

Abstract
Previous pressure-jump NMR experiments on a pressure-sensitized double mutant of ubiquitin showed evidence that its folding occurs via two parallel, comparably efficient pathways: A single barrier and a two-barrier pathway. An interrupted folding NMR experiment is introduced, where for a brief period the pressure is dropped to atmospheric conditions (1 bar), followed by a jump back to high pressure for signal detection. Conventional, forward sampling of the indirect dimension during the low-pressure period correlates the 15N or 13C′ chemical shifts of the unfolded protein at 1 bar to the 1H frequencies of both the unfolded and folded proteins at high pressure. Remarkably, sampling the data of the same experiment in the reverse direction yields the frequencies of proteins present at the end of the low-pressure interval, which include unfolded, intermediate, and folded species. Although the folding intermediate 15N shifts differ strongly from natively folded protein, its 13C′ chemical shifts, which are more sensitive probes for secondary structure, closely match those of the folded protein and indicate that the folding intermediate must have a structure that is quite similar to the native state.
Keywords: protein folding, pressure jump, folding pathway, folding intermediate, chemical shift, NMR, ubiquitin
INTRODUCTION
Proteins transition from their initially highly disordered to their well-defined native state on a time scale that is many orders of magnitude faster than would be expected from a random search of conformational space, pointing to the existence of pathways in this process.1 For many small proteins, a single barrier on such pathways, separating the unfolded from the folded state, suffices to explain a large variety of experimental data, mostly derived from rapid mixing, stopped-flow experiments. It is now well established that the actual barrier crossing event is exceedingly fast and diffusion-limited,2–5 and therefore difficult to observe directly. The presence of additional, lower barriers would result in short-lived intermediates, which would have low populations and be difficult to detect. NMR relaxation dispersion measurements, which are exquisitely sensitive for identifying such lowly populated states, have characterized a highly transient, on-pathway folding intermediate for a Fyn-SH3 mutant,6–8 suggesting that, perhaps, such intermediates are more prevalent than generally assumed. Most commonly, the protein folding process is probed by Trp fluorescence, fluorescence resonance energy transfer (FRET), or backbone amide hydrogen exchange.9–13 NMR spectroscopy has played an important role in these latter studies, yielding highly quantitative site-specific hydrogen exchange rates with a temporal resolution that is only limited by the speed at which the exchange process can be quenched.14–16 Such data yield the rates at which H-bonds form after initiation of the folding process. If differences in the hydrogen exchange protection profiles are seen for various regions of the protein, these data point to the presence of folding intermediates, i.e. multiple barriers on the folding pathway.17–19 In such work, NMR spectroscopy is used in an indirect manner, to probe the exchange-quenched sample at residue-specific resolution, and comparable results now also have become accessible by advances in mass spectrometry, thereby requiring much less sample.16, 20–21
Direct monitoring of protein folding by NMR is also possible, but due to the long time required to generate an NMR spectrum such studies initially were limited to slower folding or unfolding systems.22–25 However, improvements in technology about a decade ago have much increased the accessible time resolution.26–27
In non-equilibrium folding experiments, experimental conditions are typically changed abruptly from those favoring the unfolded state to those favoring the native state. This may be accomplished by a sudden jump in pH, dilution of denaturant, release of a co-factor, or by a sudden drop in hydrostatic pressure. The latter method, switching the pressure, is particularly benign, allowing the protein to be probed under native buffer conditions. However, a prerequisite for pressure-induced unfolding is that the protein volume of the unfolded state is substantially lower than that of the folded state, in practice by at least ~50 mL/mole. Equilibrium NMR spectra, recorded as a function of pressure, typically over the 0.1–3 kbar range, then often permit the simultaneous observation of unfolded and folded protein resonances in a slow exchange equilibrium, and can yield site-specific information on the state of each residue.28–32 Analogous to pressure-jump fluorescence experiments,33 the temporal change in the NMR spectrum following a step change in pressure can reveal kinetic information, provided the kinetics are slower than the duration of the pressure change, which often represents a challenging requirement for NMR spectroscopy.25, 34 Recently, this limitation has been addressed by the development of dedicated hardware that rapidly increases or decreases the pressure in the NMR sample cell in a manner synchronized with the data acquisition process.35–36 The ability to repeat the pressure-jump process thousands of times then provides access to processes on time scales much faster than those needed to collect an entire multi-dimensional NMR spectrum.26, 36–37
In our first application of the newly developed pressure jump NMR hardware, we studied ubiquitin, sensitized to pressure by the introduction of two Val to Ala cavity-generating mutations (V17A and V26A), while retaining the same native structure as the wild-type protein.36, 38 Below, this double mutant is referred to as VA2-ubiquitin. Ubiquitin has functioned as a model protein in numerous experimental and computational folding studies,27, 39–49 and is very well suited for detailed NMR studies, therefore providing a perfect system for technological and methodological developments. Prior work citing evidence for an intermediate in ubiquitin’s folding pathway,13, 39, 50 has been contested.51 Our work conclusively showed the presence of parallel, single-barrier and double-barrier pathways, with their relative efficiency being a strong function of temperature.36 A separate Cδ2H3 signal for Leu-50 was transiently visible for the double barrier intermediate,36 and the average 15N chemical shift during the folding process deviated from two-state behavior. This latter observation enabled estimation of the folding intermediate chemical shifts, which differed very substantially from those of both the unfolded and folded native states of the protein.52 Remarkably, the hydrogen exchange protection of the intermediate and folded states were found to be very similar, with significantly lower protection only observed for two residues in the folding intermediate.38
Here, we demonstrate that direct observation of the 15N and 13C′ chemical shifts of the folding intermediate becomes accessible by very minor modifications of a commonly used pulse scheme, where signals are digitized in a direction opposite to that conventionally used. For samples at equilibrium, the direction of sampling has no impact on the spectrum. In contrast, for the non-equilibrium, folding experiments, the forward and reverse sampled spectra are quite different. Significantly, reverse sampled spectra yield chemical shifts of the folding intermediate that are the first step towards determining its detailed molecular structure.
RESULTS AND DISCUSSION
Forward sampled pressure-jump HSQC and HNCO pulse schemes.
The NMR resonance frequencies of both the folded and unfolded states of a protein are quite sensitive to hydrostatic pressure.30, 53 Switching of the pressure during an NMR pulse scheme therefore is preferably performed during a period where the magnetization is stored along the z axis, such that small fluctuations in the timing of the switching that are invariably associated with the mechanical movement of the valve have minimal effect on the observed signal. Many NMR pulse sequences have such periods built into them naturally, and often utilize such delays to apply pulsed field gradients to remove spurious magnetization transfer pathways.54–55 For example, in the in-phase, composite-pulse-decoupled heteronuclear single quantum correlation (HSQC) experiment,56 the intervals just after the first refocused INEPT transfer of 1HN magnetization to 15N, and prior to reverse INEPT transfer back to 1HN, can be used for this purpose (SI Figure 1A). A fully analogous scheme, which is essentially a two-dimensional version of the common HNCO experiment,57 can be used to transfer magnetization from Hz to C′zNz, prior to generating transverse 13C′ magnetization by a 90° 13C′ pulse (SI Figure 1C). In our pressure-jump experiments, the pressure is dropped to 1 bar just after completing the first magnetization transfer from 1H to 15N (for HSQC), or to C′zNz (for HNCO; Figure 1). We allow the folding to occur for a low-pressure duration T, during which 15N or 13C′ chemical shift is encoded for a variable t1 evolution period. When recorded in the usual, forward manner (Figure 1B, Top), the signal intensity at the end of the low-pressure period, prior to reverse INEPT transfer to 1H, is proportional to
| 1 |
where ωX denotes the angular 15N or 13C′ frequency of the unfolded protein, assuming no residual folded protein remains present at the end of the high-pressure preparation period. [U]0 is the concentration of unfolded protein present at the start of the low-pressure period. R2U corresponds to the sum of the transverse relaxation rate of the unfolded species, R2, and the forward folding rates, kU→I and kU→F, from the unfolded to the intermediate (two-barrier pathway) and from the unfolded to the folded state (single-barrier pathway), respectively.
Figure 1.
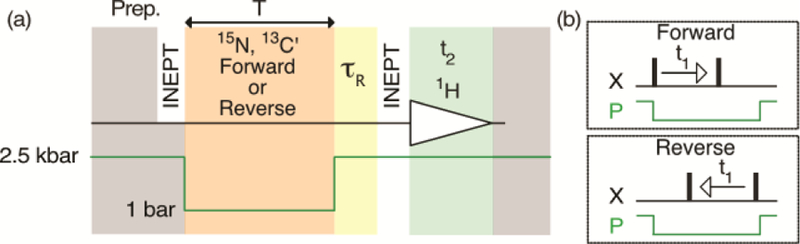
Simplified schematic of the forward and reverse detected pressure-jump 2D 1H-15N HSQC and 1H-13C′ HNCO experiments. In both schemes, the spin system is prepared (Prep.) by a long (ca. 10 s) high pressure protein denaturation interval, which is terminated by a refocused INEPT transfer to 15Nz magnetization (HSQC) or to antiphase 13C′z15Nz magnetization (HNCO). During the subsequent low pressure period (total duration T, ca. 70 ms) the 15N (HSQC) or 13C′ (HNCO) frequency is encoded either in the regular, forward manner (B, top), or in the reverse manner (B, bottom), where the second 15N (HSQC) or 13C′ (HNCO) pulse is always applied just prior to the jump back to high pressure, and the first pulse is moved stepwise to the left. Protein folding takes place during the low-pressure interval but is interrupted by the jump back to high pressure, prior to signal detection. A vibration decay delay (yellow, ~150 ms), where magnetization is stored along the z axis, precedes transfer of magnetization to 1H for detection during t2. Full details of the pulse sequence are provided in SI Figure S1.
Concentrations of the three species (plotted in Figure S2A), unfolded [U(t)], intermediate [I(t)], and folded [F(t)], at time t after the pressure drop are given by:
| 2a |
| 2b |
| 2c |
With .
While a fraction of protein will fold after the switch to low pressure and will precess with a folded chemical shift for the remainder of the t1 period, its signal will remain invisible in the HSQC spectrum as any signal with zero intensity t1=0 will have an integral of zero in the resulting spectrum. Moreover, the transverse phase of magnetization of a protein that switches from unfolded to folded at time τ after the first 90° pulse accumulates a transverse phase ωUτ and will evolve for the remainder of t1 with ωF. At the end of the t1 evolution period its phase will be ωUτ + ωF(t1 - τ) and signals from proteins that switched to the folded state at different values of τ therefore will destructively interfere with one another (unless ωU ≈ ωF) and not contribute significantly to observed signal (Figure S2A, S3). The same applies for any folding intermediate signal. Note that weak signals of the intermediate state are visible in the noise-free simulations (Figure S3B,D), but result from proteins that refolded during the “padding delay” between the pressure drop and the actual start of t1 evolution (ca. 5 ms), included in the simulations, but below the detection threshold in the experimental spectra. The final, forward-sampled spectrum therefore yields resonances that carry the unfolded 15N frequency at 1 bar in the F1 dimension of the final 2D spectrum, correlated to 1HN frequencies of both the unfolded and folded protein at high-pressure in the F2 dimension (Figure 2A and Figure S3B,D). Similarly, the forward sampled 2D HNCO spectrum shows 13C′ resonances of the unfolded protein at 1 bar (F1 dimension) correlated to 1HN frequencies of the next residue for both the unfolded and folded protein at high pressure (Figure 3A). Note that the VA2-ubiquitin unfolding rate at room temperature is slow, and proteins that switch to the folded state during the low-pressure period have insufficient time to unfold prior to 1H detection at high pressure (Figure S2A).
Figure 2.

Small regions of the (A) forward and (B) reverse sampled 2D 1H-15N HSQC spectra of a sample containing 210 mM 15N ubiquitin, pH 6.4. Full spectra are shown in Figures S5 and S6. Spectra are recorded at 600 MHz 1H frequency, using a cryogenic probehead equipped with a z-axis pulsed field gradient accessory. The temperature was regulated at 25 °C but drops by 3 °C due to adiabatic expansion during the brief low-pressure period. The forward recorded spectrum (A) primarily shows F1 dimension correlations for species present at the start of the low-pressure period, i.e. unfolded protein. These signals are correlated in the F2 dimension with unfolded 1H frequencies at 2.5 kbar for proteins that did not cross the folding barrier during the low-pressure period, or to folded 1H frequencies at 2.5 kbar for proteins that crossed the folding barrier. The reverse sampled spectrum (B) shows in the F1 dimension the 15N frequencies of all species present at the end of the low-pressure period, i.e., unfolded, intermediate, and folded, correlated with their respective 1H frequency at high pressure. Correlations are labeled by residue number, with the first superscript referring to the 15N chemical shift of the unfolded (U), Intermediate (I) or Folded (F) state at the end of the low-pressure interval, and the second superscript denoting the state of the protein during detection. No I-state 1H signals are observed because the majority of I-state proteins revert to U prior to detection; a ca. 2-fold smaller fraction crosses the barrier to the folded state, yielding resonances superscripted IF. The pattern of correlations for S57 is marked by red dashed rectangles.
Figure 3.
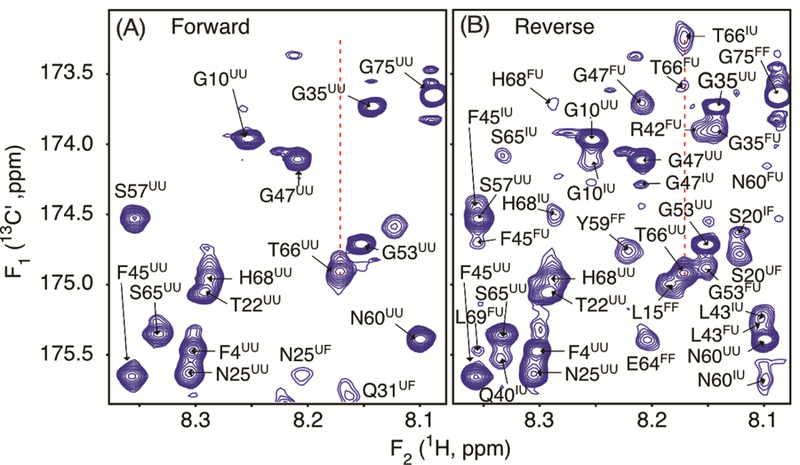
Small regions of the 600 MHz (A) forward and (B) reverse sampled 2D 1H-13C′ HNCO spectra of a sample containing 125 μM 15N/13C′/2H-VA2-ubiquitin, pH 6.4, 25 °C. Full spectra are shown in Figures S7 and S8. Fully analogous to the spectra of Figure 2, the forward spectrum (A) primarily shows F1 dimension correlations for unfolded protein, correlated to 2.5 kbar 1H F2 frequencies of either folded or unfolded protein. Analogously, the reverse sampled spectrum (B) shows in the F1 dimension the 1-bar 13C′ frequencies of U, I, and F species, correlated with 2.5 kbar 1H frequency of the amide of the next residue. Each peak number indicates the residue that is 13C′ labeled, with the first superscript referring to the state of the protein during 13C′ evolution at 1 bar, and the second superscript denoting the state during 1H detection at 2.5 kbar.
Reverse sampled, interrupted folding HSQC and HNCO pulse schemes.
Simply by changing the timing of the pulses that generate the t1 modulation, the same pulse schemes used above for detection of the unfolded 15N and 13C′ resonances at 1 bar can be used for detecting folding intermediates: By sampling the t1 evolution in the reverse direction (Figure 1B-bottom), starting from the end of the low-pressure interval, after folding intermediates and folded proteins have developed, their signals will modulate the observed spectrum (Figure S3F,H). If the folding intermediate were resistant to high pressure, such a signal would resonate at the 1H frequency of the folding intermediate during the 1H detection period (F2 dimension). However, as was observed previously in the classic interrupted folding experiments of Kiefhaber,26, 58 intermediates are commonly less stable than the folded state and rapidly revert to the unfolded state once conditions are switched back to denaturing, i.e., high pressure. Therefore, the t2-detected 1H signal of the I state is found at the high-pressure unfolded 1H frequency (Figure 2B and Figure 3B). For VA2-ubiquitin, we also find a minor fraction of I-state protein that switches to the native state prior to 1H detection. This latter fraction gives rise to signals at the I-state 15N frequency in the F1 dimension, and to the native-state, high-pressure HN frequency in the F2 dimension (Figure S3H). The intensity ratio of the I-state peaks, correlated to the unfolded and folded HN frequencies, corresponds to the ratio of the respective transition rates at high pressure kI→U / kI→F. The total amplitude of the folding intermediate signal, I, depends on the fraction of proteins that have switched to the intermediate state at time T after the drop to low pressure, but have not progressed to the folded state (Eq. 2). The integrated peak intensity of a U, I, or F resonance of the reverse-sampled time domain corresponds to the time domain point sampled at t1 = 0, i.e., to the values [U](T), [I](T), and [F](T), where T is the duration after the pressure drop at which the reverse sampling is started (Figure 1).
The decay profiles of the U-, I-, and F-state components with respect to t1 are quite different from one another, with [U](t1) only being attenuated by true transverse relaxation, whereas [I](t1) and [F](t1) drop to zero at t1=T, even in the absence of transverse relaxation, since no intermediate or folded magnetization is present immediately after the pressure drop. Details are presented in Supporting Information Figure S3. The different, non-exponential decay profiles for the [I](t1) and [F](t1) components result in non-Lorentzian line shapes after Fourier transformation, with the I and F state resonances being broader than those of the U fraction that remains unfolded during the low-pressure period (Figure S3). The differences in line shapes require that integrated peak intensities be measured to obtain accurate concentrations of the U-, I-, and F- states of ubiquitin. These intensities cannot easily be measured because of the crowded spectra obtained in our study. Nevertheless, the spectra obtained by the reverse sampling method unambiguously reveal the 15N and 13C′ frequencies of the intermediate state.
Comparison of the forward sampled 15N-1H HSQC (Figure 2A) with the reverse sampled spectrum (Figure 2B) shows a multitude of new resonances in the latter. For example, the dashed red boxes outline correlations observed for residue S57. Each cross peak is marked by two superscripts, which refer to the state of the protein during t1 evolution and during t2 detection. For instance, the cross peak labeled S57IF (top left in Figure 2B) corresponds to signal of the intermediate (I-state) 15N frequency in the F1 dimension, and the S57 1H frequency of the folded protein in the F2 dimension. Note that the 1H frequencies of the folded state at 2.5 kbar differ substantially from those at 1 bar but can easily be identified by recording a pressure jump 1H-15N 2D HSQC spectrum with preparation and 15N evolution at 1 bar, and 1H detection at 2.5 kbar (Figure S4). Figure 2A shows two intense resonances, S57UF and S57UU, from proteins that were unfolded at the end of the preparation period (marked “Prep.” in Figure 1), and either switched to the folded state during the low-pressure period (S57UF) or remained unfolded (S57UU). A very weak resonance, marked S57FF (top left Figure 2A), corresponds to the small fraction (~6%) of proteins that remained folded at the end of the high-pressure preparation period. Because unfolding of the protein at high pressure is very slow, the corresponding S57FU resonance has vanishing intensity. By contrast, the reverse sampled spectrum (Figure 2B) shows a strong S57FF peak with just above it a very weak S57IF peak from the small fraction of proteins that were in the intermediate state at the end of the low-pressure period but crossed the barrier to the folded state prior to 1H detection. A considerably stronger S57IU resonance, at the same 15N F1 frequency, is observed for I-state proteins that switched back to the unfolded state prior to 1H detection. The slow unfolding rate at high pressure causes only a very small (~10%) fraction of proteins that had reached the folded state at the end of the low-pressure period to revert back to the unfolded state, giving rise to a very weak S57FU peak (Figure 2B). A stronger S57UU resonance corresponds to proteins that remained unfolded during the low-pressure interval.
Measurement of I-state 13C′ chemical shifts.
Fully analogous to the forward and reverse sampled 15N-1H HSQC spectra, discussed above, the same forward and reverse sampling modes can be incorporated in the 2D HNCO pulse scheme (SI Figure S1C, D). The forward sampled HNCO spectrum then shows correlations between the U-state 13C′ shift of residue i and the 1HN resonance of residue i+1 in both the unfolded and folded state. For example, the dashed vertical line in Figure 3A, at the 1HN frequency of residue L67 shows a correlation to T66-13C′. At the same frequency in the reverse sampled spectrum (Figure 3B), the same T66UU signal is observed, in addition to a correlation to the intermediate state, T66IU, and a very weak resonance, T66FU from proteins that were folded at the end of the low-pressure interval but subsequently unfold during the ca. 150-ms high-pressure period that separated 15N evolution from 1H detection. Analogous T66FF and T66IF signals are present in the full spectrum, outside the region shown in Figure 3. Although the signal-to-noise ratio of the HNCO spectra is nearly two-fold lower than for the HSQC spectra, recorded in a comparable amount of time, I-state 13C′ resonances could be identified with confidence for 70 out of the potential 72 residues that are expected to give rise to such signals. 13C′ signals of three residues preceding a Pro, the C-terminal residue G76 were missing, as well as M1 due to rapid exchange of the amide of Q2, while resonance overlap precluded assignment of I23 and D52 in the crowded region of the reverse sampled HNCO spectrum (Figure 3B; Figure S8).
Chemical shift characterization of the folding intermediate.
The 15N chemical shifts of the folding intermediate measured from the folding-interrupted HSQC spectrum agree closely with values obtained previously from the pseudo-3D pressure-jump experiment (Figure 4), which measured the ensemble-averaged 15N shift during the folding process by sampling a single time point in the indirect dimension (stroboscopic observation).52 The I-state frequency was then extracted by means of a fitting procedure.52 However, the stroboscopic measurement yielded relatively large uncertainties in the ensemble-averaged 15N frequency of each residue, thereby adversely impacting the precision at which the frequency of the I-state component could be obtained. The current method yields highly precise peak positions but suffers from increased resonance overlap as the number of resonances in the reverse-sampled, folding-interrupted spectra is three-fold larger than that of the fully folded, or fully unfolded protein. Consequently, the set of residues for which unambiguous I-state 15N frequencies could be determined is somewhat smaller (59 vs. 65 residues) than in the earlier work. With a root-mean-square difference (RMSD) of only 0.37 ppm (Figure 4B), agreement with the earlier measurement is very good, and results confirm that the I-state differs most from the F-state for the C-terminal strand, β5, and its preceding loop, strand β1, and the C-terminal residues of strand β3, with β5 being sandwiched between β1 and β3 in the natively folded state (Figure 5A,B).
Figure 4.
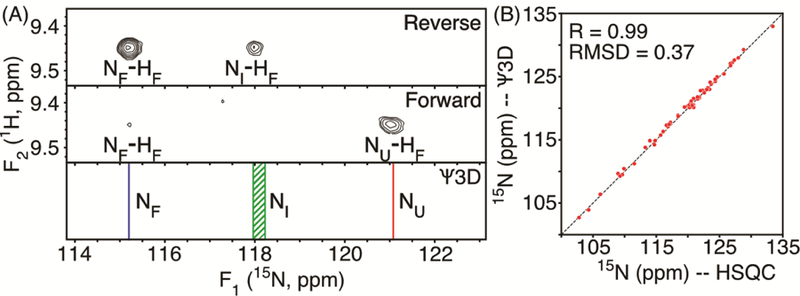
Comparison of spectral data of the E64 amide of VA2-ubiquitin, recorded with the pressure-jump 2D 1H-15N HSQC experiment of Figure 1, and 15N chemical shifts previously derived from stroboscopic observation of 15N shifts in a pseudo-3D (Ψ3D) measurement.52 (A) 15N F1 strips taken at the E64 F2 1HN frequency of folded ubiquitin at 2.5 kbar, with the bottom strip showing the results of previous fitting of the Ψ3D data of this residue. (B) Correlation plot of the 15N I-shifts extracted from the Ψ3D spectrum (y-axis) with those of the reverse-sampled pressure-jump HSQC spectrum (x-axis). The Pearson correlation coefficient equals 0.99, with an RMSD of 0.37 ppm.
Figure 5.
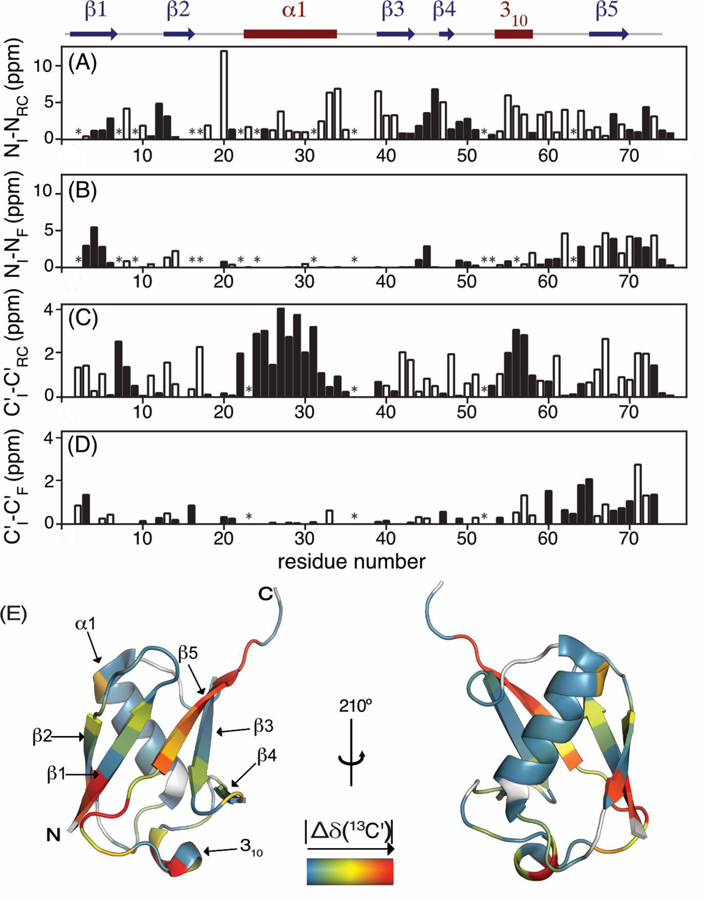
Comparison of 1-bar 15N and 13C′ chemical shifts of the VA2-ubiquitin folding intermediate with those of the folded state (δF) and with random coil values (δRC) obtained with the program POTENCI.63 Filled and open bars correspond to positive and negative values, respectively. (A) δ I(15N) - δ F(15N); (B) δ I(15N) - δRC(15N); (C) δ I(13C′) - δ F(13C′); (D) δ I(13C′) - δRC(13C′); (E) |Δδ(13C′)| =|δ I(13C′) - δ F(13C′)| color coded on a backbone ribbon structure of the native state of ubiquitin (PDB entry 2MJB). Residues for which |Δδ(13C′)| could not be determined are shown in grey.
15N chemical shifts are exquisitely sensitive to a wide range of structural parameters, including H-bond strength, sidechain rotamer positioning, backbone torsion angles, and electrostatic effects from nearby charged groups,59 complicating the interpretation of the 15N chemical shift differences in structural terms. In contrast to 15N, 13C′ chemical shifts correlate fairly well with protein secondary structure: 13C′ resonances in α-helical secondary structure are typically downfield shifted by 2–3 ppm from random coil, whereas upfield (negative) secondary shifts of 1–2 ppm are typically observed in β-sheet. Using the reverse sampled HNCO spectrum we obtained a set of 70 13C′ I-state chemical shifts (Figure 5C–E). The chemical shifts of the I-state reveal important new information on the secondary structure of this transient structure. Particularly striking is the closeness of the I-state 13C′ shifts to those of the folded state for residues F4-T55. Substantial chemical shift differences between the folded and intermediate states are observed for residues S57-L73, which correspond to the C-terminal β5 strand, its preceding loop N60-S65, and the short 310 helix preceding this loop. In addition, chemical shifts of residues Q2 and I3 in β1 differ considerably between I- and F-states. The secondary 13C′ shifts of Q2 and I3 in the I-state are ca −1.5 ppm, indicative of β-strand. Residues R72 and L73, which have close to random coil shifts in the native structure, show substantial negative and positive secondary shifts, respectively, in the I-state, indicative of well-defined structure for these residues. The chemical shift differences relative to the natively folded protein are mapped on a ribbon diagram of the folded structure (Figure 5E) and reveal more localized differences than previously seen from the 15N chemical shift differences52 (Figure 5B). Strands β3 and β1 pair with the C-terminal strand in the native structure, and it therefore appears likely that any significant difference in the N60-S65 loop, which contains two reverse turns in the native structure, could propagate into β5 and thereby β1. A structural analysis, including the collection of pressure-jump NOE data, currently in progress, will likely resolve the detailed structural differences between the intermediate and native states of the protein.
EXPERIMENTAL SECTION
Sample Preparation.
15N- and 13C-15N-2H-labeled VA2-ubiquitin was expressed and purified as described previously.38 Importantly, a final HPLC step was found necessary to prevent proteolysis at high pressure, where the denatured state of the protein is highly susceptible to proteolytic cleavage. Samples were dissolved in 25 mM potassium phosphate buffer, pH 6.4, using 1% D2O for a field frequency lock.
NMR spectroscopy.
All NMR experiments were recorded on a Bruker 600 MHz spectrometer equipped with cryogenic probehead with a z-axis pulsed field gradient accessory. A 2.8 mm ID zirconia sample cell, rated for static pressures of up to 3 kbar (Daedalus, Inc.) was used.60 The hardware enabling rapid pressure switching, under control of the Bruker pulse program, has been described previously.36 Both forward and reverse sampled pressure-jump HSQC spectra were recorded using a 210 μM uniformly 15N-enriched sample of VA2-ubiquitin. The temperature was regulated at 25°C during the high-pressure period, but briefly drops by 3 °C during the low-pressure interval which had a total duration of ca. 70 ms. The depressurization transition evolves approximately linear in time during the period in which fluid flow through the transfer tube is turbulent, and transitions to a small exponential tail as the flow becomes laminar. Forward and reverse 15N sampling was carried out for a 60 ms interval, which was initiated ca. 5 ms after the pressure drop to avoid encroaching on the tail end of the pressure drop. Inversely, the reverse sampling is initiated ca. 3 ms prior to the switch to high pressure to avoid encroaching on the high-pressure transition, which occasionally experiences ms timing jitter. A 150-ms delay separated the end of the low-pressure period from the reverse INEPT transfer of magnetization to 1H for detection. This delay serves to let decay any mechanical vibrations in the system related to the pressure switches, which have their strongest impact on the high-γ 1H signals. Spectra were recorded with 4 scans per free induction decay (FID), for a total of 120 complex t1 increments per 2D spectrum. Using a recycle delay of 9 s, the total measurement time per spectrum was ca. 2.5 h. For the forward and reverse sampled 2D HNCO spectra, 100 complex t1 increments were sampled, with 16 scans per FID, and a total measurement time of 8h per spectrum, using a 125 μM uniformly 13C/15N/2H-labeled sample.
All the NMR data were processed and analyzed using NMRPipe61 and NMRFAM-Sparky62 software.
Supplementary Material
ACKNOWLEDGMENT
We thank J. Ying, Y. Shen, and J.L. Baber for technical support, and D.A. Torchia, T.R. Alderson, and G.M. Clore for useful discussions. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.Dobson CM; Sali A; Karplus M, Protein folding: A perspective from theory and experiment. Angewandte Chemie-International Edition 1998, 37 (7), 868–893. [DOI] [PubMed] [Google Scholar]
- 2.Cellmer T; Henry ER; Hofrichter J; Eaton WA, Measuring internal friction of an ultrafast-folding protein. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (47), 18320–18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung HS; Louis JM; Eaton WA, Experimental determination of upper bound for transition path times in protein folding from single-molecule photon-by-photon trajectories. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (29), 11837–11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung HS; Gopich IV; McHale K; Cellmer T; Louis JM; Eaton WA, Extracting Rate Coefficients from Single-Molecule Photon Trajectories and FRET Efficiency Histograms for a Fast-Folding Protein. J. Phys. Chem. A 2011, 115 (16), 3642–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung HS; Eaton WA, Single-molecule fluorescence probes dynamics of barrier crossing. Nature 2013, 502 (7473), 685-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korzhnev DM; Kay LE, Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc. Chem. Res 2008, 41 (3), 442–451. [DOI] [PubMed] [Google Scholar]
- 7.Korzhnev DM; Neudecker P; Zarrine-Afsar A; Davidson AR; Kay LE, Abp1p and fyn SH3 domains fold through similar low-populated intermediate states. Biochemistry 2006, 45 (34), 10175–10183. [DOI] [PubMed] [Google Scholar]
- 8.Bezsonova I; Korzhnev DM; Prosser RS; Forman-Kay JD; Kay LE, Hydration and packing along the folding pathway of SH3 domains by pressure-dependent NMR. Biochemistry 2006, 45 (15), 4711–4719. [DOI] [PubMed] [Google Scholar]
- 9.Matouschek A; Kellis JT; Serrano L; Fersht AR, Mapping the Transition-State and Pathway of Protein Folding by Protein Engineering. Nature 1989, 340 (6229), 122–126. [DOI] [PubMed] [Google Scholar]
- 10.Kim PS; Baldwin RL, Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem 1990, 59, 631–660. [DOI] [PubMed] [Google Scholar]
- 11.Bai YW; Sosnick TR; Mayne L; Englander SW, Protein-Folding Intermediates - Native-State Hydrogen-Exchange. Science 1995, 269 (5221), 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuler B; Lipman EA; Eaton WA, Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature 2002, 419 (6908), 743–747. [DOI] [PubMed] [Google Scholar]
- 13.Vallee-Belisle A; Michnick SW, Visualizing transient protein-folding intermediates by tryptophan-scanning mutagenesis. Nat. Struct. Mol. Biol 2012, 19 (7), 731-+. [DOI] [PubMed] [Google Scholar]
- 14.Roder H, Structural characterization of protein folding intermediates by proton magnetic resonance and hydrogen exchange. Meth. Enzymol 1989, 176, 446–473. [DOI] [PubMed] [Google Scholar]
- 15.Englander SW; Mayne L, Protein Folding Studied Using Hydrogen-Exchange Labeling and 2-Dimensional Nmr. Annu. Rev. Biophys. Biomol. Struct 1992, 21, 243–265. [DOI] [PubMed] [Google Scholar]
- 16.Dyson HJ; Wright PE, How Does Your Protein Fold? Elucidating the Apomyoglobin Folding Pathway. Acc. Chem. Res 2017, 50 (1), 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eliezer D; Yao J; Dyson HJ; Wright PE, Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nature Structural Biology 1998, 5 (2), 148–155. [DOI] [PubMed] [Google Scholar]
- 18.Krishna MMG; Maity H; Rumbley JN; Englander SW, Branching in the sequential folding pathway of cytochrome c. Protein Sci 2007, 16 (9), 1946–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollien J; Marqusee S, Comparison of the folding processes of T-thermophilus and E-coli ribonucleases H. J. Mol. Biol 2002, 316 (2), 327–340. [DOI] [PubMed] [Google Scholar]
- 20.Miranker A; Robinson CV; Radford SE; Aplin RT; Dobson CM, Detection of Transient Protein-Folding Populations by Mass-Spectrometry. Science 1993, 262 (5135), 896–900. [DOI] [PubMed] [Google Scholar]
- 21.Hu WB; Walters BT; Kan ZY; Mayne L; Rosen LE; Marqusee S; Englander SW, Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (19), 7684–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoeltzli SD; Frieden C, Stopped-flow NMR spectroscopy - Real-time unfolding studies of 6-F19-tryptophan-labeled E. Coli dihydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A 1995, 92 (20), 9318–9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koide S; Dyson HJ; Wright PE, Characterization of a folding intermediate of apoplastocyanin trapped by proline isomerization. Biochemistry 1993, 32 (46), 12299–12310. [DOI] [PubMed] [Google Scholar]
- 24.Balbach J; Forge V; Vannuland NAJ; Winder SL; Hore PJ; Dobson CM, Following protein folding in real-time using NMR spectroscopy. Nature Structural Biology 1995, 2 (10), 865–870. [DOI] [PubMed] [Google Scholar]
- 25.Roche J; Dellarole M; Caro JA; Norberto DR; Garcia AE; Garcia-Moreno B; Roumestand C; Royer CA, Effect of Internal Cavities on Folding Rates and Routes Revealed by Real-Time Pressure-Jump NMR Spectroscopy. J. Am. Chem. Soc 2013, 135 (39), 14610–14618. [DOI] [PubMed] [Google Scholar]
- 26.Schlepckow K; Wirmer J; Bachmann A; Kiefhaber T; Schwalbe H, Conserved folding pathways of alpha-lactalbumin and lysozyme revealed by kinetic CD, fluorescence, NMR, and interrupted refolding experiments. J. Mol. Biol 2008, 378 (3), 686–698. [DOI] [PubMed] [Google Scholar]
- 27.Schanda P; Forge V; Brutscher B, Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (27), 11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Royer CA; Hinck AP; Loh SN; Prehoda KE; Peng XD; Jonas J; Markley JL, Effects of Amino-Acid Substitutions on the Pressure Denaturation of Staphylococcal Nuclease as Monitored by Fluorescence and Nuclear-Magnetic-Resonance Spectroscopy. Biochemistry 1993, 32 (19), 5222–5232. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi T; Yamada H; Akasaka K, Thermodynamics of Unfolding of Ribonuclease-a under High-Pressure - a Study by Proton Nmr. J. Mol. Biol 1995, 250 (5), 689–694. [DOI] [PubMed] [Google Scholar]
- 30.Kitahara R; Hata K; Li H; Williamson MP; Akasaka K, Pressure-induced chemical shifts as probes for conformational fluctuations in proteins. Prog. Nucl. Magn. Reson. Spectrosc 2013, 71, 35–58. [DOI] [PubMed] [Google Scholar]
- 31.Urbauer JL; Ehrhardt MR; Bieber RJ; Flynn PF; Wand AJ, High-resolution triple-resonance NMR spectroscopy of a novel calmodulin peptide complex at kilobar pressures. J. Am. Chem. Soc 1996, 118 (45), 11329–11330. [Google Scholar]
- 32.Inoue K; Yamada H; Akasaka K; Hermann C; Kremer W; Maurer T; Doker R; Kalbitzer HR, Pressure-induced local unfolding of the Ras binding domain of RalGDS. Nature Structural Biology 2000, 7 (7), 547–550. [DOI] [PubMed] [Google Scholar]
- 33.Dumont C; Emilsson T; Gruebele M, Reaching the protein folding speed limit with large, sub-microsecond pressure jumps. Nat. Methods 2009, 6 (7), 515–U70. [DOI] [PubMed] [Google Scholar]
- 34.Kamatari YO; Yokoyama S; Tachibana H; Akasaka K, Pressure-jump NMR study of dissociation and association of amyloid protofibrils. J. Mol. Biol 2005, 349 (5), 916–921. [DOI] [PubMed] [Google Scholar]
- 35.Kremer W; Arnold M; Munte CE; Hartl R; Erlach MB; Koehler J; Meier A; Kalbitzer HR, Pulsed Pressure Perturbations, an Extra Dimension in NMR Spectroscopy of Proteins. J. Am. Chem. Soc 2011, 133 (34), 13646–13651. [DOI] [PubMed] [Google Scholar]
- 36.Charlier C; Alderson TR; Courtney JM; Ying J; Anfinrud P; Bax A, Study of protein folding under native conditions by rapidly switching the hydrostatic pressure inside an NMR sample cell. Proc. Natl. Acad. Sci. USA 2018, 115, E4169–E4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harper SM; Neil LC; Day IJ; Hore PJ; Gardner KH, Conformational changes in a photosensory LOV domain monitored by time-resolved NMR spectroscopy. J. Am. Chem. Soc 2004, 126 (11), 3390–3391. [DOI] [PubMed] [Google Scholar]
- 38.Alderson TR; Charlier C; Torchia DA; Anfinrud P; Bax A, Monitoring Hydrogen Exchange During Protein Folding by Fast Pressure Jump NMR Spectroscopy. J. Am. Chem. Soc 2017, 139 (32), 11036–11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krantz BA; Sosnick TR, Distinguishing between two-state and three-state models for ubiquitin folding. Biochemistry 2000, 39 (38), 11696–11701. [DOI] [PubMed] [Google Scholar]
- 40.Khorasanizadeh S; Peters ID; Butt TR; Roder H, Folding and Stability of a Tryptophan-Containing Mutant of Ubiquitin. Biochemistry 1993, 32 (27), 7054–7063. [DOI] [PubMed] [Google Scholar]
- 41.Larios E; Li JS; Schulten K; Kihara H; Gruebele M, Multiple probes reveal a native-like intermediate during low-temperature refolding of ubiquitin. J. Mol. Biol 2004, 340 (1), 115–125. [DOI] [PubMed] [Google Scholar]
- 42.Herberhold H; Winter R, Temperature- and pressure-induced unfolding and refolding of ubiquitin: A static and kinetic Fourier transform infrared spectroscopy study. Biochemistry 2002, 41 (7), 2396–2401. [DOI] [PubMed] [Google Scholar]
- 43.Sosnick TR; Dothager RS; Krantz BA, Differences in the folding transition state of ubiquitin indicated by phi and psi analyses. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (50), 17377–17382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rea AM; Simpson ER; Meldrum JK; Williams HEL; Searle MS, Aromatic Residues Engineered into the beta-Turn Nucleation Site of Ubiquitin Lead to a Complex Folding Landscape, Non-Native Side-Chain Interactions, and Kinetic Traps. Biochemistry 2008, 47 (48), 12910–12922. [DOI] [PubMed] [Google Scholar]
- 45.Vallee-Belisle A; Michnick SW, Multiple tryptophan probes reveal that ubiquitin folds via a late misfolded intermediate. J. Mol. Biol 2007, 374 (3), 791–805. [DOI] [PubMed] [Google Scholar]
- 46.Piana S; Lindorff-Larsen K; Shaw DE, Atomic-level description of ubiquitin folding. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (15), 5915–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vajpai N; Nisius L; Wiktor M; Grzesiek S, High-pressure NMR reveals close similarity between cold and alcohol protein denaturation in ubiquitin. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (5), E368–E376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy G; Thirumalai D, Collapse Precedes Folding in Denaturant-Dependent Assembly of Ubiquitin. J. Phys. Chem. B 2017, 121 (5), 995–1009. [DOI] [PubMed] [Google Scholar]
- 49.Reddy G; Thirumalai D, Dissecting Ubiquitin Folding Using the Self-Organized Polymer Model. J. Phys. Chem. B 2015, 119 (34), 11358–11370. [DOI] [PubMed] [Google Scholar]
- 50.Khorasanizadeh S; Peters ID; Roder H, Evidence for a three-state model of protein folding from kinetic analysis of ubiquitin variants with altered core residues. Nature Structural Biology 1996, 3 (2), 193–205. [DOI] [PubMed] [Google Scholar]
- 51.Went HM; Benitez-Cardoza CG; Jackson SE, Is an intermediate state populated on the folding pathway of ubiquitin? FEBS Lett 2004, 567 (2–3), 333–338. [DOI] [PubMed] [Google Scholar]
- 52.Charlier C; Courtney JM; Alderson TR; Anfinrud P; Bax A, Monitoring N-15 Chemical Shifts During Protein Folding by Pressure-Jump NMR. J. Am. Chem. Soc 2018, 140 (26), 8096–8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erlach MB; Koehler J; Crusca E; Kremer W; Munte CE; Kalbitzer HR, Pressure dependence of backbone chemical shifts in the model peptides Ac-Gly-Gly-Xxx-Ala-NH2. J. Biomol. NMR 2016, 65 (2), 65–77. [DOI] [PubMed] [Google Scholar]
- 54.Bax A; Pochapsky SS, Optimized Recording of Heteronuclear Multidimensional Nmr- Spectra Using Pulsed Field Gradients. J. Magn. Reson 1992, 99 (3), 638–643. [Google Scholar]
- 55.Keeler J; Clowes RT; Davis AL; Laue ED, Pulsed-Field Gradients - Theory and Practice. Meth. Enzymol 1994, 239, 145–207. [DOI] [PubMed] [Google Scholar]
- 56.Bax A; Ikura M; Kay LE; Torchia DA; Tschudin R, Comparison of different modes of two-dimensional reverse-correlation NMR for the study of proteins. J. Magn. Reson 1990, 86, 304–318. [Google Scholar]
- 57.Ikura M; Kay LE; Bax A, A novel approach for sequential assignment of 1H, 13C, and 15N spectra of larger proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. application to calmodulin. Biochemistry 1990, 29 (19), 4659–4667. [DOI] [PubMed] [Google Scholar]
- 58.Kiefhaber T, Kinetic traps in lysozyme folding. Proc. Natl. Acad. Sci. U. S. A 1995, 92 (20), 9029–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Dios AC; Pearson JG; Oldfield E, Secondary and tertiary structural effects on protein NMR chemical shifts - an ab initio approach. Science 1993, 260 (5113), 1491–1496. [DOI] [PubMed] [Google Scholar]
- 60.Peterson RW; Wand AJ, Self-contained high-pressure cell, apparatus, and procedure for the preparation of encapsulated proteins dissolved in low viscosity fluids for nuclear magnetic resonance spectroscopy. Rev. Sci. Instrum 2005, 76 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A, NMRpipe - a multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 1995, 6 (3), 277–293. [DOI] [PubMed] [Google Scholar]
- 62.Lee W; Tonelli M; Markley JL, NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31 (8), 1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nielsen JT; Mulder FAA, POTENCI: prediction of temperature, neighbor and pH-corrected chemical shifts for intrinsically disordered proteins. J. Biomol. NMR 2018, 70, 141–175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.