogen bond with 2′-OH (5 ns; Figure 6E), which in turn destabilized the stacking interaction between the H257 imidazole ring and rC nucleobase. As a result, the critical hydrogen bonds stabilizing co-crystal structure conformation of the target ribocytidine, between the N244 sidechain and sugar, and between A258 backbone and nucleobase, were disrupted. The side chain of N244 then flipped towards rC and formed a new hydrogen bond with the rC base (10 ns; Figure 6F), and thus dislocated the rC to a position that was incompatible with the deamination reaction. rC was stable at this relocated position as it did not go back to the original catalytic position during the rest of the MD simulation (Figure 7A, red; movies of MD simulation trajectories are provided as Supplementary information). Thus, our computational results were in agreement with experimental data that A3G could deaminate dC but not rC. Furthermore, we performed similar modeling and MD simulations for A3A as a comparison since we observed binding (53) and deaminations of both dC and rC by A3A (Supplementary Figure S5). The deamination rate for rC was two orders of magnitude slower than dC in an in vitro NMR-deamination assay (Supplementary Figure S5). In agreement with experiments, the simulations showed that both dC and rC were stable in the catalytic site of A3A and maintained co-crystal structure conformation throughout the MD simulation (Figure 7D–F).
Figure 6.

Snapshots from MD simulations with deoxycytidine and ribocytidine. All panels display the expanded view of the catalytic site of A3Gctd. Zn2+ is shown as a gray sphere, and yellow dashed lines indicate possible hydrogen bonds. DNAs are shown in orange stick model, and nitrogen and oxygen atoms are colored blue and red, respectively. (A), (B) and (C) are snapshots of the 5′-TCCCAA and A3Gctd complex at 0, 5 and 10 ns time points, respectively, while (D), (E) and (F) are snapshots of the 5′-TCCrCAA and A3Gctd complex at 0, 5 and 10 ns time points, respectively.
Figure 7.

Comparison of A3Gctd and A3A in MD simulations with ssDNAs containing dC or rC. (A) A3Gctd and (D) A3A root-mean-square-deviation (RMSD) of the target deoxycytidine (dC) or ribocytidine (rC) during MD simulation. RMSD of all heavy atoms of the target deoxycytidine or ribocytidine are shown for 100 ns simulation time. dC or rC with A3Gctd is shown in blue and red respectively in (a), whereas dC or rC with A3A is shown in orange and black respectively in (d). (B, C) and (E, F) Superposition of expanded views of the catalytic site of A3Gctd (B and C), and A3A (E and F). Zn2+ molecules are shown as spheres. The snapshots at 0 ns are colored blue, whereas the snapshot at 100 ns is colored yellow. DNAs are shown in stick model, and nitrogen and oxygen atoms are colored blue and red, respectively. (B) 5′-TCCCAA and A3Gctd complex, (C) 5′-TCCrCAA and A3Gctd complex, (E) 5′-AATCGAA and A3A complex, and (F) 5′-AATrCGAA and A3A complex.
DISCUSSION
BR1 interaction distinguishes catalytic binding from non-catalytic binding
Characterizing the mechanism of substrate selection and non-substrate exclusion by A3Gctd is important for the development of inhibitors that can selectively modulate A3G activity as well as other A3 enzymes, including those with possible links to carcinogenesis such as A3A and A3B (54,55). Using a combination of experimental and computational methods, we highlight protein-ligand interactions critical for substrate binding that are absent for non-substrates. Our NMR data show that ssDNA binding interfaces of A3Gctd-2K3A form a continuous surface of the protein engaging loops 1, 3 and 7 (Figure 2D). Intense interactions with loop1 residues were observed only with substrate ssDNAs (Figure 2C). This finding supports the importance of loop1 residues for ssDNA binding that has been previously proposed based on the NMR and crystal structures of ssDNA-unbound A3Gctd (15,20), and further, Carpenter et al. provided experimental evidence by swapping loop1 regions between AID and A3Gctd (50). Our data is also consistent with our recent co-crystal structure of A3G-CTD2 and ssDNA in which W211, R213 and H216, all in loop1, form direct interactions with DNA through π–π stacking and hydrogen bonds (32). Ziegler and co-workers recently reported an interesting co-crystal structure of Pot1-fused-A3Gctd and ssDNA (56), and they suggested that loop1 and loop7 residues, including P210, W211, I314 and Y315, along with W285 interact with ssDNA nonspecifically during the search of deamination hotspots. Consistent with their suggestion, we found that W211, R215, D316 and W285 were involved in the interactions with substrate as well as non-substrate ssDNAs.
Allosteric regulation may be an effective strategy to develop inhibitors of A3 activity as competitive inhibitors have been elusive. The NMR signal perturbation data imply possible allosteric sites to target for A3Gctd. In particular, the slow exchange regime of helix 2 residues suggests that the reorientation of helix2 is an allosteric movement coupled with the target cytosine base positioning in Zn2+ containing active site, since helix 2 contains H257 and E259 which coordinate the Zn2+. Another possible allosteric site is β2′-strand (R238-L242) as the dynamic modulation of this strand is most likely helping N244 to stabilize the target deoxycytidine during catalysis (30–32,57–59).
A3Gctd suppresses the catalytic efficiency of ribocytidine through sugar conformation and 2′-OH
Two distinct attributes of RNA could be responsible for the lack of catalytic activity; the presence of the 2′-OH directly, via steric clashing or unfavorable interaction, or indirectly, via conformational impact on the structure of the ribose ring that may prevent cytosine base access to the catalytic site. ssDNAs containing a ribocytidine at the target position have been shown to be excluded from deamination by A3G in vitro (11,12). Consistently, binding of 5′-AATCCriboseCAAA to A3Gctd-2K3A-E259A resulted in a pattern of perturbations similar to that seen in 5′-AATCdeoxyUdeoxyUAAA, a non-substrate ssDNA (Figure 3B and C). We observed a slightly more extensive interaction for 5′-AATCCriboseCAAA with BR3 (residues W285, T311-E330) than seen with 5′-AATCdeoxyUdeoxyUAAA. This effect is likely due to interactions between loop7 and the 5′-CC motif present in 5′-AATCCriboseCAAA since loop7 recognizes the deoxycytidine flanking the 5′ side of the target deoxycytidine in this motif. Nonetheless, 5′-AATCCriboseCAAA binding completely lacked the characteristic BR1 interactions seen in substrate ssDNAs, suggesting that the presence of the ribose sugar prevented both the ribocytidine and the middle deoxycytidine from catalytically productive binding. Consistently, the real-time NMR deamination assay did not detect any deamination product from 5′-AATCCriboseCAAA (Figure 5B). To determine structural factors involved in DNA versus RNA differentiation, we mixed A3Gctd-2K3A-E259A with ssDNAs containing 2′-F-ANA cytidine or 2′-F-RNA cytidine at the target position. Here, fluorine serves as an isopolar and isosteric mimic of the native hydroxyl moiety in ribonucleotides, retaining similar interbond distances and similar electrostatic properties, as Pauling electronegativities are 3.44 and 3.98 for O and F, respectively (60). Based on their chemical structures, 2′-F-ANA cytidine and 2′-F-RNA cytidine are assumed to have the C2′-endo conformation and the C3′-endo conformation, respectively (61) (Figure 4). Addition of 5′-ATTCC(2′-F-RNA)CAATT resulted in moderate reduction of NMR signal intensities of A3Gctd-2K3A-E259A for BR2 and BR3 (Supplementary Figure S2E), yet lacked significant CSP and intensity change in BR1. Since 5′-ATTCC(2′-F-RNA)CAATT was not deaminated by A3Gctd-2K3A (Figure 5D), these results are consistent with 5′-AATCCriboseCAAA, suggesting the C3′-endo sugar conformation of the ribocytidine was disfavored for the catalytically productive binding. In contrast, the ssDNA containing a 2′-F-ANA cytidine exhibited interactions with BR1 (Figure 3D, Supplementary Figure S2D), and the 2′-F-ANA-cytidine was deaminated (Figure 5C), suggesting that the propensity for 2′-F-ANA to retain the C2′-endo sugar conformation of the native DNA allowed the catalytically productive binding. Furthermore, the subsequent deamination of the middle deoxycytidine in the 5′-CC(2′-F-ANA)C sequence (Figure 5C) suggested that the 2′-endo sugar conformation was also preferred for the nucleotide flanking the 3′ side of the target deoxycytidine because A3Gctd-2K3A did not deaminate the middle deoxycytidine of 5′-ATTCCriboseCAATT nor 5′-ATTCC(2′-F-RNA)CAATT (Figure 5B and D). The deamination rate for the 2′-F-ANA cytidine was 0.06 ± 0.01 reactions/minute (Supplementary Figure S4), which was over five times slower than that for deoxycytidine, suggesting that fluorine at the 2′ position negatively affected the catalytic interaction with A3Gctd. Our results extended the finding of Nabel et al. (33) to A3Gctd, and provided experimental evidence as we showed that the 2′-F-ANA (presumably 2′-endo conformation) allowed the catalytically productive binding, but 2′-F-RNA (presumably 3′-endo conformation) did not.
Since RNA is capable of adopting the 2′-endo sugar conformation, the remaining question is why the ribocytidine assuming the 2′-endo sugar conformation is not efficiently deaminated by A3Gctd. To answer this question, we investigated whether 2′-OH destabilizes the ribocytidine in the 2′-endo sugar conformation at the catalytic site of A3Gctd. MD simulations showed that 2′-OH triggered structural changes causing dislocation of the target base from the catalytic position. Therefore, 2′-OH may be another structural feature that negatively affects the deamination of ribocytidine by A3Gctd. On the other hand, A3A held ribocytidine at the catalytic position in the MD simulation, consistent with A3A’s ability to deaminate ribocytidines albeit less efficiently compared with deoxycytidine (NMR deamination assay is provided as Supplementary Figure S5). For A3A, the MD simulation showed that 2′-OH neither forms a hydrogen bond with the Zn2+-binding histidine (H70 in A3A), nor triggered subsequent structural changes of residues interacting with the target ribocytidine. The RMSD data indicated that movements of residues interacting with the ribocytidine were more restricted in A3A than A3Gctd (Supplementary Table S1); therefore, the target ribocytidine was stable at the catalytic position in A3A. Since A3A also deaminates 5-methyl-cytidine as a substrate (62–64), studying A3A further by using NMR and MD simulations to understand binding modes for DNA and RNA substrates will be enlightening.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: W.C.S. performed the protein purification, NMR data collection and NMR data analysis. W.M. performed protein purification, NMR-based deamination assays and analysis. S.H. performed MD simulations and analysis. T.K. performed MST experiments and analyzed data with R.T. W.C.S., W.M., S.H., N.K.Y., C.A.S. and H.M. interpreted results and wrote the paper. C.A.S. and H.M. conceived and oversaw the project and secured funding.
Notes
Present address: William C. Solomon, Department of Natural Sciences, Metropolitan State University, St. Paul, MN 55106, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. National Institutes of Health [P01GM091743, R01GM118474] (in part); W.C.S. was a research fellow of the NIH Training Grant [T32AI083196]; For H.M. this project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN26120080001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government; Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (in part). Funding for open access charge: Award from National Institutes of Health [R01GM118474].
Conflict of interest statement. None of the material in the submitted manuscript has been published, or is under consideration for publication elsewhere. None of the authors have any commercial conflict of interest related to the content of this manuscript.
REFERENCES
- 1. Sheehy A.M., Gaddis N.C., Choi J.D., Malim M.H.. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002; 418:646–650. [DOI] [PubMed] [Google Scholar]
- 2. Conticello S.G., Thomas C.J., Petersen-Mahrt S.K., Neuberger M.S.. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005; 22:367–377. [DOI] [PubMed] [Google Scholar]
- 3. Malim M.H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009; 364:675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiu Y.L., Greene W.C.. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008; 26:317–353. [DOI] [PubMed] [Google Scholar]
- 5. Goila-Gaur R., Strebel K.. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008; 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng Y., Baig T.T., Love R.P., Chelico L.. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 2014; 5:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bishop K.N., Holmes R.K., Sheehy A.M., Davidson N.O., Cho S.J., Malim M.H.. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004; 14:1392–1396. [DOI] [PubMed] [Google Scholar]
- 8. Yu Q., Konig R., Pillai S., Chiles K., Kearney M., Palmer S., Richman D., Coffin J.M., Landau N.R.. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004; 11:435–442. [DOI] [PubMed] [Google Scholar]
- 9. Chiu Y.L., Soros V.B., Kreisberg J.F., Stopak K., Yonemoto W., Greene W.C.. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005; 435:108–114. [DOI] [PubMed] [Google Scholar]
- 10. Khan M.A., Goila-Gaur R., Opi S., Miyagi E., Takeuchi H., Kao S., Strebel K.. Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology. 2007; 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwatani Y., Takeuchi H., Strebel K., Levin J.G.. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 2006; 80:5992–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rausch J.W., Chelico L., Goodman M.F., Le Grice S.F.. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J. Biol. Chem. 2009; 284:7047–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma S., Wang J., Alqassim E., Portwood S., Cortes Gomez E., Maguire O., Basse P.H., Wang E.S., Segal B.H., Baysal B.E.. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019; 20:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen K.M., Harjes E., Gross P.J., Fahmy A., Lu Y., Shindo K., Harris R.S., Matsuo H.. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008; 452:116–119. [DOI] [PubMed] [Google Scholar]
- 16. Harjes E., Gross P.J., Chen K.M., Lu Y., Shindo K., Nowarski R., Gross J.D., Kotler M., Harris R.S., Matsuo H.. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J. Mol. Biol. 2009; 389:819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shandilya S.M., Nalam M.N., Nalivaika E.A., Gross P.J., Valesano J.C., Shindo K., Li M., Munson M., Royer W.E., Harjes E. et al.. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure. 2010; 18:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bohn M.F., Shandilya S.M., Albin J.S., Kouno T., Anderson B.D., McDougle R.M., Carpenter M.A., Rathore A., Evans L., Davis A.N. et al.. Crystal structure of the DNA cytosine deaminase APOBEC3F: the catalytically active and HIV-1 Vif-binding domain. Structure. 2013; 21:1042–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kouno T., Luengas E.M., Shigematsu M., Shandilya S.M., Zhang J., Chen L., Hara M., Schiffer C.A., Harris R.S., Matsuo H.. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 2015; 22:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holden L.G., Prochnow C., Chang Y.P., Bransteitter R., Chelico L., Sen U., Stevens R.C., Goodman M.F., Chen X.S.. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008; 456:121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furukawa A., Nagata T., Matsugami A., Habu Y., Sugiyama R., Hayashi F., Kobayashi N., Yokoyama S., Takaku H., Katahira M.. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009; 28:440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kitamura S., Ode H., Nakashima M., Imahashi M., Naganawa Y., Kurosawa T., Yokomaku Y., Yamane T., Watanabe N., Suzuki A. et al.. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012; 19:1005–1010. [DOI] [PubMed] [Google Scholar]
- 23. Siu K.K., Sultana A., Azimi F.C., Lee J.E.. Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 2013; 4:2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Byeon I.J., Ahn J., Mitra M., Byeon C.H., Hercik K., Hritz J., Charlton L.M., Levin J.G., Gronenborn A.M.. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013; 4:1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu X., Zhang T., Xu Z., Liu S., Zhao B., Lan W., Wang C., Ding J., Cao C.. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 2015; 290:4010–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi K., Carpenter M.A., Kurahashi K., Harris R.S., Aihara H.. Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J. Biol. Chem. 2015; 290:28120–28130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Byeon I.J., Byeon C.H., Wu T., Mitra M., Singer D., Levin J.G., Gronenborn A.M.. Nuclear magnetic resonance structure of the APOBEC3B catalytic domain: structural basis for substrate binding and DNA deaminase activity. Biochemistry. 2016; 55:2944–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiao X., Li S.X., Yang H., Chen X.S.. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016; 7:12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qiao Q., Wang L., Meng F.L., Hwang J.K., Alt F.W., Wu H.. AID recognizes structured DNA for class switch recombination. Mol. Cell. 2017; 67:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kouno T., Silvas T.V., Hilbert B.J., Shandilya S.M.D., Bohn M.F., Kelch B.A., Royer W.E., Somasundaran M., Kurt Yilmaz N., Matsuo H. et al.. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017; 8:15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shi K., Carpenter M.A., Banerjee S., Shaban N.M., Kurahashi K., Salamango D.J., McCann J.L., Starrett G.J., Duffy J.V., Demir O. et al.. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017; 24:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maiti A., Myint W., Kanai T., Delviks-Frankenberry K., Sierra Rodriguez C., Pathak V.K., Schiffer C.A., Matsuo H.. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018; 9:2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nabel C.S., Lee J.W., Wang L.C., Kohli R.M.. Nucleic acid determinants for selective deamination of DNA over RNA by activation-induced deaminase. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14225–14230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T.. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000; 102:553–563. [DOI] [PubMed] [Google Scholar]
- 35. Teng B., Burant C.F., Davidson N.O.. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993; 260:1816–1819. [DOI] [PubMed] [Google Scholar]
- 36. Navaratnam N., Morrison J.R., Bhattacharya S., Patel D., Funahashi T., Giannoni F., Teng B.B., Davidson N.O., Scott J.. The p27 catalytic subunit of the apolipoprotein B mRNA editing enzyme is a cytidine deaminase. J. Biol. Chem. 1993; 268:20709–20712. [PubMed] [Google Scholar]
- 37. Mehta A., Kinter M.T., Sherman N.E., Driscoll D.M.. Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol. Cell Biol. 2000; 20:1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lellek H., Kirsten R., Diehl I., Apostel F., Buck F., Greeve J.. Purification and molecular cloning of a novel essential component of the apolipoprotein B mRNA editing enzyme-complex. J. Biol. Chem. 2000; 275:19848–19856. [DOI] [PubMed] [Google Scholar]
- 39. Larijani M., Martin A.. The biochemistry of activation-induced deaminase and its physiological functions. Semin. Immunol. 2012; 24:255–263. [DOI] [PubMed] [Google Scholar]
- 40. Casellas R., Basu U., Yewdell W.T., Chaudhuri J., Robbiani D.F., Di Noia J.M.. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat. Rev. Immunol. 2016; 16:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wienken C.J., Baaske P., Rothbauer U., Braun D., Duhr S.. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010; 1:100. [DOI] [PubMed] [Google Scholar]
- 42. Bowers K.J., Chow E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A., Klepeis J.L., Kolossvary I., Moraes M.A., Sacerdoti F.D. et al.. Proceedings of the 2006 ACM/IEEE conference on Supercomputing. 2006; NY: ACM. [Google Scholar]
- 43. Harder E., Damm W., Maple J., Wu C.J., Reboul M., Xiang J.Y., Wang L.L., Lupyan D., Dahlgren M.K., Knight J.L. et al.. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016; 12:281–296. [DOI] [PubMed] [Google Scholar]
- 44. Harjes S., Solomon W.C., Li M., Chen K.M., Harjes E., Harris R.S., Matsuo H.. Impact of H216 on the DNA binding and catalytic activities of the HIV restriction factor APOBEC3G. J. Virol. 2013; 87:7008–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Autore F., Bergeron J.R., Malim M.H., Fraternali F., Huthoff H.. Rationalisation of the differences between APOBEC3G structures from crystallography and NMR studies by molecular dynamics simulations. PLoS One. 2010; 5:e11515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chelico L., Sacho E.J., Erie D.A., Goodman M.F.. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008; 283:13780–13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nowarski R., Britan-Rosich E., Shiloach T., Kotler M.. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 2008; 15:1059–1066. [DOI] [PubMed] [Google Scholar]
- 48. Chiba J., Kouno T., Aoki S., Sato H., Zhang J., Matsuo H., Inouye M.. Electrochemical direct detection of DNA deamination catalyzed by APOBEC3G. Chem. Commun. (Camb.). 2012; 48:12115–12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rathore A., Carpenter M.A., Demir O., Ikeda T., Li M., Shaban N.M., Law E.K., Anokhin D., Brown W.L., Amaro R.E. et al.. The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J. Mol. Biol. 2013; 425:4442–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carpenter M.A., Rajagurubandara E., Wijesinghe P., Bhagwat A.S.. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair (Amst.). 2010; 9:579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chelico L., Pham P., Calabrese P., Goodman M.F.. APOBEC3G DNA deaminase acts processively 3′ → 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006; 13:392–399. [DOI] [PubMed] [Google Scholar]
- 52. Williams A.A., Darwanto A., Theruvathu J.A., Burdzy A., Neidigh J.W., Sowers L.C.. Impact of sugar pucker on base pair and mispair stability. Biochemistry. 2009; 48:11994–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Silvas T.V., Hou S., Myint W., Nalivaika E., Somasundaran M., Kelch B.A., Matsuo H., Kurt Yilmaz N., Schiffer C.A.. Substrate sequence selectivity of APOBEC3A implicates intra-DNA interactions. Sci. Rep. 2018; 8:7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B. et al.. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013; 494:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y., Schmitt K., Guo K., Santiago M.L., Stephens E.B.. Role of the single deaminase domain APOBEC3A in virus restriction, retrotransposition, DNA damage and cancer. J. Gen. Virol. 2016; 97:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ziegler S.J., Liu C., Landau M., Buzovetsky O., Desimmie B.A., Zhao Q., Sasaki T., Burdick R.C., Pathak V.K., Anderson K.S. et al.. Insights into DNA substrate selection by APOBEC3G from structural, biochemical, and functional studies. PLoS One. 2018; 13:e0195048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Betts L., Xiang S., Short S.A., Wolfenden R., Carter C.W. Jr. Cytidine deaminase. The 2.3 A crystal structure of an enzyme: transition-state analog complex. J. Mol. Biol. 1994; 235:635–656. [DOI] [PubMed] [Google Scholar]
- 58. Xiang S., Short S.A., Wolfenden R., Carter C.W. Jr. Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry. 1995; 34:4516–4523. [DOI] [PubMed] [Google Scholar]
- 59. Xiang S., Short S.A., Wolfenden R., Carter C.W. Jr. The structure of the cytidine deaminase-product complex provides evidence for efficient proton transfer and ground-state destabilization. Biochemistry. 1997; 36:4768–4774. [DOI] [PubMed] [Google Scholar]
- 60. Batsanov S.S. Van der Waals radii of elements. Inorg. Mater. 2001; 37:871–885. [Google Scholar]
- 61. Watts J.K., Choubdar N., Sadalapure K., Robert F., Wahba A.S., Pelletier J., Pinto B.M., Damha M.J.. 2 ‘-Fluoro-4 ’-thioarabino-modified oligonucleotides: conformational switches linked to siRNA activity. Nucleic Acids Res. 2007; 35:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wijesinghe P., Bhagwat A.S.. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic. Acids. Res. 2012; 40:9206–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suspene R., Aynaud M.M., Vartanian J.P., Wain-Hobson S.. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One. 2013; 8:e63461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nabel C.S., Jia H., Ye Y., Shen L., Goldschmidt H.L., Stivers J.T., Zhang Y., Kohli R.M.. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012; 8:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.