

Abstract
Vitiligo is an autoimmune disease that results in patches of depigmented skin and hair. Previous genome-wide association studies (GWASs) of vitiligo have identified 50 susceptibility loci. Variants at the associated loci are generally common and have individually small effects on risk. Most vitiligo cases are “simplex,” where there is no family history of vitiligo, though occasional family clustering of vitiligo occurs, and some “multiplex” families report numerous close affected relatives. Here, we investigate whether simplex and multiplex vitiligo comprise different disease subtypes with different underlying genetic etiologies. We developed and compared the performance of several different vitiligo polygenic risk scores derived from GWAS data. By using the best-performing risk score, we find increased polygenic burden of risk alleles identified by GWAS in multiplex vitiligo cases relative to simplex cases. We additionally find evidence of polygenic transmission of common, low-effect-size risk alleles within multiplex-vitiligo-affected families. Our findings strongly suggest that family clustering of vitiligo involves a high burden of the same common, low-effect-size variants that are relevant in simplex cases. We furthermore find that a variant within the major histocompatibility complex (MHC) class II region contributes disproportionately more to risk in multiplex vitiligo cases than in simplex cases, supporting a special role for adaptive immune triggering in the etiology of multiplex cases. We suggest that genetic risk scores can be a useful tool in analyzing the genetic architecture of clinical disease subtypes and identifying subjects with unusual etiologies for further investigation.
Keywords: vitiligo, autoimmunity, polygenic inheritance, genetic risk score, family clustering, genetic architecture
Introduction
Vitiligo is an autoimmune disease in which destruction of skin melanocytes leads to patches of depigmented skin and hair. Although most vitiligo cases are “simplex,” wherein there is no family history of vitiligo, about 9% of cases are “multiplex,” belonging to families with more than one close affected relative. Inheritance of vitiligo within multiplex-affected families typically appears non-Mendelian, and early segregation analyses of such families suggested additive, polygenic inheritance with heritability between 50% and 75%.1, 2, 3
A polygenic nature of vitiligo is also supported by the results of subsequent genome-wide association studies (GWASs) in European-derived whites; these studies have identified 50 contributory loci, and each locus contributes a small amount to overall vitiligo heritability.4, 5, 6 Like in most other genetically complex diseases, most vitiligo GWAS loci involve common, low-to-moderate effect-size variants (Figure 1). And, like in other autoimmune diseases, the corresponding identified genes involve the regulation of immune cells and apoptosis, as well as melanocyte components that can act as autoantigens.
Figure 1.

Distribution of Risk Allele Frequency and Estimated Odds Ratio for 48 Autosomal Variants Previously Identified in Vitiligo GWAS123
Each dot represents the most-associated variant for a vitiligo susceptibility locus, and each locus is labeled with a gene in close proximity to the most-associated variant in the locus. Dot and label colors represent the likely functional category of the locus, designated by manual review of each locus: orange = apoptosis, green = immune regulation, blue = functional component of the melanocyte, and pink = unknown function. ∗ denotes an HLA-DQB1 locus specifically associated in early-onset vitiligo cases;12 the effect size used for the HLA-DQB1 variant here was derived from all vitiligo cases, regardless of the age of onset.
We have assembled a highly curated collection of over 400 multiplex-vitiligo-affected families and over 4,000 simplex-vitiligo-affected subjects of European descent. On the basis of loci identified in previous GWASs, we investigated whether the genetic risk factors for multiplex and simplex vitiligo are fundamentally similar or different. We considered two alternative hypothetical models of genetic architecture in multiplex-vitiligo-affected families versus simplex-affected subjects. First, we considered that in multiplex-affected families, rare, high-penetrance alleles might be the major driver of vitiligo risk; such alleles would have little impact on risk for simplex vitiligo cases and would be difficult to identify by GWAS. This hypothesis was supported by the identification of several loci in previous genetic linkage analysis of multiplex-vitiligo-affected families; none of these loci overlap any vitiligo GWAS loci.7, 8 Alternatively, we considered that multiplex-vitiligo-affected families might segregate a high burden of the same common, low-to-moderate effect-size risk alleles detected by vitiligo GWAS. This hypothesis was supported by previous segregation analyses of multiplex-affected families that suggested polygenic inheritance.3, 9, 10 To explore these alternative possibilities, we developed several different polygenic risk scores based on data from previous vitiligo GWASs. We then used the best-performing risk score to compare polygenic risk in simplex-affected subjects versus in the probands of multiplex-affected families. Our results suggest that multiplex-vitiligo-affected families do in fact segregate a high burden of common, low-to-moderate effect-size risk alleles identified by GWAS. Thus, genetic risk of vitiligo in both multiplex-affected families and simplex-affected subjects appears to be generally similar, largely involving the same common risk alleles and thus involving the same underlying pathobiological processes.
Subjects and Methods
Study Subjects
All subjects were from North America and Europe and were of self-described non-Hispanic/Latino European-derived white ancestry. All cases met diagnostic criteria for generalized vitiligo.11 We obtained written informed consent from all participants, and the study was approved by the institutional review board at each participating center with oversight from the Colorado Multiple Institutional Review Board (COMIRB).
Subjects consisted of 4,668 unrelated vitiligo-affected individuals and 39,436 controls from our three previous vitiligo GWASs (referred to in sum as GWAS123): GWAS1,4 ncases = 1,377, ncontrols = 14,401; GWAS2,5 ncases = 412, ncontrols = 5,189; GWAS3,6 ncases = 1,052, ncontrols = 17,665), as well as an independent replication study6 (ncases = 1,827, ncontrols = 2,181). Subjects in the replication study cohort were genotyped for the most-associated variant for all genome-wide significant loci (Table S1).
Genotyping and Quality Control
Some individuals in GWAS123 were excluded on the basis of pairwise identity-by-descent sharing 0.0884; affected subjects were retained over controls, and individuals with the highest SNP call rate were retained otherwise; identity-by-descent analysis was performed with KING12 version 1.4. For each GWAS, variants with minor allele frequency (MAF) > 0.01, Hardy-Weinberg equilibrium , and INFO scores > 0.5 were retained. A total of 8,615,281, 8,632,074, and 8,616,642 autosomal variants passed the QC process for GWAS1, 2, and 3, respectively.
Vitiligo Multiplex-Affected Family Cohort
Multiplex-affected families were identified by manual, post hoc review of all European-derived vitiligo-affected subjects and relatives in our collection. When a single multiplex “proband” was included in a prior GWAS or replication study, this subject was virtually always a “true” proband (the initial contact subject from the family that enrolled in the study).
Diagnoses of vitiligo in multiplex-affected subjects and reportedly unaffected family members were verified by manual review of all available phenotype information for each subject. Subjects with apparent segmental vitiligo or with an uncertain diagnosis were assigned an unknown phenotype (nunknown = 83). In total, we identified 444 unique multiplex-affected families with at least two relatives with generalized vitiligo and for which at least one subject had a DNA sample. Across all 444 multiplex-affected families, we identified a total 1,332 subjects with generalized vitiligo and 6,476 relatives who reported being unaffected with vitiligo.
After aligning all available genotype data from the previous GWASs and replication studies with pedigree data, we identified 324 unrelated multiplex-affected family probands with genome-wide data from GWAS123, plus an additional 78 unrelated multiplex-affected probands genotyped at all confirmed vitiligo GWAS loci in our replication study. Thus, 8.6% (402/4,668) of the combined GWAS123 and replication study vitiligo-affected subjects are multiplex probands.
Extended Multiplex-Affected Family Analysis
In our previous vitiligo GWASs and replication studies, only a single proband from each multiplex-affected family was genotyped. However, in a previous family-based association study,4 we genotyped tagSNPs for 14 of the 50 confirmed vitiligo GWAS loci (Table S2) in all available members of 330 multiplex-vitiligo-affected families. This family-based study thus provided limited relevant genotype information for extended families of multiplex-affected probands. We used these data to investigate the transmission of polygenic risk within families. From the total set of 330 multiplex-affected families, we retained trios that consisted of a vitiligo-affected proband and two unaffected parents for whom genotype data were available for all trio members. We additionally removed subjects with >1 missing genotype among the 14 loci. This resulted in 107 affected subjects and 148 unaffected parents from 71 multiplex-affected families whom we used to assess polygenic transmission with the 14-SNP “FAMILY” risk score described below.
Calculation of Ancestry-Based Principal Components
Genetic sub-structure for each of the three GWAS cohorts was determined by Spectral-GEM.14 We used best-guess genotypes if the genotype posterior probability was ≥0.9; otherwise, we set genotypes to missing. Variants with call rate ≤ 0.99 were excluded. We performed linkage disequilibrium (LD) pruning by using PLINK v.1.07, resulting in 113,595 variants ( in sliding windows of 500 variants) after excluding four genomic regions (MHC, LCT, chromosome 8, and chromosome 11 inversion regions). Sparser sets of markers were created by random sampling from the set of 113,595 variants, yielding additional datasets with 15,000, 40,000, 65,000, and 90,000 variants. We performed spectral-GEM cluster analyses separately for GWAS1, GWAS2, and GWAS3, and we used the pseudo-F statistics computed from the GEM-derived clusters to determine the variant set that best describes genetic substructure (90,000 variants). Spectral-GEM principal components (PCs) with logistic regression family-wise error rate < 0.1 were included as covariates in case-control analyses (GWAS1: PC2, PC8; GWAS2: PC1, PC2, PC3, PC4; and GWAS3: PC1, PC3, PC4, PC5, PC7, PC10, PC12, and PC14).
The vitiligo-affected subjects were relatively homogeneous with respect to European ancestry (Figure S1); however, we generated affected-only PCs by combining affected subjects from GWAS123 with 1000 Genomes Project (1KGP) European-derived (EUR) subjects at all variants genotyped in both groups, excluding ambiguous (A/T or G/C) SNPs. Variants with an overall missing genotype rate >0.05 in the combined set of subjects were removed. Variants were then LD-pruned with PLINK-1.9 command–indep-pairwise 50 5 0.2. Some 1KGP subjects were excluded on the basis of pairwise identity-by-descent sharing ≥ 0.0884, resulting in a total of 444 EUR 1KGP subjects. The top ten PCs were calculated from these EUR 1KGP individuals, and the vitiligo-affected subjects from GWAS123 were then projected onto the principal component analysis space. Only PC3 was significantly associated (p < 0.05) with the CONFIRMED risk score (described below) in affected subjects; accordingly, PC3 was included in affected-only analyses of the CONFIRMED risk score.
Construction and Comparison of Vitiligo Polygenic Risk Scores
We used 10-fold cross-validation to estimate risk score functions and to compare the performance of polygenic risk scores by using four different approaches in the GWAS123 affected-control cohorts described above. Cross-validation sets were defined by randomly assigning individuals in each GWAS cohort to one of ten groups of approximately equal size. Within each GWAS, a training set, , was formed by leaving out a cross-validation set. We then performed vitiligo GWAS for each training set by logistic regression using dosage values as genotypes and including as covariates the ancestry-based PCs described above. We the combined effect estimates over GWAS1, GWAS2, and GWAS3 by using inverse variance weighting,15 generating meta-analysis summary statistics for each of the ten training sets.
A risk score function was defined based on the variant effect estimates derived from the meta-analysis summary statistics from each training set as , where is the vector of best-guess genotypes for variants selected for inclusion in the score function, is the vector of variant effect sizes from training set , and indexes an individual. Best-guess genotypes were called if a genotype had posterior probability of at least 0.499 and otherwise were set to missing. After hard-calls, 89,158, 81,217, and 84,031 SNPs were excluded on the basis of >2% missingness for GWAS1, 2, and 3, respectively. When genotype data were missing, we used the expected genotype value based on allele frequency, 2p2 + 2p (1−p), where p is the risk allele frequency for the variant with missing data, estimated in both affected subjects and controls for each GWAS study cohort separately.
By using the test set left out of cross-validation set , we estimated the probability of disease for each individual by logistic regression including the risk score and ancestry-based PCs. The area under the curve (AUC) for the receiver operator characteristic (ROC) curve was derived based on the probability of disease. We calculated AUC, with standard error, for each score function for each GWAS by using the pROC package version 1.14.0 in R version 3.6.0. To combine AUC over GWAS1, GWAS2, and GWAS3, we used inverse-variance weighting, resulting in an AUC for each cross-validation set.
We compared AUC for polygenic risk scores by four different approaches: (1) ALL, (2) CLUMPED, (3) CONFIRMED, and (4) FAMILY. For risk scores ALL and CLUMPED, variants were filtered to those falling below different p value thresholds (αT), and AUC was compared at each threshold; p value thresholds considered were 5 × 10−8, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, 0.007, 0.008, 0.009, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, and 1. For the risk scores denoted “ALL,” all variants below the p value threshold were included. For the risk scores denoted “CLUMPED,” we reduced the set of variants considered by LD clumping, implemented in PLINK. Using a greedy algorithm, clumps start with a lead variant defined by the smallest p value and other variants are assigned to a clump on the basis of r2 > 0.2 within 250 KB of the lead variant. For the risk score denoted “CONFIRMED,” only the 48 autosomal variants that exceeded genome-wide significance (p < 5 × 10−8) in previous vitiligo GWASs and confirmed by independent replication6 were included (Table S1). The two MHC Class II variants (dbSNP: rs145954018 and dbSNP: rs9271597) included in the CONFIRMED risk score are correlated;13 we therefore applied a special coding strategy for these two variants: if 1 or 2 copies of the dbSNP: rs145954018 risk allele were present, we used (reflecting a dominant effect), but if 0 copies of the dbSNP: rs145954018 risk allele were present, we used .
For the risk score denoted “FAMILY,” we used a set of 14 variants (Table S2) identified in GWAS1.4 The 14 variants in the FAMILY risk score represent those with the highest MAFs and effect sizes identified by vitiligo GWASs and included in the CONFIRMED risk score.
To assess the extent of upward bias in CONFIRMED risk score effect sizes estimated from the GWAS123 variant-discovery cohort, we fit a model assuming normal errors with
where represents the normalized CONFIRMED risk score (mean centered and scaled on the basis of GWAS123 controls), represents the reference group of GWAS123 controls, is 1 if the subject is a vitiligo-affected individual and 0 if the subject is a control, is 1 if the subject was genotyped in the replication study and 0 if the subject was genotyped in GWAS123, and represents an interaction term. To test the hypothesis that , we used a two-sided Wald test and considered p < 0.05 to represent a significant difference in the relationship between the CONFIRMED risk score and vitiligo affliction in GWAS123 versus in the independent replication study.
We estimated odds ratios (ORs) of developing vitiligo for subjects belonging to the upper tails of the CONFIRMED and FAMILY risk score distributions (i.e., above the 80th, 90th, 95th, or 99th percentile of risk score among controls) by fitting a logistic regression model:
where and was coded as 1 if the subject belonged to the high risk group and 0 otherwise. corresponds to the Spectral-GEM PCs described above. PCs were not included for replication study subjects because genome-wide genotype data were not available.
Affected-Only Comparisons of the CONFIRMED Risk Score
We calculated risk scores by using the best-performing score, the CONFIRMED risk score (denoted SC), in all vitiligo cases. For CONFIRMED risk score calculation, we used the variants and effect sizes, defined in Table S1, from the published GWAS123 meta-analysis.6 We compared the CONFIRMED risk score between multiplex-affected probands and simplex-vitiligo-affected subjects by linear regression, assuming normal errors, with
where is the normalized (mean centered and scaled in GWAS123 controls) CONFIRMED risk score, represents the reference simplex-affected group, is coded 0 if the case is simplex and 1 if the case is multiplex, and represents ancestry-based PC3 described above. To test the hypothesis that , we used a two-sided Wald test, where p < 0.05 represented a significant difference in CONFIRMED risk score between simplex and multiplex cases.
We categorized GWAS probands into five different groups by total number of affected relatives in the family: simplex, 2 affected, 3 affected, 4 affected, and ≥5 affected. We fit a linear regression model, assuming normal errors:
where was coded in an ordinal fashion (simplex = 0; 2 affected = 1; 3 affected = 2; 4 affected = 3; ≥ 5 affected = 4). We used a two-sided Wald test to test the hypothesis , where p < 0.05 represented a significant relationship between the polygenic risk score and the number of affected relatives.
We analyzed the transmission of polygenic risk within multiplex-affected families by using the polygenic transmission disequilibrium test (pTDT)16 in the Extended Multiplex-Affected Family cohort described above. We used the FAMILY risk score because genotypes were only available for 14 of the confirmed loci in probands’ relatives. We considered pTDT p < 0.05 to represent significant average deviation between the unaffected, mid-parent risk score and affected offspring’s risk score.
We compared the single variant components of the CONFIRMED risk score in multiplex-affected probands versus simplex-vitiligo-affected subjects. Given an individual’s genotype, , at variant k and the effect size, , we calculated the proportion of their CONFIRMED risk score explained as = . We then compared the proportion of the CONFIRMED risk score explained by each of the 48 variants in simplex- and multiplex-vitiligo-affected probands by fitting a linear regression with normal errors:
To test the hypothesis that , we used a two-sided Wald test and controlled false discovery to a rate of 5%.
For individual variants that explained significantly different proportions of the CONFIRMED risk score in multiplex- versus simplex-affected probands, we assessed the allele frequency in multiplex- versus simplex-affected probands by fitting a logistic regression model:
where and represents the risk allele counts (0, 1, or 2) of the variant. We used a two-sided Wald test to test the hypothesis = 0, where p < 0.05 represented a significant difference in allele frequency in multiplex and simplex cases.
Results
Selection of the Best-Performing Polygenic Risk Score
Figure 2 shows the mean AUC from 10-fold cross-validation as a function of the p value threshold for inclusion of a variant in each polygenic risk score. Results are given for each of the four approaches that we used for calculating risk score (ALL, CLUMPED, CONFIRMED, and FAMILY). AUC varied from 0.76 (SE = 0.0046) to 0.84 (SE = 0.0033) over the risk scores considered. The CONFIRMED risk score produced the highest AUC.
Figure 2.

Comparison of AUC for Four Different Approaches for Vitiligo Risk Score Calculation at Multiple P Value Thresholds (∝T)
Four different approaches are shown: ALL (orange), CLUMPED (green), CONFIRMED (blue), and FAMILY (pink). The solid line represents the mean AUC across the 10-fold cross-validation sets, and the shaded region represents the standard error of the mean AUC.
As shown in Figure S2, the CONFIRMED risk score distribution in affected subjects and controls appears similar in GWAS123 and in the independent replication study; however, there is significant (p = 0.013, -0.096 SD) inflation of the estimated association between risk score and vitiligo in GWAS123 versus the replication study. Such inflation is expected because the variants and effect sizes we used in calculating the CONFIRMED score were derived from GWAS123. Thus, the ORs comparing the upper tails of the CONFIRMED risk score to the rest of the distribution might be more accurately represented by the independent replication study.
The CONFIRMED risk score performs better in predicting vitiligo risk than does vitiligo family history. If an individual has a sibling with vitiligo, the empirical risk of that individual developing vitiligo is 0.061,3 and the corresponding AUC17 based on sibling family history is 0.53. Thus, the CONFIRMED risk score (AUC 0.84) substantially outperforms empirical risk prediction based on family history alone.
Vitiligo risk is extremely high among subjects with CONFIRMED risk score above the 80th percentile (Table 1). Indeed, the risk of developing vitiligo among individuals in the top percentile of the CONFIRMED risk score (replication OR99 = 8.79) is substantially higher than for many other genetically complex diseases, including coronary artery disease (OR99 = 4.83), atrial fibrillation (OR99 = 4.63), type II diabetes (OR99 = 3.30), inflammatory bowel disease (OR99 = 3.87), and breast cancer (OR99 = 3.36).18
Table 1.
Risk of Vitiligo Associated with High CONFIRMED and FAMILY Risk Scores
| High-Risk Score Groupa | Reference Group |
CONFIRMED Risk Score |
FAMILY Risk Scorec |
|||
|---|---|---|---|---|---|---|
| Minimum Risk Scoreb | GWAS123 OR (95% CI) | Replication OR (95% CI) | Minimum Risk Scoreb | GWAS123 OR (95% CI) | ||
| Top 20% of distribution | Remaining 80% | 0.84 | 5.32 (4.90–5.77) | 4.87 (4.21–5.64) | 0.84 | 3.20 (2.95–3.47) |
| Top 10% of distribution | Remaining 90% | 1.29 | 6.21 (5.70–6.76) | 5.26 (4.42–6.29) | 1.30 | 3.50 (3.19–8.84) |
| Top 5% of distribution | Remaining 95% | 1.66 | 7.33 (6.64–8.08) | 6.20 (4.96–7.79) | 1.69 | 3.83 (3.42–4.29) |
| Top 1% of distribution | Remaining 99% | 2.35 | 11.10 (9.41–13.10) | 8.79 (5.85–13.78) | 2.39 | 4.51 (3.63–5.59) |
Abbreviations are OR - odds ratio and CI = confidence interval.
Risk score distribution defined by GWAS123 controls.
Minimum normalized risk score to be assigned to the high-risk group; the normalized score represents the standard deviations above the mean in GWAS123 controls.
FAMILY risk score cannot be calculated in the replication study because replication samples were not genotyped at all of the variants included in the FAMILY risk score.
Comparison of Polygenic Risk Score in Simplex- and Multiplex-Vitiligo-Affected Probands
To assess whether the elevated burden of common variants identified by GWAS contributes to family clustering of vitiligo cases, we first compared the CONFIRMED risk score in 391 multiplex- and 4,102 simplex-vitiligo-affected probands. As shown in Figure 3, the CONFIRMED risk score was significantly higher (p = 2.62 × 10−4, = 0.21 SD) in multiplex-affected probands than in simplex-affected subjects, suggesting that the common risk variants identified by vitiligo GWASs represent an important component of genetic risk in multiplex-affected families.
Figure 3.
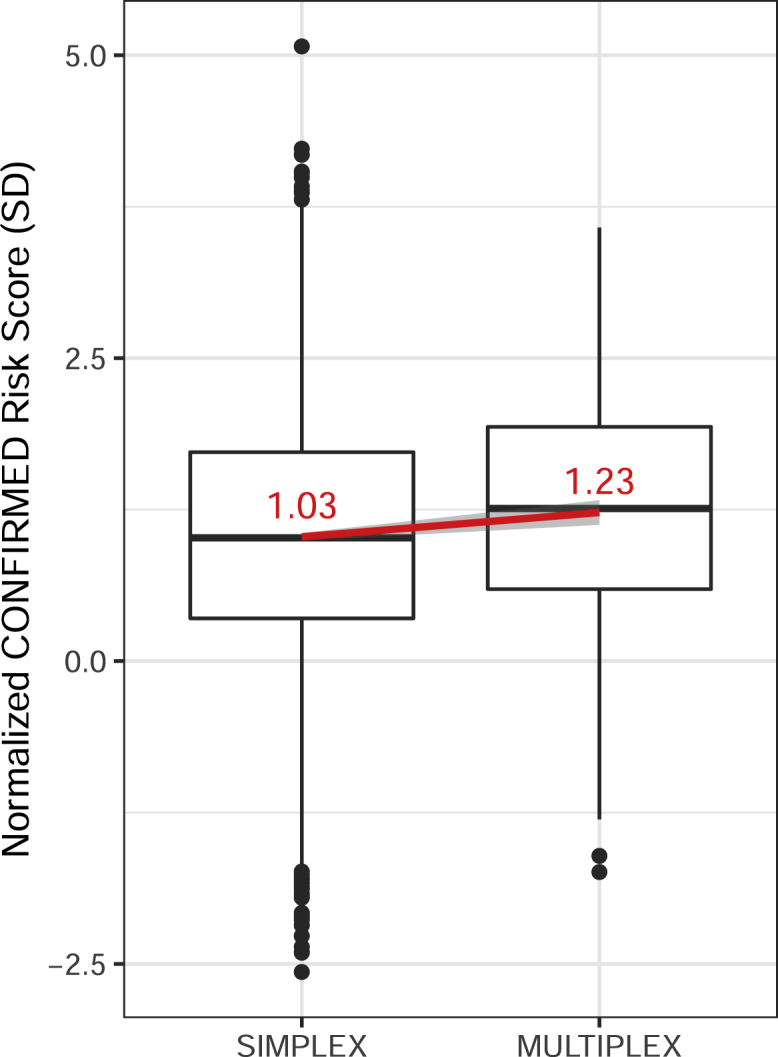
Comparison of CONFIRMED Risk Score in Simplex-Affected Subjects and Multiplex-Vitiligo-Affected Probands in GWAS123 + Replication Data
The y axis represents the normalized CONFIRMED risk score; units represent standard deviations (SDs) difference from the mean risk score in controls. Center horizontal lines denote the medians, and the value of the mean is shown in red text. The red line shows the fit linear regression line, and the corresponding 95% CI surrounds the line in gray. Boxes denote first through third quartiles. Each vertical bar extends from the box to the largest or smallest value, no farther than 1.5 times the inter-quartile range. Data beyond the vertical bars are considered outliers and are plotted individually.
To determine how the burden of common vitiligo risk alleles relates to the number of affected relatives in multiplex-affected families, we assessed the relationship between a proband’s CONFIRMED risk score and the total number of affected relatives in their family. As shown in Figure 4, we found a significant positive relationship between increasing risk score and increasing number of affected relatives (p = 1.85 × 10−4, = 0.10 SD per number of affected relatives). This suggests that the burden of common vitiligo risk alleles is an important underlying cause of family clustering, including in families with many affected relatives.
Figure 4.

Comparison of CONFIRMED Risk Score in Vitiligo Cases Categorized by the Number of Affected Relatives Reported by the Proband
The y axis represents the normalized CONFIRMED risk score; units represent standard deviations (SDs) difference from the mean risk score in controls. Horizontal lines denote the median, and the value of the mean is shown in red text. The red line shows the fit linear regression line, and the corresponding 95% CI surrounds the line in gray. Boxes denote first through third quartiles. Each vertical bar extends from the box to the largest or smallest value, no farther than 1.5 times the inter-quartile range. Data beyond the vertical bars are considered outliers and are plotted individually.
To confirm polygenic inheritance of common, low-to-moderate effect size loci in vitiligo family clustering, we performed pTDT by using the FAMILY risk score in 71 multiplex-affected families. We found that higher polygenic risk as defined by the FAMILY risk score is significantly over-transmitted from unaffected parents to affected offspring (p = 0.011, mean = 0.25 SD, SE = 0.087 SD). Altogether, these findings strongly support a polygenic inheritance model in which a high burden of common, low-to-moderate effect risk alleles constitutes a major driver of vitiligo risk in multiplex-affected families.
Polygenic Risk Score is High in a Multiplex-Vitiligo-Affected Family Segregating a Rare, High-Penetrance Variant
The largest vitiligo multiplex-affected family in our collection includes 13 close relatives affected by vitiligo. In this large family, we previously demonstrated genetic linkage of vitiligo to chromosome 1p31.37 and discovered a rare functional variant (dbSNP: rs41285370G>T, MAFgnomAD = 6.4 × 10−4) in the promoter region of FOXD3, which encodes a master regulator of melanocyte development.19 The rare T allele of dbSNP: rs41285370 is present in all 13 of the vitiligo-affected subjects, and the LOD score maximizes at a penetrance of 52%.7 Integrating the family pedigree with each individual’s FAMILY risk score and dbSNP: rs41285370 genotype (Figure 5), most affected family members have high polygenic risk scores. Most affected subjects with available genotype data (8/11) fell in the 80th percentile of the FAMILY risk score or higher and thus have polygenic risk corresponding to an OR of at least 3.2 (Table 1). Altogether, our findings in this family suggest that the effect of the rare FOXD3 promoter variant combines with the effects of common, low-penetrance risk alleles to drive especially high genetic risk of vitiligo in this remarkable family cluster.
Figure 5.

Annotated Pedigree for the Multiplex-Vitiligo-Affected Family with the Largest Number of Affected Relatives in Our Collection
In the largest multiplex-vitiligo-affected family in our collection, 13 individuals were affected. Symbols filled with black indicate individuals affected with vitiligo. The black arrow indicates the proband, who was genotyped genome-wide in GWAS123. Each individual is annotated with available genotype information; the top number represents the normalized FAMILY risk score (the units of the risk score are standard deviations from the mean in GWAS123 controls). ∗ denotes individuals whose risk scores fall in the top 20th percentile and ∗∗ denotes individuals whose risk scores fall in the top 10th percentile, as defined in Table 1. The second annotation is the individual’s genotype for the FOXD3 promotor variant dbSNP: rs41285370; the rare risk allele of dbSNP: rs41285370 (T) is shown in red. Most dbSNP: rs41285370 genotypes were inferred based on linkage data.7 Unk. = unknown, where genotype or risk score could not be inferred or calculated based on available data.
Individual Variant Contributions to the CONFIRMED Risk Score in Multiplex versus Simplex Cases
To determine whether the increased CONFIRMED risk score in multiplex cases is driven by a particular subset of variants, we assessed each variant’s average contribution to the score (Figure S3 and Table S3). Two variants, dbSNP: rs9271597 (FDR-adjusted p = 0.0046) and dbSNP: rs6059655 (FDR-adjusted p = 0.020), contributed significantly different proportions to the CONFIRMED risk score in multiplex versus simplex cases. dbSNP: rs9271597, which tags the MHC class II HLA-DRB1/DQA1 locus, contributed an average 4.8% in multiplex cases versus 4.1% in simplex cases. Furthermore, the frequency of the dbSNP: rs9271597 risk allele is significantly higher (p = 2.07 × 10−5, = 0.29) in multiplex versus simplex cases. The dbSNP: rs6059655/ASIP locus contributed an average 7.5% to the risk score in multiplex cases versus 7.8% in simplex cases; however, there is no significant difference (p = 0.20, = −0.21) in the allele frequency of dbSNP: rs6059655 in multiplex versus simplex cases. On the basis of the allele frequency differences, we conclude that elevated contribution of dbSNP: rs9271597 to the score in multiplex versus simplex cases is biologically meaningful, whereas that of dbSNP: rs6059655 is not, simply balancing the proportional increase of risk from dbSNP: rs9271597 (as the proportion of risk score explained by dbSNP: rs9271597 goes up, the proportion of risk score explained by other variants must go down).
Even when dbSNP: rs9271597 (as well as a correlated MHC class II variant, dbSNP: rs145954018) and the ASIP SNP (dbSNP: rs6059655) are removed from the CONFIRMED risk score, the score still remains significantly higher in multiplex cases than in simplex cases (p = 0.0064, = 0.15 SD). These results indicate that, although the HLA-DRB1/DQA1 locus might play a special role in vitiligo pathobiology in multiplex-affected families, the other confirmed vitiligo susceptibility loci additionally contribute to family case clustering.
Discussion
Genetic risk scores have attracted considerable attention for their potential applications as predictive or diagnostic tools, though there is some controversy surrounding their clinical utility in this regard.20 Here, we developed and applied several different polygenic risk scores as tools to compare genetic architecture underlying two potentially different subtypes of vitiligo: simplex and multiplex. We first defined the best-performing score by comparing four different approaches for calculating a vitiligo polygenic risk score from dense genotyping data. We obtained best performance with the CONFIRMED risk score, which consists of only the most-associated GWAS variants and which outperformed risk scores that included a larger number of variants. This suggests that a large proportion of overall genetic risk for vitiligo is described by a relatively small number of variants, many captured by the lead variants at the GWAS loci and in many cases known to be pathologically causal. Thus, much of the genetic architecture of vitiligo might be defined by a limited number of variants, rather than following an infinitesimal or omnigenic model,21 under which inclusion of a large number of additional variants would likely improve risk score performance. This further suggests that risk scores comprised of only the most-associated variant at each locus should be considered when comparing possible risk scores for other genetically complex diseases.
By using the best-performing CONFIRMED risk score, we find that the burden of common vitiligo risk alleles is significantly higher in multiplex-affected probands than in simplex-affected subjects and that polygenic risk is over-transmitted from unaffected parents to vitiligo-affected subjects in multiplex families. Together, these findings indicate that multiplex-vitiligo-affected families segregate a high burden of the common, low-effect risk alleles identified by GWASs. Furthermore, the risk score is roughly proportional to the number of affected relatives within a family, suggesting that high polygenic risk is an important contributor to overall genetic risk for vitiligo in both smaller and larger multiplex-affected families. High polygenic burden also appears to contribute to family clustering of other traits, including bipolar disorder,22 migraine,23 and testicular cancer,24 suggesting this phenomenon might be widespread among genetically complex diseases.
Although polygenic risk appears to be a major component of overall risk in multiplex-affected families, additional variants might also contribute. In our largest multiplex-affected family with 13 affected relatives, a rare, high-penetrance functional promoter variant in FOXD3, encoding a master regulator of melanocyte development, is carried by all affected family members.19 Nevertheless, even within this especially large family, most affected relatives additionally have a polygenic risk score in the 80th percentile or higher, suggesting that the effect of the rare, high-penetrance variant combines with high polygenic risk to drive overall vitiligo risk. Although all vitiligo-affected subjects in this family carry the high-penetrance FOXD3 variant, and most also have a high polygenic risk score, three affected subjects in this family had a relatively low risk score. These three subjects demonstrate that we are unable to capture all the components of vitiligo risk, even in this special family. Furthermore, such subjects with low risk scores lend support to concerns that genetic risk scores, even those with large effects, might not be as clinically predictive at the level of the individual as previously hoped.20
Interestingly, we find that the MHC class II vitiligo susceptibility locus enhancer variant dbSNP: rs9271597A is significantly over-represented in multiplex-affected probands compared to simplex-affected subjects. We previously showed that dbSNP: rs9271597A is associated with increased surface expression of cell-surface HLA-DR and HLA-DQ on immune cells.25 This suggests that HLA-related triggering of adaptive immune responses might be an especially important component of vitiligo pathobiology in multiplex-affected families. Despite apparent special importance of the MHC class II locus, the polygenic burden of the other vitiligo susceptibility loci also contributes to the phenomenon of family clustering. This has important clinical implications; whereas only about 8.6% of vitiligo cases derive from multiplex-affected families, the pathobiology of vitiligo appears to be basically the same in both familial and simplex cases. Accordingly, vitiligo therapies applicable to simplex-affected subjects are also likely to be applicable to multiplex-affected families.
We thus find that, for vitiligo, increased polygenic burden appears to contribute to the phenomenon of family clustering. Furthermore, the same set of common risk alleles that are relevant in simplex-affected subjects appear to contribute similarly to vitiligo risk in multiplex-affected families. Altogether, our findings suggest that the genetic architecture of vitiligo in multiplex-affected families is likely similar to that of sporadically-occurring vitiligo, though with additional polygenic burden. Nevertheless, not all members of multiplex-affected families have high risk scores, suggesting that some vitiligo family clusters might reflect enrichment of risk factors that are not captured by the current risk score. As such, multiplex-affected families with low risk scores seem especially likely to harbor rare, high-penetrance variants that could be identified by genetic linkage analysis. Alternatively, multiplex-vitiligo-affected families with low polygenic risk scores might reflect shared exposure to a strong environmental trigger, which could be identified by follow-up epidemiologic analysis. In either case, we suggest that families with low polygenic risk scores are atypical, and further investigation of these families might facilitate discovery of novel risk factors.
Declaration of Interests
The authors declare no competing interests.
Acknowledgments
This work was supported by grants R01AR056292, R01AR057212, R01AR065951, and P30AR057212 from the National Institutes of Health. G.H.L.R. was supported by training grant T32 AR007411-31A1 from the National Institutes of Health.
Published: June 18, 2019
Footnotes
Supplemental Data can be found online at https://doi.org/10.1016/j.ajhg.2019.06.013.
Accession Numbers
Case genotype and phenotype data for GWAS1, GWAS2, and GWAS3 subjects have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under accession numbers phs000224.v1.p1, phs000224.v2.p1, phs000224.v3.p2, and phs000224.v4.p2 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000224.v3.p2). GWAS summary statistics have been deposited in the NHGRI-EBI Catalog of published genome-wide association studies (https://www.ebi.ac.uk/ega/studies/phs000224.v1.p1).
Web Resources
Database of Genotypes and Phenotypes, https://www.ncbi.nlm.nih.gov
NHGRI-EBI Catalog of Published Genome-Wide Association Studies, https://www.ebi.ac.uk
PLINK-1.9, https://www.cog-genomics.org/plink2/
1KG Phase 3, http://www.internationalgenome.org/category/phase-3/
pROC, https://cran.r-project.org/web/packages/pROC/index.html
Supplemental Data
References
- 1.Hafez M., Sharaf L., Abd el-Nabi S.M. The genetics of vitiligo. Acta Derm. Venereol. 1983;63:249–251. [PubMed] [Google Scholar]
- 2.Das S., Majumder P.P., Majumdar T., Haldar B., Rao D.J.G.e. Studies on vitiligo. II. Familial aggregation and genetics. Genet. Epidemiol. 1985;2:255–262. doi: 10.1002/gepi.1370020303. [DOI] [PubMed] [Google Scholar]
- 3.Alkhateeb A., Fain P.R., Thody A., Bennett D.C., Spritz R.A. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16:208–214. doi: 10.1034/j.1600-0749.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 4.Jin Y., Birlea S.A., Fain P.R., Gowan K., Riccardi S.L., Holland P.J., Mailloux C.M., Sufit A.J., Hutton S.M., Amadi-Myers A. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N. Engl. J. Med. 2010;362:1686–1697. doi: 10.1056/NEJMoa0908547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Y., Birlea S.A., Fain P.R., Ferrara T.M., Ben S., Riccardi S.L., Cole J.B., Gowan K., Holland P.J., Bennett D.C. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat. Genet. 2012;44:676–680. doi: 10.1038/ng.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin Y., Andersen G., Yorgov D., Ferrara T.M., Ben S., Brownson K.M., Holland P.J., Birlea S.A., Siebert J., Hartmann A. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 2016;48:1418–1424. doi: 10.1038/ng.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alkhateeb A., Stetler G.L., Old W., Talbert J., Uhlhorn C., Taylor M., Fox A., Miller C., Dills D.G., Ridgway E.C. Mapping of an autoimmunity susceptibility locus (AIS1) to chromosome 1p31.3-p32.2. Hum. Mol. Genet. 2002;11:661–667. doi: 10.1093/hmg/11.6.661. [DOI] [PubMed] [Google Scholar]
- 8.Spritz R.A., Gowan K., Bennett D.C., Fain P.R. Novel vitiligo susceptibility loci on chromosomes 7 (AIS2) and 8 (AIS3), confirmation of SLEV1 on chromosome 17, and their roles in an autoimmune diathesis. Am. J. Hum. Genet. 2004;74:188–191. doi: 10.1086/381134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majumder P.P., Das S.K., Li C.C. A genetical model for vitiligo. Am. J. Hum. Genet. 1988;43:119–125. [PMC free article] [PubMed] [Google Scholar]
- 10.Nath S.K., Majumder P.P., Nordlund J.J. Genetic epidemiology of vitiligo: Multilocus recessivity cross-validated. Am. J. Hum. Genet. 1994;55:981–990. [PMC free article] [PubMed] [Google Scholar]
- 11.Picardo M., Taieb A. Springer; Heidelberg, New York: 2019. Vitiligo. [Google Scholar]
- 12.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y., Roberts G.H.L., Ferrara T.M., Ben S., van Geel N., Wolkerstorfer A., Ezzedine K., Siebert J., Neff C.P., Palmer B.E. Early-onset autoimmune vitiligo associated with an enhancer variant haplotype that upregulates class II HLA expression. Nat. Commun. 2019;10:391. doi: 10.1038/s41467-019-08337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee A.B., Luca D., Klei L., Devlin B., Roeder K. Discovering genetic ancestry using spectral graph theory. Genet. Epidemiol. 2010;34:51–59. doi: 10.1002/gepi.20434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartung J., Knapp G., Sinha B.K. John Wiley & Sons; 2011. Statistical meta-analysis with applications. [Google Scholar]
- 16.Weiner D.J., Wigdor E.M., Ripke S., Walters R.K., Kosmicki J.A., Grove J., Samocha K.E., Goldstein J.I., Okbay A., Bybjerg-Grauholm J., iPSYCH-Broad Autism Group. Psychiatric Genomics Consortium Autism Group Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017;49:978–985. doi: 10.1038/ng.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jostins L., Barrett J.C. Genetic risk prediction in complex disease. Hum. Mol. Genet. 2011;20(R2):R182–R188. doi: 10.1093/hmg/ddr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khera A.V., Chaffin M., Aragam K.G., Haas M.E., Roselli C., Choi S.H., Natarajan P., Lander E.S., Lubitz S.A., Ellinor P.T., Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alkhateeb A., Fain P.R., Spritz R.A. Candidate functional promoter variant in the FOXD3 melanoblast developmental regulator gene in autosomal dominant vitiligo. J. Invest. Dermatol. 2005;125:388–391. doi: 10.1111/j.0022-202X.2005.23822.x. [DOI] [PubMed] [Google Scholar]
- 20.Wald N.J., Old R. The illusion of polygenic disease risk prediction. Genet. Med. 2019;2019:12. doi: 10.1038/s41436-018-0418-5. [DOI] [PubMed] [Google Scholar]
- 21.Boyle E.A., Li Y.I., Pritchard J.K. An expanded view of complex traits: From polygenic to omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fullerton J.M., Koller D.L., Edenberg H.J., Foroud T., Liu H., Glowinski A.L., McInnis M.G., Wilcox H.C., Frankland A., Roberts G., Bipolar High Risk Study Group, BiGS Consortium Assessment of first and second degree relatives of individuals with bipolar disorder shows increased genetic risk scores in both affected relatives and young At-Risk Individuals. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2015;168:617–629. doi: 10.1002/ajmg.b.32344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gormley P., Kurki M.I., Hiekkala M.E., Veerapen K., Häppölä P., Mitchell A.A., Lal D., Palta P., Surakka I., Kaunisto M.A., 23andMe Research Team. International Headache Genetics Consortium (IHGC) Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron. 2018;99:1098. doi: 10.1016/j.neuron.2018.08.029. [DOI] [PubMed] [Google Scholar]
- 24.Loveday C., Law P., Litchfield K., Levy M., Holroyd A., Broderick P., Kote-Jarai Z., Dunning A.M., Muir K., Peto J., UK Testicular Cancer Collaboration, The PRACTICAL Consortium Large-scale analysis demonstrates familial testicular cancer to have polygenic aetiology. Eur. Urol. 2018;74:248–252. doi: 10.1016/j.eururo.2018.05.036. [DOI] [PubMed] [Google Scholar]
- 25.Cavalli G., Hayashi M., Jin Y., Yorgov D., Santorico S.A., Holcomb C., Rastrou M., Erlich H., Tengesdal I.W., Dagna L. MHC class II super-enhancer increases surface expression of HLA-DR and HLA-DQ and affects cytokine production in autoimmune vitiligo. Proc. Natl. Acad. Sci. USA. 2016;113:1363–1368. doi: 10.1073/pnas.1523482113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.