


Abstract
Guanine quadruplexes (G4s) are non-canonical nucleic acids structures common in important genomic regions. Parallel-stranded G4 folds are the most abundant, but their folding mechanism is not fully understood. Recent research highlighted that G4 DNA molecules fold via kinetic partitioning mechanism dominated by competition amongst diverse long-living G4 folds. The role of other intermediate species such as parallel G-triplexes and G-hairpins in the folding process has been a matter of debate. Here, we use standard and enhanced-sampling molecular dynamics simulations (total length of ∼0.9 ms) to study these potential folding intermediates. We suggest that parallel G-triplex per se is rather an unstable species that is in local equilibrium with a broad ensemble of triplex-like structures. The equilibrium is shifted to well-structured G-triplex by stacked aromatic ligand and to a lesser extent by flanking duplexes or nucleotides. Next, we study propeller loop formation in GGGAGGGAGGG, GGGAGGG and GGGTTAGGG sequences. We identify multiple folding pathways from different unfolded and misfolded structures leading towards an ensemble of intermediates called cross-like structures (cross-hairpins), thus providing atomistic level of description of the single-molecule folding events. In summary, the parallel G-triplex is a possible, but not mandatory short-living (transitory) intermediate in the folding of parallel-stranded G4.
INTRODUCTION
DNA G-quadruplexes (G4s) are non-canonical secondary structures formed by stacked G-tetrads and stabilized by central coordinating cation. As indicated by high-throughput sequencing there are over 700 000 potential G4 forming sites in the human genome (1). They are notably concentrated in telomeric and centromeric regions as well as in promoter regions of protein-coding genes, particularly in those of proto-oncogenes (2,3). These findings agree with experimental observations that G4 structures may contribute to maintaining genome integrity of centromeric and telomeric DNA regions (4–10). A number of studies suggested G4 formation at oncogene promoters; their role in transcription regulation then highlighted G4s as promising therapeutic targets in a broad range of human diseases including cancer (11–21). Considerable effort has been directed towards the identification of small molecules enabling modulation of transcription of (onco)genes via binding and stabilization of the G4 structures in a specific and controlled way (22–26).
To understand and potentially modulate the biochemical roles of G4 molecules, it is essential to comprehend their folding properties (27–57). Analysis of available in vitro experimental and computational data indicates that DNA G4-forming sequences have very complex free-energy (folding) landscapes kinetically dominated by a set of deep free-energy basins, i.e. substates formed by molecular structures with very long lifetimes (58). This phenomenon is generally known as kinetic partitioning of the folding landscape (39,58–61), and the DNA G4 folding landscape has been suggested to be an extreme case of kinetic partitioning (58). The deep (long-living, metastable) free-energy basins likely correspond to diverse G4 folds and typically act with respect to each other as competing off-pathway folding intermediates (58). Such extremely rugged free-energy landscape naturally explains the conformational polymorphism of many G4 sequences, i.e. the ability of a single G4-forming sequence to form several distinct G4 folds at the thermodynamic equilibrium under different experimental conditions (and/or with different flanking sequences) or even detectable co-existence of different folds in a given experiment (46,62–72). Diverse G4 folds differ by virtue of strand orientations, conformation of glycosidic bonds of Gs within the individual G-tetrads and loop types (73). Even for those DNA G4 molecules that at first sight do not appear as polymorphic, non-native G4 folds may be transiently populated during the folding process (39,41,42,58,74). Slow folding occurs when the kinetically first-accessed population of G4 folds (or other long-lived species) is metastable but does not match the one at free-energy minimum (39,58–61).
The fact that reaching equilibrium state for a number of known G4 systems requires a long time (up to days) (39,61,75) in in vitro experiments generally appears incompatible with the time-scale of biologically relevant processes, e.g. transcription operates on seconds-to-minutes time-scale. This indicates a possibility that biologically active conformations could be those that arise in the early stages of the folding process (i.e. are kinetically preferred) rather than those corresponding to the global thermodynamic minima (44,75,76). Obviously, we cannot rule out that a fast folding into a single G4 topology under in vivo conditions could occur because the initial unfolded ensemble is somehow dominantly funnelled to just one folded G4 basin, thus bypassing the kinetic partitioning (see below) (58). Nevertheless, the idea that the biochemical roles of G4-forming sequences could be under kinetic control is plausible. However, in analogy to other biomolecular targets such as proteins and RNAs, many of which fold fast into the thermodynamically most stable structure (77,78), the search for the G4 stabilizing ligands (drug-like molecules) has been focused on targeting of most stable G4 conformations known from the in vitro experiments (24,79). In the last two decades, numerous ligands that do bind and stabilize these conformations have been discovered (79,80). While these compounds generally display strong interaction phenotype with thermodynamically most stable G4 structures, this phenotype often does not directly translate into their ability to modulate G4-structure-coupled biological processes. In this respect, cumulating experimental evidence suggests that G4 action may be controlled kinetically rather than thermodynamically and indicates that established strategies for the development of G4-targeting drugs should be broadened (76,81). Either short-lived non-G4 intermediates (see below) or some initial distribution of G4 folds (a mixture of G4s) existing in the early stages of the G4 folding process might represent a relevant target for G4 ligands. While this suggests that the investigation of folding intermediates is essential, only a little information is currently available about the initial stages of the G4 folding.
The free-energy landscape of G4 molecules can contain a wide range of structures with diverse kinetic accessibilities and lifetimes. As noted above, the essence of kinetic partitioning of the G4 folding landscape is the capability of the G4-forming sequences to fold into different G4 topologies readily (39,41,42,49,58,82). They are associated with different syn and anti glycosidic orientations of guanosines. The presence of 2N possible syn–anti combinations, where N is the number of guanosines in the sequence, together with the slow interconversion between different G4 folds with their specific syn–anti patterns poses the main obstacle for fast reaching the equilibrium during the folding process (49,51,58). Due to the presumably fast (compared to the whole folding process) fluctuations of the glycosidic orientations of Gs in the unstructured states, we hypothesize that the folding process has two stages (phases): (i) the initial unfolded ensemble of molecules quickly converts into some initial distribution of G4 folds, which (ii) then slowly rearranges towards the final distribution of G4 folds (Figure 1). The time-scale of the first stage is not known. Because folding times of a biopolymer from a disordered (denatured) conformation into a deep basin on the free-energy landscape depend on the polymer length (60), we suggest that the initial stage could occur in seconds or even faster. It could be within the dead-time of many experimental techniques or correspond to the fastest detectable processes (37,38,47). A truly unstructured ensemble would have only very short lifetime, before being converted into the early ensemble of longer living structures.
Figure 1.
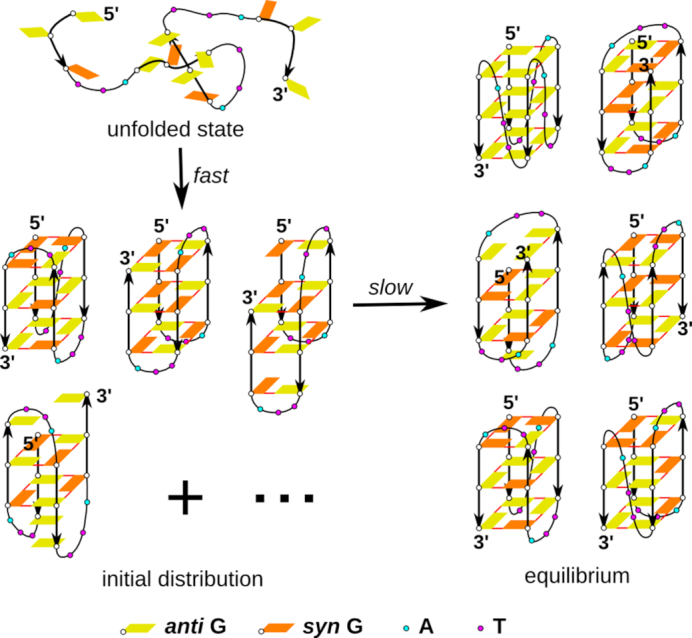
Suggested stages of G4 folding initiated from a truly unstructured (denatured) ensemble. The first stage is represented by fast multiple-pathway folding into an initial ensemble distribution of long-living metastable species (probably misfolded G4s and a fraction of native G4s) that is the most easily accessible from a given unfolded ensemble. It is then followed by a slow transformation into the equilibrium ensemble distribution of G4 species. All conformational rearrangements must be realized through some transitory ensembles. The involvement of cations is not shown.
The G4 folding landscape must also contain other types of structures, such as G-triplexes and G-hairpins. The available primary experimental data on G4 folding have been in the past often interpreted via simple few-state models in which G-hairpins and mainly G-triplexes play key roles (27,29,32,35,37,38,45,83–85). For example, antiparallel and hybrid G4s with at least three tetrads have been often suggested to fold via antiparallel G-hairpins, from which the process proceeds either through antiparallel G-triplexes or via G-hairpins merge (27–32,35,38,45). It has also been proposed that folding of hybrid G4s proceeds through antiparallel G-hairpins and G-triplexes, with the propeller loop being formed at the end, with some of the suggested pathways including an antiparallel chair-type G4 structure (31,35,38,42,45,48,50,85). Other recent studies disputed significant participation of G-triplexes in G4 folding (39,41,42,58,82,86). One of the main arguments against their role as key metastable intermediates is that they do not seem to have sufficiently long lifetimes to act as the kinetically trapped basins on the landscape (58,82). An alternative pathway including a parallel G-hairpin, but avoiding G-triplex, has been suggested for the folding of the hybrid-2 human telomeric G4 in a recent computational study (55).
Many literature reports on the role of G-triplexes in G4 folding are based on structural interpretations of experimental techniques that do not allow unambiguous structural interpretation of the primary experimental data. On the other hand, complete characterization of the folding landscape by contemporary computational chemistry tools is entirely inaccessible due to the fundamental disparity between the simulation time-scales and the time-scale of the real folding processes (58). Thus, while experiments capture the whole folding process but have a limited structural resolution, computations can model atomistic structures but only in very confined regions of the folding landscape. These limitations make unambiguous clarification of the roles of G-triplexes and G-hairpins in the process of G4 folding very difficult. Nevertheless, various G-hairpins, G-triplexes and likely many other so far unnoticed structures (such as compacted coil-like structures) could play key roles as transitory ensembles in both above-noted stages of the folding process (Figure 1), i.e. in navigating the unfolded molecules into the initial distribution of the G4 folds, as well as during the subsequent structural rearrangements from one G4 structure to another, when progressing towards the equilibrium distribution of G4 folds. The roles of G-triplexes and other transient structures may be further enhanced when, e.g. not all G-tracts are released from the DNA duplex at the same instance. Thus, even if the G-triplexes do not correspond to the dominant basins on the G4 folding landscapes in the in vitro studies, they may be relevant in potential in vivo G4-related regulatory processes.
An important feature of complex folding (free-energy) landscapes is that they can lead to diverse folding processes. First, the landscape may be modulated by external (experimental) conditions and sequence contexts. Second, the actual folding process may depend on the initial distribution of the molecules on the landscape. The initial distribution of structures (the specific denatured ensemble in a given experiment) directly determines the fast phase of the folding in Figure 1 and then indirectly also the subsequent slow equilibration. Thus, a given folding landscape may be associated with a diverse spectrum of actual folding processes (58). Unfolded ensembles at different temperatures, ionic strengths, upon stretching by a force, etc. are not the same. Likewise, temporal availability of individual Gs (e.g. due to their consecutive release from the duplex state), conditions changing in the course of time and interactions with other molecules may affect the folding mechanisms. Thus, in vivo folding pathways of G4-forming sequences may substantially differ from those indicated by in vitro experiments and may be tuneable. In addition, metastable species (prefolded structures, intermediates or alternative G4 folds) that might be present under given experimental conditions, but hidden to a given experimental technique, might be unconsciously considered as part of the unfolded ‘unstructured’ ensemble. It can affect the measurements and their interpretations. Thereby experiments performed even at quite similar conditions might lead to different and sometimes even contradictory conclusions. ‘Hidden’ structured species can also affect the thermodynamic parameters that characterize the difference between the initial and final states, because a given change of sequence or external conditions may affect stability of both the initial and final ensembles in the experiment.
We report MD simulation study of two possible short-lived intermediates of G4 folding, namely, the parallel-stranded DNA G-triplex and G-hairpin. Short-lived structures can contribute to the overall folding process provided they occur in bottlenecks of the folding pathways to the main kinetic basins. We have focused on the parallel species since our earlier computational studies suggested that the formation of parallel G-hairpins/triplexes is considerably more difficult compared to their antiparallel counterparts. We have even argued that the short lifetime of parallel-stranded G-triplexes might preclude them from any participation in the folding process (51,58,86). In this work, we partially revisit the latter assessment. We propose that parallel G-triplexes could act as transitory species in rearrangements between more stable structures on the landscape. However, their formation may still be bypassed by other types of structures even during folding of parallel G4 folds. We suggest that the parallel G-triplex is a rather broad ensemble of triplex-like species instead of being a single well-defined structure. However, well-defined parallel G-triplex species can be substantially stabilized by the interface between the G-triplex and double-helix or by flanking nucleotides. Even much larger stabilization may be achieved by stacking of flat aromatic ligands. Thus, our data reveal the influence of the surrounding structural context on the stability and free-energy landscape of the G-triplexes. In addition, we describe multiple folding pathways of the parallel G-hairpin from unfolded structures. All these pathways meet at the point of formation of a cross-hairpin structure, in which two G-tracts are hydrogen-bonded, but mutually rotated in a roughly perpendicular manner. It is then able to transform into the parallel G-hairpin; nevertheless, the cross-hairpin seems entropically more stabilized than the parallel G-hairpin. The results support the view that at the atomistic level of description a ‘single-molecule’ folding event of parallel-stranded G4 may proceed from cross-hairpin structures directly to cross-like triplexes or cross-like G4s and that the fully folded isolated parallel G-hairpins play only a minor role. Their formation during the process of folding event is not necessary. The data bring new ‘atomistic-level’ pieces of information into the G4 folding puzzle, mainly regarding the principles of transitions between the unfolded and folded molecules and amongst the central kinetic basins on the free-energy landscape. We have also attempted to complement the MD simulation data by NMR, PAGE and CD experiments, but our efforts confirmed that experimental detection of the target molecular structures remains very challenging.
MATERIALS AND METHODS
Starting structures for standard simulations
The structural stability of three types of systems was investigated in standard MD simulations. First, we studied parallel-stranded triplexes d(GGGTGGGTGGG), d(GGGAGGGAGGG) and d(GGGTTAGGGTTAGGG), herein referred to as T-T, T-A, and T-TTA, respectively. The T-A structure was derived from the G4 structure of d(TAGGGCGGGAGGGAGGGAA) (PDB ID: 2LEE (87); the triplex is underlined). T-T was modelled from the same G4 structure by mutation of the loops to T. T-TTA comprises two systems differing in the propeller loop conformation. T-TTA1 was derived directly from the G4 structure of d(AGGGTTAGGGTTAGGGTTAGGG) (PDB ID: 1KF1 (63)). T-TTA2 was extracted from a simulation of the full G4 1KF1.
Second, we investigated triplex interfacing with adjacent nucleotides, either a double helical DNA region or two flanking nucleotides. The duplex was emulated by ultra-stable antiparallel mini-hairpin d(GCGAAGC) (PDB ID: 1PQT (88)). The hairpin was linked directly to the 5′-terminus of the triplex and was stacked on its adjacent triad. The four triplexes described in the previous paragraph have been used in the hybrid constructs referred to as HT-T (Figure 2), HT-A, HT-TTA1 and HT-TTA2 (i.e. hairpin-triplex construct HT with corresponding bases in the triplex loops; HT-TTA1 and HT-TTA2 are often collectively referred to as HT-TTA). To study the effect of two flanking nucleotides linked to the 5′-terminus of the triplex, we dissected two thymines stacked on the G4 core of the hybrid-1 G4 (PDB ID: 2GKU (89), first two Ts) and connected them to the parallel triplex while keeping the orientation of the two Ts like in the 2GKU structure. We again used the four triplexes described above and the resulting structures are designated T2T-T, T2T-A, T2T-TTA1 and T2T-TTA2 (T2 stands for the flanking thymines). See Supporting Data for atomic coordinates of the constructs.
Figure 2.
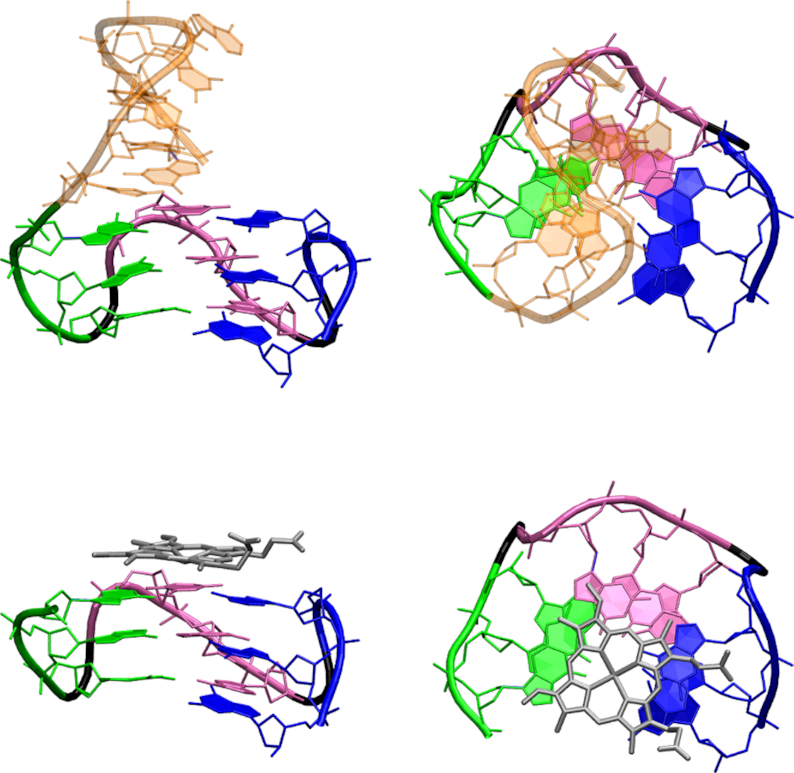
Top row: side view (left) and top view (right) of the starting structure of the hairpin-triplex system HT-T. Bottom row: side view (left) and top view (right) of the starting structure of the hemin-triplex system hemT-T. The hairpin part is orange, hemin is silver, the first G-tract of the triplex is green, the second one is purple and the third one is blue. The backbone of the propeller loops is black. The propeller loop nucleotides and two cations in the channel are not shown. Structures of the other constructs are analogous.
Third, we studied G-triplex with stacked hemin (protoporphyrin IX with FeIII). The hemin molecule was stacked at the 5′-terminus and did not contain a chloride ligand (Figure 2). We took the starting structures from 5 μs long explicit solvent MD simulations of the hemin–G4 complex, where G4 was either 1KF1 or 2LEE (both without the flanking bases). In the case of hemin-1KF1, we removed the last six nucleotides to obtain the triplex sequence identical to T-TTA and the resulting complex is referred to as hemT–TTA. To match the sequences of T-A and T-T, we took the hemin–2LEE complex, removed the first four nucleotides and took the structure as was or mutated the loop As into Ts; the complexes are labelled as hemT–A and hemT–T, respectively. The mutations should not affect the binding mode of hemin, as it is stacked on the 5′-terminal tetrad, while the mutated single-nucleotide loop As aim into the solvent. See Supporting Data for atomic coordinates of the models.
Standard MD simulations
Two ions were placed manually inside the triplex channel between the triplets, mimicking the G4-binding. The solute was solvated in a truncated octahedral box of water molecules with a minimal distance of 10 Å from the box border. TIP3P (90) and SPC/E (91) water models were used. For each water model, the system was neutralized by the addition of Na+ or K+ counter-ions and 0.15 M NaCl or KCl was added then. Joung and Cheatham ion parameters for TIP3P and SPC/E waters were used accordingly (92). The preparation was done in the xleap module of AMBER 16 (93).
MD runs
Each system (a combination of DNA + ions + water model) was simulated with two slightly different recent AMBER DNA force-field versions. One simulation has been performed with bsc0χOL4ϵζOL1 (which is parmbsc0 (94) + parmχOL4 (95) + parmϵζOL1 (96)) force field and one simulation with OL15 force field (bsc0χOL4ϵζOL1 further extended by parmβOL1 refinement (97)). OL15 is a complete reparametrization of all DNA backbone dihedrals compared with the Cornell et al. force field (98) and provides optimal (although not flawless) performance for both B-DNA (99) and G4 molecules (58). Both force-field versions appear to provide equivalent results for the studied systems and the investigated properties. Hemin was described by GAFF parameters (100) with charges derived by Shahrokh et al. (101).
All systems were equilibrated using a standard protocol (see Supporting Data for full details). Simulations were carried out using periodic boundary conditions, electrostatic interactions were treated by the PME algorithm and the non-bonded cutoff was set to 9 Å (102,103). The temperature was held at 300 K and the pressure at 1 atm using the Berendsen weak-coupling thermostat and barostat, respectively (104). The integration time step was set to 4 fs utilizing the hydrogen mass repartitioning (105). Covalent bonds involving hydrogen atom were constrained using the SHAKE (106) and SETTLE (107) algorithms for solute and solvent, respectively.
Triplex and hairpin-triplex systems were once simulated with the unmodified force field for 500 ns and once with the Hydrogen Bond fix (HBfix) potential function (108,109) for 1000 ns. HBfix is an additional locally acting potential increasing stability of the native GG hydrogen bonds (see Supporting Data for full details). Systems with two flanking Ts and hemin were always simulated with HBfix. The complete list of simulations of triplexes, hairpin-triplex, T2-triplex and hemin-triplex constructs is in Supplementary Tables S1–S4.
Replica exchange MD simulations
We used Replica Exchange Solute Tempering (REST2) (110,111) and Temperature Replica Exchange MD (T-REMD) (112). REST2 was used to study the folding of d(GGGAGGGAGGG), a potential G-triplex-forming sequence, and d(GGGAGGG) and d(GGGTTAGGG) corresponding to G-hairpin-forming sequences. Starting topologies and coordinates were prepared using the tLEaP module of AMBER 14 (113). Starting structures were constructed as ssDNA with the anti orientations of glycosidic torsions of all residues and solvated using a rectangular box with a 10 Å thick layer of SPC/E water molecules surrounding the solute (91). The simulations were performed in 0.15 M NaCl salt excess (114,115). Before the simulation, all replicas were minimized and equilibrated using standard equilibration protocol as described in Supporting Data. The simulations were carried out using AMBER 14 with bsc0χOL4ϵζOL1 force field (94–96) at a temperature of 298 K with 12, 12 and 16 replicas of GGGAGGG, GGGTTAGGG and GGGAGGGAGGG, respectively. The scaling factor (λ) values ranged from 1 to 0.6 for all systems, and were chosen to maintain an exchange rate of ∼20%. The effective solute temperature thus ranged from 298 to ∼500 K. The hydrogen mass repartitioning with a 4 fs integration time step was used. Each replica was simulated for ∼10 μs, obtaining a cumulative time of all REST2 simulations ∼400 μs (Supplementary Table S5). T-REMD simulations were used to study d(GGGAGGG) and d(GGGAGGGAGGG), with a cumulative simulation time of ∼380 μs (Supplementary Table S6). A detailed description of the T-REMD simulations is given in Supporting Data.
Native GG hydrogen bonding in GGGAGGGAGGG, GGGAGGG and GGGTTAGGG was supported using HBfix (see Supporting Data). Further, all enhanced-sampling simulations except the GGGAGGGAGGG T-REMD run were performed with restraints keeping the Gs in the anti region, referred to as the anti-G restraints. The aim was to prevent flipping of Gs to the syn orientation, and thus restricting the conformational space to target folding of the parallel-stranded structures. The anti-G restraint reduces kinetic partitioning by avoiding other types of the G-hairpin/triplexes on the free-energy landscape. The restraining potential is described in Supporting Data.
Clustering and cation binding analysis
The trajectories were clustered based on the ϵRMSD metrics (116) by a custom clustering algorithm (108,117). This metric is well-suited for monitoring differences in base-pairing and stacking and has a considerably better resolution than coordinate-based RMSD. In addition, we monitored cation binding inside the channel of different clusters. Details of the clustering procedure and cation coordination number evaluation are given in Supporting Data.
Experimental methods
Details about our (unsuccessful) attempt to determine the structure of T and HT constructs by 1H NMR and CD spectroscopy are given in Supporting Data.
RESULTS
Types of structures identified on the free-energy landscape
We have carried out a set of standard and enhanced-sampling simulations with the primary aim to study the potential role of parallel three-layer G-triplexes and G-hairpins in the folding of G4 molecules. Let us first provide an overview of key structures that will be discussed. Clustering of the standard simulations suggested eight triplex-like cluster families with similar structural features, i.e. structure types (Figure 3). By triplex-like structures, we mean molecular structures that possess at least some similarity with the fully paired parallel (‘native’) G-triplex. The identified structure types correspond to quite broad ensembles of structures rather than to a single conformation, perhaps with the exception of the G-triplex itself.
Figure 3.

Principal structures discussed in this work. (A) Schemes summarizing the key structural features of eight triplex-like cluster families seen in the standard simulations; the conformations depicted represent broader ensembles of structures. Gs are depicted as yellow rectangles, those in the back are darker. Other nucleotides are not shown. If Gs are stacked or roughly parallel, they have the same colour of the edge. The G4-like Hoogsteen GG base pairs are shown as a solid red line, while the dashed red line depicts any other hydrogen bonding. The overview of the cluster families and their populations is given in Supplementary Tables S7 and S8. Some sequences do not sample all the clusters families in the simulations, though this observation should not be over-interpreted in view of the limited simulation time-scale, due to which some structures can be missed. (B) Important intermediates with only two H-bonded G-tracts; the term ‘cross’ (‘cross-hairpin’) is analogously used also in the description of the replica exchange simulations of G-hairpins. (C) Examples of common non-triplex structures. Cross-hairpin structure (3′-cross) (left), antiparallel hairpin (middle) and circular structure with stacked 5′ and 3′ ends (right). First G-tract is green, the second one is purple and the third one is blue. The loop nucleotides are black.
In the first cluster family, the G-triplex, the first G-tract (1) is hydrogen bonded to the second G-tract (2), and G-tract 2 is also hydrogen bonded to the third G-tract (3). There is no bonding between G-tracts 1 and 3. The central channel characteristic for G4 remains intact in the G-triplex. The G-triplex resembles parallel G4 with one G-tract removed, and thus may be considered as the ‘native’ G-triplex. The second cluster family, the 1-3-2-G-triplex, also has three WC-Hoogsteen paired parallel G-tracts and the central channel. However, G-tract 3 binds both G-tracts 1 and 2, while there is no interaction between G-tract 1 and 2 (therefore the designation ‘1-3-2’, the G-triplex would be labelled ‘1-2-3’). The loop connecting G-tracts 1 and 2 spans a longer distance and runs through the space that would be occupied by a G-tract in a full G4. It could be called a propeller-diagonal loop; although this arrangement is not compatible with a complete G4 structure, it may be populated in triplex-like intermediates. Analogous 2-1-3-G-triplex was not sampled in our simulations.
The family of structures intermediate between the G-triplex and the 1-3-2 G-triplex is named the symmetric triplex. It has direct hydrogen bonding of all three G-tracts and may contain symmetric triads (86). The symmetric triplex ensemble has a bit variable channel, which may accommodate cations, depending on the exact type of base pairing between the bases. The last significant cluster family closely related to the G-triplex is the G-triplex-1|51. It is essentially the G-triplex with G-tract 1 slipped by one base in the 5′ direction (G-tract 1 | 5′ direction, 1 base).
All four above-described cluster families feature all three G-tracts in parallel orientation and utilize mostly Hoogsteen hydrogen bonding. The remaining triplex cluster families have at least one G-tract in a different position, and they are thus called cross-like triplexes. The most common cross-like arrangement is the 3-cross. In this structure, G-tracts 1 and 2 remain Hoogsteen paired like in the G-triplex, while G-tract 3 is rotated and uses Hoogsteen or Watson–Crick edge of its Gs to form hydrogen bonds with the first two G-tracts. Gs in G-tract 3 are not strictly parallel, so one of the Gs in G-tract 3 sometimes forms a native G-triad with Gs from G-tracts 1 and 2. This ensemble has poorly developed central ion channel. Analogously, the 1-cross family has G-tracts 2 and 3 parallel, while G-tract 1 is rotated. In the family called 2-cross-3|51, G-tracts 1 and 3 are parallel and Hoogsteen paired with G-tract 2 rotated, but G-tract 3 is slipped by one base in the 5′ direction relative to G-tract 1. In addition, the last G of G-tract 3 is stacked below the last G of G-tract 2. The most loosely defined cluster family, the cross-cross, contains structures in which all three G-tracts are mutually rotated, and each G-tract is hydrogen bonded to the other two by either Hoogsteen or Watson–Crick edges. There is no central channel.
Besides the above-described triplex-like structures, many other arrangements were observed in the simulations. They include intermediates of G-triplex unfolding in standard MD simulations, as well as intermediates in folding found in the enhanced sampling simulations. The most prominent ones are cross-hairpin structures, i.e. a cross-like arrangement of two G-tracts with the third G-tract unbound. Cross-hairpins may be formed by G-tracts 1 and 2 (designated as 5′-cross) or by G-tracts 2 and 3 (3′-cross) (Figure 3; note the different meaning of 3′-cross [cross-hairpin] and 3-cross [cross-like triplex]). The two crossed G-tracts have at least two stacked Gs each that are mutually rotated and hydrogen-bonded through their WC or Hoogsteen edge so that the overall structure resembles a cross. Though the structure with perpendicular orientation of the G-tracts is common, their arrangement is flexible and they often adopt a more parallel-like orientation, resulting in a broad ensemble of structures rather than a single well-defined conformation. Upon complete folding of the propeller loop, the 5′-cross forms a parallel G-hairpin with G-tract 3 unbound, called the 5′-G-hairpin (Figure 3). Similarly, the 3′-cross folds into the 3′-G-hairpin.
Other observed structures include the ss-helix and various non-G4-resembling arrangements, such as antiparallel hairpins or circular structures with two ends stacked together (Figure 3). All clusters found in all the simulations are visualized in Supporting Data in sections with a detailed description of each system (see below). Numerous other conformations did not pass the 1% cluster threshold. Their description is outside the scope of this work, though they illustrate the overwhelming richness of the free-energy landscape of the studied molecules.
Standard simulations of isolated G-triplexes
Let us first analyse standard simulations starting from the isolated folded G-triplex structures (Figure 4 – top, Supplementary Figure S1 and Supplementary Table S1). All results reported in the main text use the HBfix potential to support the native GG base pairing. Without using HBfix, the native G-triplex structures disintegrate quickly (Supplementary Tables S7 and S9). This is consistent with underestimation of stability of the GG base pairing by the standard force field reported by us (58,86,118) and confirmed later by others (119). We thus describe the simulations without HBfix only in Supporting Data. HBfix increases structural stability (lifetime) of the triplex-like structures sufficiently, and we assume in the right direction.
Figure 4.

Triplex clusters sampled in simulations of the T-T, T2T-T, HT-T and hemT-T systems with HBfix; see Supplementary Figures S1–S4 for data obtained for the other systems. Two stripes are shown for each simulation. The upper shows cluster type (cf. Figure 3; not all triplex-like structures are sampled) at a given time, while the lower denotes the number of cations with coordination number to guanine O6 atoms equal or greater than four. The molecules are able to interconvert between different clusters during the course of the simulations. For the simulation numbering see Supplementary Tables S1–S4.
The average total triplex population ranged from 70% to 90%; by total triplex population, we mean aggregate sampling of all cluster families from Figure 3A. The 3-cross was the most populated cluster family for T-T (Figure 4, top) and T-A, accounting for 64% and 77% of all their conformations, respectively. With the T-TTA system, the 3-cross was adopted in 34% of simulation time. The cross-cross was populated by ∼8% regardless of the loop sequence. The G-triplex populated ∼9% in T-T and T-A simulations, and ∼23% in T-TTA simulations. Importantly, the G-triplex could form throughout the whole simulation course, reversibly partially unfolding and refolding (Figure 4, Supplementary Figure S1 and Supplementary Table S9). The T-TTA system marginally sampled also the symmetric triplex (1%) and the 1-cross (4%). The average populations display high variance (even dozens of %; Supplementary Table S8), demonstrating the stochastic nature of the individual simulations (Figure 4 and Supplementary Figure S1). More simulation details are given in Supporting Data. We did not attempt to prolong these standard simulations due to the complementary insights obtained by the enhanced sampling simulations (see below). We emphasize that the populations discussed in this paragraph do not correspond to equilibrium populations and merely summarize that structures were sampled on the 1 μs time-scale when starting from the G-triplex structure. The simulations would indicate overall lifetime of the triplex-like ensembles around 1 μs; for example, for the T-T system shown in Figure 4 three simulations out of eight are unfolded after 1 μs. However, the absolute lifetime values indicated by our MD should be taken with a great care.
Duplex-triplex junction and flanking nucleotides visibly stabilize the G-triplex ensemble
Hairpin-triplex (HT) constructs (Figure 2), which emulate protrusion of G-rich ssDNA from dsDNA, are profoundly more stable (i.e. have considerably longer lifetime) than the isolated triplexes (cf. Figure 4 HT-T versus T-T, Supplementary Figure S2 versus S1, Supplementary Tables S8 and S10). The average total triplex ensemble population in the hairpin-triplex constructs exceeded 90% for all loop sequences (Supplementary Table S8), and with only low variance, which illustrates that triplex families had a high population in most of the simulations. The most populated structure was the native G-triplex, accounting for about half of the simulation time for all loop sequences. The population of the 3-cross is similar for HT-T and HT-TTA (25% and 29%, respectively), while it is higher for HT-A (45%). On the other hand, while the cross-cross is populated similarly by HT-T and HT-TTA (13% and 10%, respectively), it is not sampled by HT-A at all. Similarly to T-TTA, HT-TTA can adopt the conformation of the 1-cross (5%). The simulations are able to reversibly interconvert between different cluster families (Figure 4 and Supplementary Figure S2).
5′-Flanking nucleotides have a similar stabilizing effect, though more loop-sequence-dependent (Figure 4, Supplementary Figure S3 and Supplementary Table S11). The population of the G-triplex in T2T-T and T2T-A is 40% and 28%, respectively, while it reaches 68% in T2T-TTA (Supplementary Table S8). On the other hand, the population of the 3-cross is 39% and 51% in T2T-T and T2T-A, respectively, but only 10% in T2T-TTA. Other minor clusters populated by all the T2T constructs are the 1-cross and cross-cross. The G-triplex stabilization in T2T-TTA can be attributed to formation of base pairs between the flanking Ts and easily accessible loop nucleotides. Such base pairs stacked on the terminal G-triplet and supported the whole G-triplex. Thus, while the flanking duplex of HT systems stabilizes the triplex ensembles quite uniformly by stacking, flanking nucleotides of the T2T systems affect the triplex also via sequence-dependent alignments with loop nucleotides. Further details are given in Supporting Data.
Hemin binding leads to profound stabilization of native G-triplex
Even much larger stabilizing effect was achieved by hemin stacking. It resulted in essentially 100% population of the G-triplex (Figure 4; Supplementary Figure S4 and Supplementary Table S8). Hemin remained stacked on the triplex in all the simulations regardless of the loop type; however, the dominant hemin position was different from the starting G4 one. The initial position can be characterized by the coaxial alignment of the channel cations and the FeIII cation, while in the dominant G-triplex conformation hemin is slid sideways (Figure 5). It increases the stacking overlap between hemin and the G-triplex. One carboxylic group of hemin forms two hydrogen bonds with the available WC edge of G beneath it, and one of the two oxygen atoms of the carboxylic group is also able to coordinate the upper channel cation, mimicking O6 of a fourth G in full G-tetrad (Figure 5). When hemin is not in the dominant conformation, i.e. it is not hydrogen bonded to the G, it rotates while remaining stacked.
Figure 5.
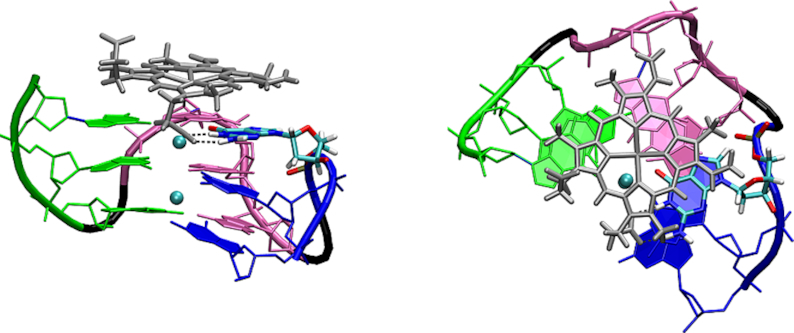
Side (left) and top view (right) of the dominant hemT-A complex conformation. One carboxylic group of hemin forms two hydrogen bonds the first G of the last G-tract (two black dashed lines, the G is highlighted). Hemin is silver, the first G-tract of the triplex is green, the second one is purple, the third one is blue, the loop backbone is black and the channel cations are cyan. The dominant structure of hemT-T and hemT–TTA is analogous.
Cation binding
In the simulations, the G-triplex channel binds mostly one or two cations in the G4-like manner. When fluctuations of the G-triplex are attenuated (e.g. by hemin), binding of two cations dominates. Binding of two cations to other triplex-like structures is scarce. The 3-cross and cross-cross either bind one or no cation (Supplementary Table S12). More details are given in Supporting Data.
G-triplex folding is not observed in GGGAGGGAGGG REST2 simulation, but parallel G-hairpin folding events are common
To study the folding of G-triplex, we ran a REST2 simulation starting from the unfolded ss-helical structure. Full G-triplex folding was not reached, nor did we observe the formation of any of the triplex clusters shown in Figure 3A. Nevertheless, there was a clear tendency to form G-hairpin arrangements within the context of the GGGAGGGAGGG sequence, namely, the cross-hairpin structures (Figures 3B and 6). A broad ensemble of the 3′-cross accounted for 22.4% population of the reference replica. The 5′-cross had a population of 2.7%. Importantly, we have even detected the formation of parallel native G-hairpins from these cross-hairpins. 3′-G-hairpin and 5′-G-hairpin (Figures 3B and 6) formed 0.3% and 0.1% populations, respectively, of the reference replica. The remaining population of the reference replica corresponded to various non-G4-supporting antiparallel-hairpin-like and ss-helix-like conformations. Structures across all replicas were divided into 13 clusters, and 58% of them were not assigned (Supplementary Figure S5, Supplementary Tables S13 and S14).
Figure 6.
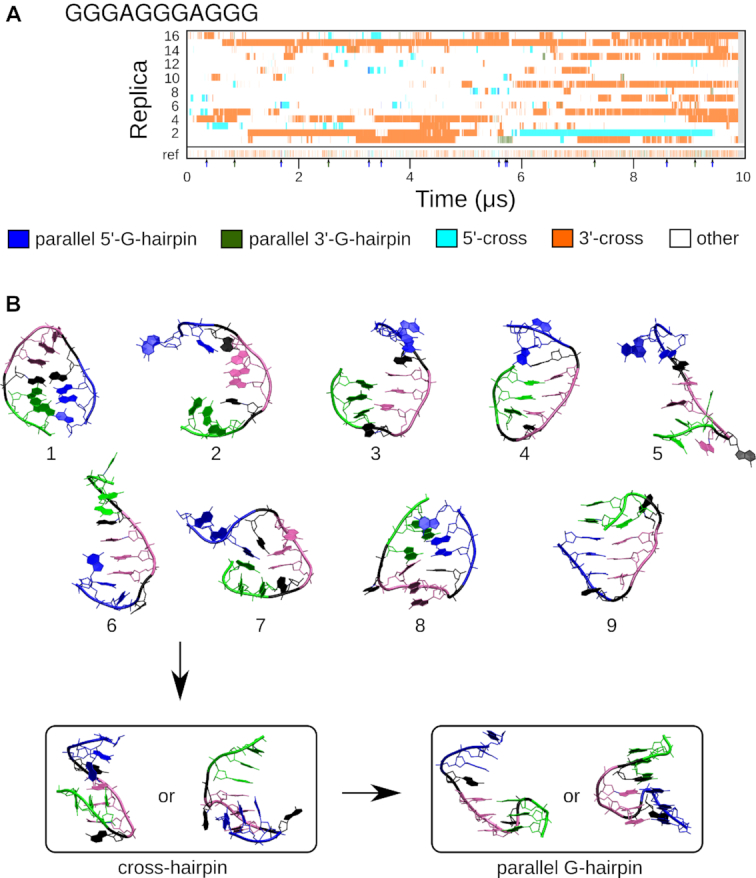
(A) G-hairpin clusters populated in the REST2 GGGAGGGAGGG simulation. Each numbered line represents one continuous (demultiplexed, coordinate-following) replica; the discontinuous reference replica collecting the unbiased populations is at the bottom of the graph. Parallel G-hairpin occurrences in the reference replica are highlighted by arrows below the reference replica line. (B) G-hairpin folding pathways. Nine different structures that led to folding of parallel G-hairpin through cross-hairpin structures involving two G-tracts. The structures are briefly described in the text. The first G-tract is green, the second one is purple, the third one is blue and the loops are black.
Observed G-hairpin folding events (Figure 6) always involved prior formation of the cross-hairpin structure. The last step of the folding mechanism consists of counter-rotation of two G-tracts of the cross-hairpin structure directly into the parallel G-hairpin. Nine different precursors of the cross-hairpin structures were identified (Figure 6): (i) long antiparallel hairpin with crossed G-tracts 1 and 3; (ii) open structure with G-tract 1 approaching G-tract 2 from side; (iii) antiparallel hairpin, (iv) antiparallel hairpin with a base-phosphate (BPh) interaction between G3 and G5; (v) fully unfolded structure; (vi) ss-helix-like structure; (vii) antiparallel hairpin enclosed in a shortened circular structure; (viii) long antiparallel hairpin without well-defined base pairs; (ix) antiparallel hairpin enclosed in a circular structure with 5′ and 3′ ends stacked together.
Although we did not see the formation of the full parallel G-triplex, the simulation shows a clear trend to form cross-hairpin structures between two consecutive G-tracts and parallel G-hairpins. The lack of a folding event to the G-triplex may be due to limited simulation time and the need to fold both parallel G-hairpins simultaneously; the issue will be commented more in ‘Discussion’ section.
Propeller loop folding observed in REST2 simulations of GGGAGGG and GGGTTAGGG
We finally studied simpler systems containing only two G-tracts, thus capable of G-hairpin formation. For these systems, we expected better sampling of the conformational space. The most populated state of the REST2 simulation of the GGGAGGG sequence, was the cross-hairpin ensemble (two crossed G-tracts like in the triplex sequence simulation, but obviously without a third G-tract), which accounted for 44% of all observed structures of the reference (unbiased) replica (Figure 7; Supplementary Figure S6, Supplementary Tables S15 and S16). About half of the remaining structures are ss-helices. The parallel G-hairpin represents 0.6% of the unbiased population collected at the reference replica. Despite the low population, the folding is statistically significant, because we observed at least one complete G-hairpin folding event in every continuous replica (Figure 7). Nevertheless, the parallel G-hairpin always quickly returned into the cross-hairpin ensemble. Given this and the populations in the reference replica, the parallel G-hairpin is (in the force field approximation) less stable than the cross-hairpin ensemble.
Figure 7.
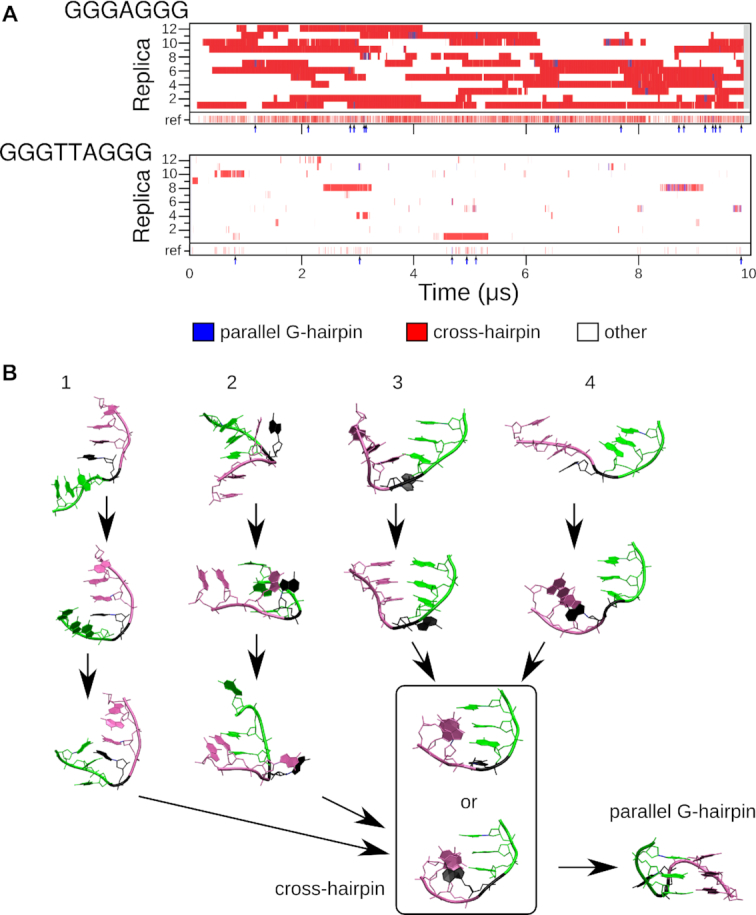
(A) G-hairpin clusters populated in REST2 simulations. Each numbered line represents one continuous (demultiplexed, coordinate-following) replica; the discontinuous reference replica collecting the unbiased populations is at the bottom of each graph. Parallel G-hairpin occurrences in the reference replica are highlighted by arrows below each reference replica line. (B) Four independent pathways leading to cross-hairpins and then to the parallel G-hairpin in the REST2 simulation of GGGAGGG. The first G-tract is green, the second one is purple, and the loop is black.
The folding events are analogous to those described for GGGAGGGAGGG (Figure 6). The last step is the formation of the parallel G-hairpin from the cross-hairpin ensemble. We identified four mechanistically different pathways leading to the cross-hairpins (Figure 7). In pathway 1, a slipped antiparallel hairpin is formed as an intermediate between the unfolded state and the cross-hairpin. Pathway 2 starts from an ss-helix-like structure with G5 and A4 bulged out. This structure transforms into the cross-hairpin, initially with the participation of only G2, G3, G6 and G7 bases. Nevertheless, G1 and G5 eventually stack on the other Gs, and a proper cross-hairpin structure is formed. Pathway 3 begins by the formation of an antiparallel hairpin with one base pair, followed by an antiparallel hairpin with three base pairs. Its G-tracts then rotate into the cross-hairpin. Finally, pathway 4 starts by bending of the ss-helix, initiated by the formation of a BPh interaction between the G3 amino group and G5 phosphate. After the bending, the BPh interaction is lost, and the structure reaches the cross-hairpin ensemble. Two different cross-hairpin structures are possible intermediates, one with its loop A stacked with the first G-tract, and the other one with the A stacked with the second G-tract.
The results obtained by the T-REMD simulations of GGGAGGGAGGG and GGGAGGG also show the key role of the cross-hairpin ensembles and thus corroborate the findings from the REST2 simulations. Details about the T-REMD simulations are given in Supporting Data.
Conformational space sampled by GGGTTAGGG is broader than that of GGGAGGG, resulting in decreased populations of the cross-hairpin ensemble and G-hairpin (Figure 7; Supplementary Figure S7, Supplementary Tables S17 and S18). Still, the G-hairpin folding mechanisms were similar to those found in GGGAGGG (Supplementary Figure S8). The REST2 simulation is described in more detail in Supporting Data. Obviously, the anti-G restraints purposely prevented the GGGTTAGGG sequence from the formation of antiparallel G-hairpins, i.e. we focused the simulation on the folding funnel pertinent to the parallel G-hairpin. We have demonstrated elsewhere that without these restraints the sequence prefers folding into diverse antiparallel hairpins (51).
NMR and CD spectroscopy
To mimic the design of solution experiments used for structural characterization of the thrombin binding aptamer triplex (120,121), we attempted to characterize the studied T and HT constructs using NMR and CD spectroscopy (Supplementary Figures S9 and S10). However, with a single exception represented by HT-TTA, our efforts were halted by apparent oligomerization of the studied constructs, as shown by control native PAGE experiments (Supplementary Figure S11). The imino region of the 1D 1H NMR spectrum of HT-TTA (Supplementary Figure S9) was marked by strong signal(s) from the GCGAAGC hairpin and by rather diffuse, broad and overlapped multiple signals of very low intensity in the region specific for imino protons involved in Hoogsteen base pairing spanning from ∼10.5 to 12 ppm. The overall appearance of this spectral region indicates co-existence of multiple, dynamic species stabilized by Hoogsteen type of hydrogen bonding. In all other cases, the structural interpretation of these experiments would only be highly speculative, so we refrain from drawing any conclusions. Despite that our attempts to experimentally characterize potential triplex intermediates were unsuccessful, we report the results to highlight the major challenges in the experimental characterization of transient intermediates that may participate in G4 folding. See Supporting Data for further information.
DISCUSSION
We have carried out a series of MD simulations (cumulative time 929 μs) to investigate properties of three-layer parallel-stranded DNA G-triplexes and G-hairpins, which are expected to contribute to free-energy landscapes of three-tetrad G4 systems. The standard simulations aim to probe structural stability of the G-triplexes once already formed, while the enhanced-sampling REST2 and T-REMD simulations seek for the folding pathway of parallel G-hairpins and G-triplexes from the unfolded state. We have focused on parallel G-triplexes with two propeller loops since they represent a considerably more challenging system than antiparallel G-triplexes. This assessment is based on previous MD studies which revealed that parallel G-triplexes are structurally less stable (have shorter lifetime) than antiparallel G-triplexes, while the formation of propeller loops in simulations is considerably more difficult compared to lateral and diagonal loops (58,86).
The parallel G-triplex is represented by a broad ensemble of rapidly interconverting structures
The simulations have shown that the triplex-forming sequences can populate a broad ensemble of parallel-triplex-like conformations. We have identified eight such structural families (Figure 3). The most important are the ‘native’ G-triplex, which has its three G-tracts paired like in a G4, and the 3-cross, which differs from the G-triplex by the position of the third G-tract, being rotated to the other two. We have also observed other less populated cluster families, closely related to either the G-triplex or other cross-like triplex structures. Our simulations thus predict that the local part of the free-energy landscape pertinent to parallel G-triplexes corresponds to a broad range of quickly interconverting structures rather than to a well-structured native-like G-triplex. Analysis of ion binding has shown that cross-like triplexes could be the folding state at which one albeit weakly interacting cation gets bound to the forming channel.
Adjacent DNA sequences and ligands can substantially stabilize the parallel G-triplex
All simulations indicate that parallel G-triplexes and G-hairpins would be only marginally populated when being isolated (Figures 4 and 6; Supplementary Figure S1 and Supplementary Table S8). However, standard simulations show that flanking DNA elements (canonical duplex – G-triplex and flanking T2 – G-triplex junctions in our study) and mainly interaction with ligands can significantly decrease unfolding rate (and thus perhaps increase populations) of parallel G-triplexes compared to their isolated forms. The effect of the flanking elements or ligand-triplex interface is demonstrated by substantially prolonged lifetimes of the starting G-triplex structure in standard simulations (Figure 4; Supplementary Figures S2–S4 and Supplementary Tables S9–S11). The presence of a flanking canonical base pair or a base pair formed between a flanking nucleotide and a sufficiently long loop at the 5′ end of the triplex stabilizes the triplex by stacking with the terminal triad; the stacking remains preserved in simulations not only for the ideal (native) parallel G-triplex but also for many other triplex-like cluster families depicted in Figure 3.
Hemin binding dramatically increases the structural stability of the G-triplex. Its stacking and formation of two hydrogen bonds with the G-triplex provided such stabilization that the G-triplex was entirely stable in all simulations (Figure 4 and Supplementary Figure S4), i.e. its lifetime is already well beyond the simulation time-scale. Similar stabilization can also be expected for other flat aromatic ligands.
Based on these results we suggest that parallel G4-like arrangements of G-tracts with propeller loops can, in general, be stabilized and substantially populated when embedded into longer DNA strands when additional DNA nucleotides provide a stacking platform. The stacking platform could also be a double-helix or a flat aromatic ligand, similar to those employed for stabilization of parallel-stranded G4s. It is possible that some ligands could alter the G4 folding process by benefiting states with the parallel-stranded G-hairpin or G-triplex and decreasing the probability of formation of alternative G4 structures. However, although the data suggest that dsDNA or ligands stabilize parallel-stranded G-triplex, the same factors may also stabilize other G-triplex folds and even some G4-unrelated structures. In other words, we cannot distinguish kinetic (longer lifetime) and thermodynamic (full competition with other states) stabilization of the parallel-stranded G-triplex by the presented MD simulations. Experimental studies are scarce and do not provide detailed insights. Stabilization effect of dsDNA on the antiparallel chair-like G4 structure (122) and the effect of proximal dsDNA on G4 folding and shifting G4 equilibrium distribution in general have been documented (45). The sequence d[(GGGTT)3GGG] speeds up its conversion from an antiparallel to parallel G4 in Na+ environment upon K+ addition and also its folding into the parallel G4 from the unfolded state when flanking Ts are attached to the sequence (123). The promotional effect of ligands on the formation of G4 by stabilization of supposedly antiparallel G-hairpin and (2+1)hybrid G-triplex intermediates has been suggested by DNA origami experiments (76).
Parallel G-triplex could contribute to the DNA G4 folding landscape as transitory species, but is not an obligatory intermediate
Earlier MD studies analysing potential intermediates in folding of tetramolecular and intramolecular parallel G4 DNA structures identified the tendency of the G-tracts to form cross-like interactions rather than the perfectly parallel G4-like arrangements (49,124). Thus, cross-hairpin structures, diverse cross-like triplexes and cross-like four-stranded ensembles have been tentatively suggested as transient species populated during the G4 folding. Various cross-like G-tract interactions may participate in diverse hypothetical coil-like ensembles (ensembles of collapsed compacted structures lacking well-defined G-tetrads (58,124)), from which the folding process may proceed towards fully structured G4 via a series of continuous rearrangements (conformational diffusion) of the H-bond networks (56,58).
The present simulations provide a considerably refined picture of these potential folding intermediates with substantially improved force-field description and several orders of magnitude better sampling. We suggest that parallel G-hairpin and G-triplex DNA structures, once formed, would correspond to dynamic ensemble rather than to precisely defined intermediate conformations of the folding. We propose that the cross-like triplexes and parallel G-triplex (Figure 3) readily interconvert and both can participate in nucleation towards the G4 structures in the context of the full G4-forming sequence. Approaching of the fourth G-tract to the cross-like triplexes might result in the formation of a cross-like G4 ensemble, which we have observed in our previous G4 unfolding study (49). The cross-like G4 ensemble can subsequently convert into a G4 upon G-tract rotations, concurrent stabilization by ions and possibly strand slippages. Figure 8 depicts a simplified late-stage folding pathway network in the vicinity of the G-triplex that is consistent with the MD data. If the fourth G-tract binds to the G-triplex, a slipped or full G4 could be formed directly. Similarly, approaching of the fourth G-tract to the G-triplex-1|51 would lead to the formation of native G4 structure upon strand slippage (49,124). However, the G-triplex intermediate can be easily bypassed via other structures, which are in the figure represented by one of the cross-like G4 structures, which itself could form by merging two cross-hairpins. Cation binding is not shown in Figure 8, though our data suggest that cross-like triplex structures are capable of cation binding in the forming central channel; the G-triplex is able to bind two channel cations but is also stable with only one (Figure 4; Supplementary Figures S1–S4 and Supplementary Table S12). The picture of triplex-like structures likely being among the first intermediates that capture cations specifically is in agreement with earlier simulation and experimental data (42,50,86,125). We emphasize that the possible mechanism in Figure 8 is still very simplified and many more structures may appear on the landscape. For example, no syn–anti dynamics of the interacting Gs is assumed. In reality, the single-molecule G4 folding events may proceed through an overwhelmingly broad and multidimensional spectrum of micro-pathways and do not need to dominantly include any salient parallel structures with G4-like triads and tetrads. Balance between the individual micro-pathways likely depends on sequence and environmental conditions.
Figure 8.

Example of possible late-stage folding transitions leading to the parallel-stranded G4. The 3-cross structure is selected as a representative of the cross-like triplex structures. The figure depicts a part of the folding landscape connecting cross-hairpin structures to the target parallel G4 via diverse pathways including those bypassing the parallel G-triplex. The presence of cross-like and slipped G4 structures, although not directly simulated in this work, is fully consistent with the simulations. These structures have been documented in earlier unfolding simulations of parallel-stranded G4 stems (49). See the legend to Figure 3 for the colour explanation.
Our REST2 and T-REMD folding simulations were relatively abundant in parallel G-hairpin folding events (Figures 6 and 7). This indicates that parallel G-hairpins can be straightforwardly accessed from the unfolded state, though we have suppressed the competition of all structures with different syn–anti G-tract patterns. However, the population of parallel G-hairpins was low, unlike the much more abundant ensemble of cross-hairpin structures. When considering the simulations and also their limitations (see below), we suggest the following interpretation of the data. The G-triplex state would correspond to a broad spectrum of fast-interconverting conformations rather than to a fully paired G4-like G-triplex (Figure 3). When comparing with simulations of complete cation-stabilized G4 folds (58), the parallel G-triplex has very short lifetimes and thus does not seem to be capable of forming major kinetic traps. Therefore, parallel three-layer G-triplex-like ensembles can only act as transitory intermediates during folding into some G4 basins from the unfolded state and during transitions between different G4 folds (cf. Figure 1). This suggestion (58,86) is consistent with the latest fluorescence-force experiments, which indicate that if there is the G-triplex present during the folding, it does not correspond to a deep-enough free-energy minimum, and that its lifetime is shorter than 20 ms (82). Nevertheless, the triplexes could still contribute to the folding rate to the individual G4 basins, when contributing to the bottleneck of the process, and their role may be sensitive to the sequence context, ligand binding and other external factors; our simulations with hemin show that lifetime of parallel G-triplexes can be increased perhaps even by several orders of magnitude. In general, the ensemble of structures corresponding to the unfolded state of G4 molecules can be dramatically modulated by the environment.
An intramolecular parallel G4 could fold via cross-hairpin structures and then cross-like triplexes or the G-triplex structure (Figure 8). A bimolecular parallel G4 might prefer folding by a merge of two cross-hairpins into a cross-like G4 state. Nevertheless, the presence of a suitable ligand could modulate the free-energy landscape also for the bimolecular and tetramolecular G4s and promote folding via the parallel-stranded G-triplex. In conclusion, this work suggests that the parallel G-triplex may transiently participate during the parallel G4 folding. However, it is not an obligatory intermediate (or transitory) structure for the process (Figure 8), and its actual participation and lifetime may vary based on other circumstances.
The past simulation studies have revealed that antiparallel G-triplexes and G-hairpins supporting lateral and diagonal loops are structurally more stable and more easily accessible than the parallel G-hairpins/triplexes supporting propeller loops (51,58,86,120). Thus, although this study has been focused on parallel G-triplex, we suggest that also antiparallel G-triplex ensembles may readily participate as transitory species in folding pathways of other G4-folds and are more populated than the parallel G-triplex ensembles.
The simulation technique alone is not capable of providing complete coverage of the G4 folding. Nevertheless, the richness of structures and structural transitions already documented in MD simulations (38,47,49–53,55–57,86,119,120,126–128) caution against interpreting the G4 folding mechanisms using too simple models based on a few straightforward and structurally appealing intermediates.
Limitations of MD simulations
The MD simulation technique is not capable of providing a complete picture of G4 folding, due to a number of limitations (58,109). However, we have carefully taken all these limitations into account while analysing the data, and we propose that all our suggestions regarding properties and possible roles of parallel G-triplexes are justified. First limitation is sampling. Although our standard simulations cannot provide comparison of stabilities in the thermodynamic sense as the simulations are only able to explore the conformational space near the G-triplex, they are sufficiently long to estimate lifetime (1/koff) of the G-triplex in different structural contexts. Converged REST2 and T-REMD simulations would be able to provide equilibrium populations (correct thermodynamics) at each replica of the replica ladder (and thus even indicate molecule’s melting profile), but due to hardware limitations this goal is currently unachievable (51,56,109,129).
Second source of limitations is the force field. We have used local HBfix potential function to moderately bias the simulations in favour of the native GG H-bonds in parallel G-triplexes and G-hairpins, as these interactions are known to be underestimated by the force field (58,86,118). In four out of five enhanced-sampling simulations, we also prevented sampling of the syn conformation of Gs to reduce competition with the antiparallel structures. Still, we assume that the simulations underestimate the stability (lifetimes) of the studied parallel-stranded structures, as we did not use any means to stabilize the propeller loops. Excessive instability of propeller loops in MD has been broadly discussed in the literature though its origin is not yet unambiguously clarified (51,58,86,130). MD description of cation binding in G4 channels is also imperfect, mainly due to lack of polarization (58,131–133). While this issue does not significantly affect simulations of fully folded G4 molecules due to their extraordinary long lifetimes (58), the G-triplex might be under-stabilized in MD simulations. Also binding of the ions is assumed to be underestimated, especially in case of binding of two or more closely-spaced ions (131). More thorough discussion of all the limitations is presented in Supporting Data.
The ambiguity of experimental data
The complexity of intramolecular G4 folding, experimentally manifested by co-existence of multiple quasi-stable (pre-) folded species that transit at time-scales ranging from sub-microsecond to hours, generally prevents isolation and structural characterisation of individual intermediates. Although the G4 folding has been extensively studied under near-to-physiological conditions by CD and NMR spectroscopy, FRET, DNA origami with AFM, and optical or magnetic tweezers (27,29,32,34–41,43,45,47,76,84,85,134), these studies have been unable to provide unambiguous prove for existence of G-triplex and/or G-hairpin due to either inherently low-structural resolution (CD, FRET, AFM, tweezers), need for covalent tagging or binding that can potentially influence G4 folding pathway (FRET, DNA origami), or due to inability to observe low-populated species and limited time-resolution (NMR). The only resolved atomistic G-triplex structure thus far is the antiparallel two-tetrad one derived from the thrombin binding aptamer G4 (120,121). Interpretations of some published primary experimental data may have been affected by prejudice in favour of the G-triplex or other straightforward, structurally appealing intermediates. Experiments attempted in the framework of our study were meant to shed light on the possibility of formation of monomeric secondary (G-triplex-like) structures. Instead, they demonstrate existing difficulties encountered in studies of G4 folding. This, however, is not surprising. Experimental characterization of transitory ensembles populated during the G4 folding events is even more challenging than studies of metastable states (species) for both ensemble and single-molecule experiments. MD simulations indicate extreme structural richness of such ensembles with very fast structural interconversions. Their monitoring would require an experimental method capable to resolve time-development of populations of specific GG base pairs (i.e. atomic resolution) within heterogeneous ensembles or during very fast transitions. We reiterate that the richness of the spectrum of structures and transitions suggested by the MD simulations is still likely significantly underestimated compared to the real molecules, due to the sampling limits. Despite their elusiveness, the transitory ensembles on the free-energy landscapes of G-quadruplexes are essential for the folding process and its sensitivity to external conditions, ions, ligands, etc. Although they are populated in only short time periods, they correspond to those processes where the molecules undergo the conformational changes, while in the more easily detectable and more populated metastable states they stably reside in just one usually well-defined conformation. Thus, for example, the network of individual atomistic folding pathways decides about the (initial kinetic) probability with which different metastable G-quadruplex folds emerge from a given unfolded state. MD method, despite all its limitations, is currently the best tool to obtain at least some information about structural properties of the transitory ensembles. More discussion can be found in Supporting Data.
CONCLUSIONS
Parallel-stranded DNA G4s are the most common G4 types. However, their folding pathways remain not fully understood. In this study, we used MD simulations to assess the structural stability and variability of the putative parallel-stranded G4 folding intermediates, namely the parallel G-triplex and G-hairpin. By accumulating almost one millisecond of MD simulations, we characterized the conformational state in the vicinity of the parallel G-hairpin and G-triplex, as well as visualized their connection with the unstructured part of the unfolded ensemble. The simulations suggest that the parallel G-hairpin and G-triplex belong to a broader and dynamical ensemble of structures that also includes a wide spectrum of imperfectly paired structures with not fully parallel or even perpendicular arrangement of the G-tracts, with a fast exchange of conformations within the ensemble. In addition, parallel all-anti G-hairpins/triplexes appear in simulations less stable than the corresponding antiparallel structures with alternating syn and anti G orientations. On the other hand, the parallel G-triplex can be decisively stabilized by a stacked aromatic ligand and to a certain extent also by a flanking duplex region or flanking nucleotides. Overall, the simulations are consistent with possible but rather transient role of parallel G-triplexes in the folding of parallel G4. In addition, well-structured G-triplex is not an obligatory folding intermediate or transitory species since it can be bypassed by transitions via other less ordered structures in a multi-pathway process.
Supplementary Material
ACKNOWLEDGEMENTS
We thank to Martin Gajarsky for providing us the data used for Supplementary Figure S30, acquired during the study of the Saccharomyces G-hairpin sequence (135). Ministry of Education, Youth and Sports of the Czech Republic is acknowledged for their support of access to research infrastructure [CIISB-LM2015043].
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Czech Science Foundation [16-13721S to P.S., J.S.; 17-12075S to L.V., L.T.]; project SYMBIT: European Regional Development Fund [CZ.02.1.01/0.0/0.0/15_003/0000477 to L.T., J.S.]; ERDF/ESF project ‘Nanotechnologies for Future' [CZ.02.1.01/0.0/0.0/16_019/0000754 to P.K., P.B., M.O.]; project CEITEC 2020 [LQ1601] with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Programme II. Funding for open access charge: Institute of Biophysics of the Czech Academy of Sciences, v.v.i.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-Quadruplex structures in the human genome. Nat. Biotech. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 2. Huppert J.L., Balasubramanian S.. G-Quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007; 35:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huppert J.L. Structure, location and interactions of G-Quadruplexes. FEBS J. 2010; 277:3452–3458. [DOI] [PubMed] [Google Scholar]
- 4. Lipps H.J., Rhodes D.. G-Quadruplex Structures: In vivo evidence and function. Trends Cell Biol. 2009; 19:414–422. [DOI] [PubMed] [Google Scholar]
- 5. Mendez-Bermudez A., Hills M., Pickett H.A., Phan A.T., Mergny J.-L., Riou J.-F., Royle N.J.. Human telomeres that contain (CTAGGG)(n) repeats show replication dependent instability in somatic cells and the male germline. Nucleic Acids Res. 2009; 37:6225–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juranek S.A., Paeschke K.. Cell cycle regulation of G-Quadruplex DNA structures at telomeres. Curr. Pharm. Des. 2012; 18:1867–1872. [DOI] [PubMed] [Google Scholar]
- 7. Rizzo A., Salvati E., Porru M., D’Angelo C., Stevens M.F., D’Incalci M., Leonetti C., Gilson E., Zupi G., Biroccio A.. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009; 37:5353–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Postberg J., Tsytlonok M., Sparvoli D., Rhodes D., Lipps H.J.. A Telomerase-associated RecQ Protein-like helicase resolves telomeric G-quadruplex structures during replication. Gene. 2012; 497:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rice C., Skordalakes E.. Structure and function of the telomeric CST complex. Comput. Struct. Biotechnol. J. 2016; 14:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoffmann R.F., Moshkin Y.M., Mouton S., Grzeschik N.A., Kalicharan R.D., Kuipers J., Wolters A.H.G., Nishida K., Romashchenko A.V., Postberg J. et al.. Guanine quadruplex structures localize to heterochromatin. Nucleic Acids Res. 2016; 44:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai J., Chen D., Jones R.A., Hurley L.H., Yang D.. NMR solution structure of the Major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 2006; 34:5133–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agrawal P., Hatzakis E., Guo K., Carver M., Yang D.. Solution structure of the Major G-quadruplex formed in the human VEGF promoter in K+: Insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013; 41:10584–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernando H., Reszka A.P., Huppert J., Ladame S., Rankin S., Venkitaraman A.R., Neidle S., Balasubramanian S.. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry. 2006; 45:7854–7860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Greco M.L., Kotar A., Rigo R., Cristofari C., Plavec J., Sissi C.. Coexistence of two main folded G-Quadruplexes within a single G-Rich domain in the EGFR promoter. Nucleic Acids Res. 2017; 45:10132–10142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Simonsson T., Pecinka P., Kubista M.. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998; 26:1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H.. Direct evidence for a G-Quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grand C.L., Bearss D.J., Von Hoff D.D., Hurley L.H.. Quadruplex formation in the c-MYC promoter inhibits protein binding and correlates with in vivo promoter activity. Eur. J. Cancer. 2002; 38:S106–S107. [Google Scholar]
- 18. Wei D., Parkinson G.N., Reszka A.P., Neidle S.. Crystal structure of a c-kit promoter quadruplex reveals the structural role of metal ions and water molecules in maintaining loop conformation. Nucleic Acids Res. 2012; 40:4691–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cogoi S., Xodo L.E.. G-Quadruplex formation within the promoter of the KRAS Proto-Oncogene and its effect on transcription. Nucleic Acids Res. 2006; 34:2536–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dexheimer T.S., Sun D., Hurley L.H.. Deconvoluting the structural and Drug-Recognition complexity of the G-Quadruplex-Forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 2006; 128:5404–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun D., Guo K., Rusche J.J., Hurley L.H.. Facilitation of a structural transition in the Polypurine/Polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-Quadruplex-Interactive agents. Nucleic Acids Res. 2005; 33:6070–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huppert J.L. Four-stranded nucleic acids: structure, function and targeting of G-Quadruplexes. Chem. Soc. Rev. 2008; 37:1375–1384. [DOI] [PubMed] [Google Scholar]
- 23. Monchaud D., Teulade-Fichou M.-P.. A Hitchhiker’s guide to G-Quadruplex ligands. Org. Biomol. Chem. 2008; 6:627–636. [DOI] [PubMed] [Google Scholar]
- 24. Balasubramanian S., Neidle S.. G-quadruplex nucleic acids as therapeutic targets. Curr. Opin. Chem. Biol. 2009; 13:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balasubramanian S., Hurley L.H., Neidle S.. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy. Nat. Rev. Drug Discover. 2011; 10:261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang S., Wu Y., Zhang W.. G-Quadruplex structures and their interaction diversity with ligands. ChemMedChem. 2014; 9:899–911. [DOI] [PubMed] [Google Scholar]
- 27. Boncina M., Lah J., Prislan I., Vesnaver G.. Energetic basis of human telomeric DNA folding into G-quadruplex structures. J. Am. Chem. Soc. 2012; 134:9657–9663. [DOI] [PubMed] [Google Scholar]
- 28. Koirala D., Mashimo T., Sannohe Y., Yu Z.B., Mao H.B., Sugiyama H.. Intramolecular folding in three tandem guanine repeats of human telomeric DNA. Chem. Commun. 2012; 48:2006–2008. [DOI] [PubMed] [Google Scholar]
- 29. Gray R.D., Buscaglia R., Chaires J.B.. Populated intermediates in the thermal unfolding of the human telomeric quadruplex. J. Am. Chem. Soc. 2012; 134:16834–16844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buscaglia R., Gray R.D., Chaires J.B.. Thermodynamic characterization of human telomere quadruplex unfolding. Biopolymers. 2013; 99:1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koirala D., Ghimire C., Bohrer C., Sannohe Y., Sugiyama H., Mao H.B.. Long-Loop G-Quadruplexes are misfolded population minorities with fast transition kinetics in human telomeric sequences. J. Am. Chem. Soc. 2013; 135:2235–2241. [DOI] [PubMed] [Google Scholar]
- 32. Jiang H.-X., Cui Y., Zhao T., Fu H.-W., Koirala D., Punnoose J.A., Kong D.-M., Mao H.. Divalent cations and molecular crowding buffers stabilize G-Triplex at physiologically relevant temperatures. Sci. Rep. 2015; 5:9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marchand A., Ferreira R., Tateishi-Karimata H., Miyoshi D., Sugimoto N., Gabelica V.. Sequence and solvent effects on telomeric DNA bimolecular G-Quadruplex folding kinetics. J. Phys. Chem. B. 2013; 117:12391–12401. [DOI] [PubMed] [Google Scholar]
- 34. Long X., Parks J.W., Bagshaw C.R., Stone M.D.. Mechanical unfolding of human telomere G-quadruplex DNA probed by integrated fluorescence and magnetic tweezers spectroscopy. Nucleic Acids Res. 2013; 41:2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W., Hou X.-M., Wang P.-Y., Xi X.-G., Li M.. Direct measurement of sequential folding pathway and energy landscape of human telomeric G-quadruplex structures. J. Am. Chem. Soc. 2013; 135:6423–6426. [DOI] [PubMed] [Google Scholar]
- 36. You H.J., Zeng X.J., Xu Y., Lim C.J., Efremov A.K., Phan A.T., Yan J.. Dynamics and stability of polymorphic human telomeric G-Quadruplex under tension. Nucleic Acids Res. 2014; 42:8789–8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li Y., Liu C., Feng X.J., Xu Y.Z., Liu B.F.. Ultrafast microfluidic mixer for tracking the early folding kinetics of human telomere G-Quadruplex. Anal. Chem. 2014; 86:4333–4339. [DOI] [PubMed] [Google Scholar]
- 38. Gray R.D., Trent J.O., Chaires J.B.. Folding and unfolding pathways of the human telomeric G-Quadruplex. J. Mol. Biol. 2014; 426:1629–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bessi I., Jonker H.R., Richter C., Schwalbe H.. Involvement of Long-Lived intermediate states in the complex folding pathway of the human telomeric G-Quadruplex. Angew. Chem. Int. Ed. 2015; 54:8444–8448. [DOI] [PubMed] [Google Scholar]
- 40. Noer S.L., Preus S., Gudnason D., Aznauryan M., Mergny J.-L., Birkedal V.. Folding dynamics and conformational heterogeneity of human telomeric G-quadruplex structures in Na+ solutions by single molecule FRET microscopy. Nucleic Acids Res. 2016; 44:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aznauryan M., Søndergaard S., Noer S.L., Schiøtt B., Birkedal V.. A direct view of the complex Multi-Pathway folding of telomeric G-Quadruplexes. Nucleic Acids Res. 2016; 44:11024–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marchand A., Gabelica V.. Folding and misfolding pathways of G-Quadruplex DNA. Nucleic Acids Res. 2016; 44:10999–11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boncina M., Vesnaver G., Chaires J.B., Lah J.. Unraveling the thermodynamics of the folding and interconversion of human telomere G-Quadruplexes. Angew. Chem. Int. Ed. 2016; 55:10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rigo R., Dean W.L., Gray R.D., Chaires J.B., Sissi C.. Conformational profiling of a G-Rich sequence within the c-KIT promoter. Nucleic Acids Res. 2017; 45:13056–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hou X.-M., Fu Y.-B., Wu W.-Q., Wang L., Teng F.-Y., Xie P., Wang P.-Y., Xi X.-G.. Involvement of G-Triplex and G-Hairpin in the Multi-Pathway folding of human telomeric G-Quadruplex. Nucleic Acids Res. 2017; 45:11401–11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. You J., Li H., Lu X.-M., Li W., Wang P.-Y., Dou S.-X., Xi X.-G.. Effects of monovalent cations on folding kinetics of G-Quadruplexes. Biosci. Rep. 2017; 37:BSR20170771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gray R.D., Trent J.O., Arumugam S., Chaires J.B.. Folding landscape of a parallel G-Quadruplex. J. Phys. Chem. Lett. 2019; 10:1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mashimo T., Yagi H., Sannohe Y., Rajendran A., Sugiyama H.. Folding pathways of human telomeric Type-1 and Type-2 G-quadruplex structures. J. Am. Chem. Soc. 2010; 132:14910–14918. [DOI] [PubMed] [Google Scholar]
- 49. Stadlbauer P., Krepl M., Cheatham T.E. 3rd, Koca J., Sponer J.. Structural dynamics of possible Late-Stage intermediates in folding of quadruplex DNA studied by molecular simulations. Nucleic Acids Res. 2013; 41:7128–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bian Y., Tan C., Wang J., Sheng Y., Zhang J., Wang W.. Atomistic picture for the folding pathway of a Hybrid-1 type human telomeric DNA G-quadruplex. PLoS Comput. Biol. 2014; 10:e1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stadlbauer P., Kuhrova P., Banas P., Koca J., Bussi G., Trantirek L., Otyepka M., Sponer J.. Hairpins participating in folding of human telomeric sequence quadruplexes studied by standard and T-REMD simulations. Nucleic Acids Res. 2015; 43:9626–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Islam B., Stadlbauer P., Krepl M., Koca J., Neidle S., Haider S., Sponer J.. Extended molecular dynamics of a c-kit promoter quadruplex. Nucleic Acids Res. 2015; 43:8673–8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luo D., Mu Y.. Computational insights into the stability and folding pathways of human telomeric DNA G-Quadruplexes. J. Phys. Chem. B. 2016; 120:4912–4926. [DOI] [PubMed] [Google Scholar]
- 54. Stadlbauer P., Mazzanti L., Cragnolini T., Wales D.J., Derreumaux P., Pasquali S., Sponer J.. Coarse-Grained simulations complemented by atomistic molecular dynamics provide new insights into folding and unfolding of human telomeric G-Quadruplexes. J. Chem. Theory Comput. 2016; 12:6077–6097. [DOI] [PubMed] [Google Scholar]
- 55. Bian Y., Ren W., Song F., Yu J., Wang J.. Exploration of the folding dynamics of human telomeric G-Quadruplex with a hybrid atomistic structure-based model. J. Chem. Phys. 2018; 148:204107. [DOI] [PubMed] [Google Scholar]
- 56. Havrila M., Stadlbauer P., Kuhrova P., Banas P., Mergny J.-L., Otyepka M., Sponer J.. Structural dynamics of propeller Loop: Towards folding of RNA G-Quadruplex. Nucleic Acids Res. 2018; 46:8754–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li H., Cao E.H., Gisler T.. Force-Induced unfolding of human telomeric G-quadruplex: A steered molecular dynamics simulation study. Biochem. Biophys. Res. Commun. 2009; 379:70–75. [DOI] [PubMed] [Google Scholar]
- 58. Sponer J., Bussi G., Stadlbauer P., Kuhrova P., Banas P., Islam B., Haider S., Neidle S., Otyepka M.. Folding of guanine quadruplex Molecules–Funnel-Like mechanism or kinetic partitioning? An overview from MD simulation studies. Biochim. Biophys. Acta Gen. Subj. 2017; 1861:1246–1263. [DOI] [PubMed] [Google Scholar]
- 59. Thirumalai D., Klimov D.K., Woodson S.A.. Kinetic partitioning mechanism as a unifying theme in the folding of biomolecules. Theor. Chem. Acc. 1997; 96:14–22. [Google Scholar]
- 60. Thirumalai D., O’Brien E.P., Morrison G., Hyeon C.. Theoretical perspectives on protein folding. Annu. Rev. Biophys. 2010; 39:159–183. [DOI] [PubMed] [Google Scholar]
- 61. Long X., Stone M.D.. Kinetic partitioning modulates human telomere DNA G-Quadruplex structural polymorphism. PLoS One. 2013; 8:e83420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang Y., Patel D.J.. Solution structure of the human telomeric repeat d[AG(3)(T(2)AG(3))3] G-tetraplex. Structure. 1993; 1:263–282. [DOI] [PubMed] [Google Scholar]
- 63. Parkinson G.N., Lee M.P.H., Neidle S.. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002; 417:876–880. [DOI] [PubMed] [Google Scholar]
- 64. Ambrus A., Chen D., Dai J.X., Bialis T., Jones R.A., Yang D.Z.. Human telomeric sequence forms a Hybrid-Type intramolecular G-Quadruplex structure with mixed Parallel/Antiparallel strands in potassium solution. Nucleic Acids Res. 2006; 34:2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dai J., Punchihewa C., Ambrus A., Chen D., Jones R.A., Yang D.. Structure of the intramolecular human telomeric G-quadruplex in potassium Solution: A novel adenine triple formation. Nucleic Acids Res. 2007; 35:2440–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dai J., Carver M., Punchihewa C., Jones R.A., Yang D.. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ Solution: Insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007; 35:4927–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Z.J., Dai J.X., Veliath E., Jones R.A., Yang D.Z.. Structure of a Two-G-Tetrad intramolecular G-Quadruplex formed by a variant human telomeric sequence in K+ Solution: Insights into the interconversion of human telomeric G-Quadruplex structures. Nucleic Acids Res. 2010; 38:1009–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lim K.W., Ng V.C.M., Martin-Pintado N., Heddi B., Phan A.T.. Structure of the human telomere in Na+ Solution: An antiparallel (2+2) G-quadruplex scaffold reveals additional diversity. Nucleic Acids Res. 2013; 41:10556–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dailey M.M., Miller M.C., Bates P.J., Lane A.N., Trent J.O.. Resolution and characterization of the structural polymorphism of a single Quadruplex-Forming sequence. Nucleic Acids Res. 2010; 38:4877–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brcic J., Plavec J.. ALS and FTD linked GGGGCC-Repeat containing DNA oligonucleotide folds into two distinct G-Quadruplexes. Biochim. Biophys. Acta Gen. Subj. 2017; 1861:1237–1245. [DOI] [PubMed] [Google Scholar]
- 71. Palacky J., Vorlickova M., Kejnovska I., Mojzes P.. Polymorphism of human telomeric quadruplex structure controlled by DNA Concentration: A raman study. Nucleic Acids Res. 2013; 41:1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Renciuk D., Kejnovska I., Skolakova P., Bednarova K., Motlova J., Vorlickova M.. Arrangements of human telomere DNA quadruplex in physiologically relevant K+ solutions. Nucleic Acids Res. 2009; 37:6625–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Karsisiotis A.I., O’Kane C., da Silva M.W.. DNA quadruplex folding Formalism - A tutorial on quadruplex topologies. Methods. 2013; 64:28–35. [DOI] [PubMed] [Google Scholar]
- 74. Gabelica V. A Pilgrim’s guide to G-quadruplex nucleic acid folding. Biochimie. 2014; 105C:1–3. [DOI] [PubMed] [Google Scholar]
- 75. Xue Y., Liu J.-Q., Zheng K.-W., Kan Z.-Y., Hao Y.-H., Tan Z.. Kinetic and thermodynamic control of G-Quadruplex folding. Angew. Chem. Int. Ed. 2011; 50:8046–8050. [DOI] [PubMed] [Google Scholar]
- 76. Rajendran A., Endo M., Hidaka K., Teulade-Fichou M.-P., Mergny J.-L., Sugiyama H.. Small molecule binding to a G-hairpin and a G-triplex: A new insight into anticancer drug design targeting G-rich regions. Chem. Commun. 2015; 51:9181–9184. [DOI] [PubMed] [Google Scholar]
- 77. Bryngelson J.D., Onuchic J.N., Socci N.D., Wolynes P.G.. Funnels, pathways, and the energy landscape of Protein-folding - A synthesis. Proteins: Struct. Funct. Genet. 1995; 21:167–195. [DOI] [PubMed] [Google Scholar]
- 78. Dill K.A., Chan H.S.. From levinthal to pathways to funnels. Nat. Struct. Mol. Biol. 1997; 4:10–19. [DOI] [PubMed] [Google Scholar]
- 79. Neidle S. The structures of quadruplex nucleic acids and their drug complexes. Curr. Opin. Struct. Biol. 2009; 19:239–250. [DOI] [PubMed] [Google Scholar]
- 80. Neidle S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2010; 277:1118–1125. [DOI] [PubMed] [Google Scholar]
- 81. Zhang A.Y.Q., Balasubramanian S.. The kinetics and folding pathways of intramolecular G-Quadruplex nucleic acids. J. Am. Chem. Soc. 2012; 134:19297–19308. [DOI] [PubMed] [Google Scholar]
- 82. Mitra J., Makurath M.A., Ngo T.T.M., Troitskaia A., Chemla Y.R., Ha T.. Extreme mechanical diversity of human telomeric DNA lby Fluorescence-Force spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:8350–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. You H., Zeng X., Xu Y., Lim C.J., Efremov A.K., Phan A.T., Yan J.. Dynamics and stability of polymorphic human telomeric G-Quadruplex under tension. Nucleic Acids Res. 2014; 42:8789–8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rajendran A., Endo M., Hidaka K., Sugiyama H.. Direct and Single-Molecule visualization of the Solution-State structures of G-Hairpin and G-Triplex intermediates. Angew. Chem. 2014; 126:4191–4196. [DOI] [PubMed] [Google Scholar]
- 85. Okamoto K., Sannohe Y., Mashimo T., Sugiyama H., Terazima M.. G-Quadruplex structures of human telomere DNA examined by single molecule FRET and BrG-Substitution. Bioorg. Med. Chem. 2008; 16:6873–6879. [DOI] [PubMed] [Google Scholar]
- 86. Stadlbauer P., Trantirek L., Cheatham T.E. 3rd, Koca J., Sponer J.. Triplex intermediates in folding of human telomeric quadruplexes probed by Microsecond-scale molecular dynamics simulations. Biochimie. 2014; 105:22–35. [DOI] [PubMed] [Google Scholar]
- 87. Trajkovski M., da Silva M.W., Plavec J.. Unique structural features of interconverting monomeric and dimeric G-quadruplexes adopted by a sequence from the intron of the N-myc gene. J. Am. Chem. Soc. 2012; 134:4132–4141. [DOI] [PubMed] [Google Scholar]
- 88. Padrta P., Stefl R., Kralik L., Zidek L., Sklenar V.. Refinement of d(GCGAAGC) hairpin structure using One- and Two-Bond residual dipolar couplings. J. Biomol. NMR. 2002; 24:1–14. [DOI] [PubMed] [Google Scholar]
- 89. Luu K.N., Phan A.T., Kuryavyi V., Lacroix L., Patel D.J.. Structure of the human telomere in K+ Solution: An intramolecular (3+1) G-Quadruplex scaffold. J. Am. Chem. Soc. 2006; 128:9963–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 91. Berendsen H.J.C., Grigera J.R., Straatsma T.P.. The missing term in effective pair potentials. J. Phys. Chem. 1987; 91:6269–6271. [Google Scholar]
- 92. Joung I.S., Cheatham T.E.. Determination of Alkali and Halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008; 112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Case D.A., Betz R.M., Botello-Smith W., Cerutti D.S., Cheatham T.E. III, Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W. et al.. Amber 16. 2016; San Francisco: University of California. [Google Scholar]
- 94. Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., Orozco M.. Refinenement of the AMBER force field for Nucleic Acids: Improving the description of Alpha/Gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Krepl M., Zgarbova M., Stadlbauer P., Otyepka M., Banas P., Koca J., Cheatham T.E., Jurecka P., Sponer J.. Reference simulations of noncanonical Nucleic Acids with different chi variants of the AMBER force Field: Quadruplex DNA, quadruplex RNA, and Z-DNA. J. Chem. Theory Comput. 2012; 8:2506–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zgarbova M., Luque F.J., Sponer J., Cheatham T.E., Otyepka M., Jurecka P.. Toward improved description of DNA Backbone: Revisiting epsilon and zeta torsion force field parameters. J. Chem. Theory Comput. 2013; 9:2339–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zgarbova M., Sponer J., Otyepka M., Cheatham T.E., Galindo-Murillo R., Jurecka P.. Refinement of the Sugar–Phosphate backbone torsion beta for AMBER force fields improves the description of Z- and B-DNA. J. Chem. Theory Comput. 2015; 11:5723–5736. [DOI] [PubMed] [Google Scholar]
- 98. Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A.. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995; 117:5179–5197. [Google Scholar]
- 99. Galindo-Murillo R., Robertson J.C., Zgarbova M., Sponer J., Otyepka M., Jurecka P., Cheatham T.E.. Assessing the current state of AMBER force field modifications for DNA. J. Chem. Theory Comput. 2016; 12:4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A.. Development and testing of a general amber force field. J. Comput. Chem. 2004; 25:1157–1174. [DOI] [PubMed] [Google Scholar]
- 101. Shahrokh K., Orendt A., Yost G.S., Cheatham T.E.. Quantum mechanically derived AMBER-Compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012; 33:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Darden T., York D., Pedersen L.. Particle mesh Ewald - An N.log(N) method for ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 103. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 104. Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R.. Molecular-Dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 105. Hopkins C.W., Le Grand S., Walker R.C., Roitberg A.E.. Long-Time-Step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015; 11:1864–1874. [DOI] [PubMed] [Google Scholar]
- 106. Ryckaert J.P., Ciccotti G., Berendsen H.J.C.. Numerical integration of cartesian equations of motion of a system with Constraints - Molecular dynamics of N-alkans. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 107. Miyamoto S., Kollman P.A.. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992; 13:952–962. [Google Scholar]
- 108. Kuhrova P., Best R.B., Bottaro S., Bussi G., Sponer J., Otyepka M., Banas P.. Computer folding of RNA Tetraloops: Identification of key force field deficiencies. J. Chem. Theory Comput. 2016; 12:4534–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sponer J., Bussi G., Krepl M., Banas P., Bottaro S., Cunha R.A., Gil-Ley A., Pinamonti G., Poblete S., Jurecka P. et al.. RNA structural dynamics as captured by molecular Simulations: A comprehensive overview. Chem. Rev. 2018; 118:4177–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu P., Kim B., Friesner R.A., Berne B.J.. Replica exchange with solute Tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:13749–13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wang L., Friesner R.A., Berne B.J.. Replica exchange with solute Scaling: A more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B. 2011; 115:9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sugita Y., Okamoto Y.. Replica-Exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999; 314:141–151. [Google Scholar]
- 113. Case D.A., Babin V., Berryman J.T., Betz R.M., Cai Q., Cerutti D.S., Cheatham T.E. III, Darden T.A., Duke R.E., Gohlke H. et al.. Amber 14. 2014; San Francisco: University of California. [Google Scholar]
- 114. Smith D.E., Dang L.X.. Computer simulations of NaCl association in polarizable water. J. Chem. Phys. 1994; 100:3757–3766. [Google Scholar]
- 115. Dang L.X., Kollman P.A.. Free energy of association of the K+18-crown-6 complex in Water - A new molecular dynamics study. J. Phys. Chem. 1995; 99:55–58. [Google Scholar]
- 116. Bottaro S., Di Palma F., Bussi G.. The role of nucleobase interactions in RNA structure and dynamics. Nucleic Acids Res. 2014; 42:13306–13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rodriguez A., Laio A.. Clustering by fast search and find of density peaks. Science. 2014; 344:1492–1496. [DOI] [PubMed] [Google Scholar]
- 118. Kuhrova P., Mlynsky V., Zgarbova M., Krepl M., Bussi G., Best R.B., Otyepka M., Sponer J., Banáš P.. Improving the performance of the amber RNA force field by tuning the Hydrogen-Bonding interactions. J. Chem. Theory Comput. 2019; 15:3288–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yang C., Kulkarni M., Lim M., Pak Y.. Insilico direct folding of Thrombin-Binding aptamer G-Quadruplex at All-Atom level. Nucleic Acids Res. 2017; 45:12648–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Limongelli V., De Tito S., Cerofolini L., Fragai M., Pagano B., Trotta R., Cosconati S., Marinelli L., Novellino E., Bertini I. et al.. The G-Triplex DNA. Angew. Chem. Int. Ed. 2013; 52:2269–2273. [DOI] [PubMed] [Google Scholar]
- 121. Cerofolini L., Amato J., Giachetti A., Limongelli V., Novellino E., Parrinello M., Fragai M., Randazzo A., Luchinat C.. G-Triplex structure and formation propensity. Nucleic Acids Res. 2014; 42:13393–13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lim K.W., Khong Z.J., Phan A.T.. Thermal stability of DNA Quadruplex–Duplex hybrids. Biochemistry. 2014; 53:247–257. [DOI] [PubMed] [Google Scholar]
- 123. Largy E., Marchand A., Amrane S., Gabelica V., Mergny J.-L.. Quadruplex Turncoats: Cation-Dependent folding and stability of Quadruplex-DNA double switches. J. Am. Chem. Soc. 2016; 138:2780–2792. [DOI] [PubMed] [Google Scholar]
- 124. Stefl R., Cheatham T.E., Spackova N., Fadrna E., Berger I., Koca J., Sponer J.. Formation pathways of a Guanine-Quadruplex DNA revealed by molecular dynamics and thermodynamic analysis of the substates. Biophys. J. 2003; 85:1787–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bergues-Pupo A.E., Arias-Gonzalez J.R., Moron M.C., Fiasconaro A., Falo F.. Role of the central cations in the mechanical unfolding of DNA and RNA G-quadruplexes. Nucleic Acids Res. 2015; 43:7638–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Stadlbauer P., Mazzanti L., Cragnolini T., Wales D.J., Derreumaux P., Pasquali S., Sponer J.. Coarse-Grained simulations complemented by atomistic molecular dynamics provide new insights into folding of human telomeric G-Quadruplexes. J. Chem. Theory Comput. 2016; 12:6077–6097. [DOI] [PubMed] [Google Scholar]
- 127. Yang C., Jang S., Pak Y.. Multiple stepwise pattern for potential of mean force in unfolding the thrombin binding aptamer in complex with Sr2+. J. Chem. Phys. 2011; 135:225104. [DOI] [PubMed] [Google Scholar]
- 128. Cragnolini T., Chakraborty D., Sponer J., Derreumaux P., Pasquali S., Wales D.J.. Multifunctional energy landscape for a DNA G-Quadruplex: An evolved molecular switch. J. Chem. Phys. 2017; 147:152715. [DOI] [PubMed] [Google Scholar]
- 129. Portella G., Orozco M.. Multiple routes to characterize the folding of a small DNA hairpin. Angew. Chem. Int. Ed. 2010; 49:7673–7676. [DOI] [PubMed] [Google Scholar]
- 130. Islam B., Stadlbauer P., Gil-Ley A., Perez-Hernandez G., Haider S., Neidle S., Bussi G., Banas P., Otyepka M., Sponer J.. Exploring the dynamics of propeller loops in human telomeric DNA quadruplexes using atomistic simulations. J. Chem. Theory Comput. 2017; 13:2458–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gkionis K., Kruse H., Platts J.A., Mladek A., Koca J., Sponer J.. Ion binding to quadruplex DNA Stems. Comparison of MM and QM descriptions reveals sizable polarization effects not included in contemporary simulations. J. Chem. Theory Comput. 2014; 10:1326–1340. [DOI] [PubMed] [Google Scholar]
- 132. Havrila M., Stadlbauer P., Islam B., Otyepka M., Sponer J.. Effect of monovalent ion parameters on molecular dynamics simulations of G-Quadruplexes. J. Chem. Theory Comput. 2017; 13:3911–3926. [DOI] [PubMed] [Google Scholar]
- 133. Salsbury A.M., Lemkul J.A.. Molecular dynamics simulations of the c-kit1 promoter G-Quadruplex: Importance of electronic polarization on stability and cooperative ion binding. J. Phys. Chem. B. 2019; 123:148–159. [DOI] [PubMed] [Google Scholar]
- 134. Lu X.-M., Li H., You J., Li W., Wang P.-Y., Li M., Dou S.-X., Xi X.-G.. Folding dynamics of parallel and antiparallel G-Triplexes under the influence of proximal DNA. J. Phys. Chem. B. 2018; 122:9499–9506. [DOI] [PubMed] [Google Scholar]
- 135. Gajarsky M., Zivkovic M.L., Stadlbauer P., Pagano B., Fiala R., Amato J., Tomaska L., Sponer J., Plavec J., Trantirek L.. Structure of a stable G-Hairpin. J. Am. Chem. Soc. 2017; 139:3591–3594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.