

Abstract
We discuss a decision-theoretic approach to building a panel-based, preemptive genotyping program. The method is based on findings that a large percentage of patients are prescribed medications that are known to have pharmacogenetic associations, and over time, a substantial proportion are prescribed additional such medication. Preemptive genotyping facilitates genotype-guided therapy at the time medications are prescribed; panel-based testing allows providers to reuse previously collected genetic data when a new indication arises. Because it is cost-prohibitive to conduct panel-based genotyping on all patients, we describe a three-step approach to identify patients with the highest anticipated benefit. First, we construct prediction models to estimate the risk of being prescribed one of the target medications using readily available clinical data. Second, we use literature-based estimates of adverse event rates, variant allele frequencies, secular death rates, and costs to construct a discrete event simulation that estimates the expected benefit of having an individual’s genetic data in the electronic health record after an indication has occurred. Finally, we combine medication prescription risk with expected benefit of genotyping once a medication is indicated to calculate the expected benefit of preemptive genotyping. For each patient-clinic visit, we calculate this expected benefit across a range of medications and select patients with the highest expected benefit overall. We build a proof of concept implementation using a cohort of patients from a single academic medical center observed from July 2010 through December 2012. We then apply the results of our modeling strategy to show the extent to which we can improve clinical and economic outcomes in a cohort observed from January 2013 through December 2015.
Keywords: precision medicine, risk prediction, pre-emptive genotyping, electronic health records, discrete event simulation
It is well known that genetic variation can influence patients’ responsiveness to medications and risk for toxicity,1 and more than 250 genetic variants have accumulated within Food and Drug Administration (FDA) medication label inserts.2 Over the past 10 years, the popularity of acquiring pharmacogenomic (PGx) data to guide prescribing has increased significantly, driven by dramatic reductions in the cost of the technology and accumulating evidence for clinical utility. Many insurers, institutions, and organizations are developing programs to preemptively test their populations for genetic variants, expecting that clinical value and return on investment will be accrued as the information is used for delivering care. Previously, we estimated that over a 5-year timeframe (2005–2010), in a medical home population at our institution approximately 65% of patients were prescribed at least one medication with an FDA label insert and 23% of patients were prescribed at least three such medications.3,4 Preemptive genotyping allows physicians to proactively apply genetic data at the time of prescribing, and multiplexing (with a panel that contains hundreds to thousands of relevant genetic variants) permits cost efficiencies when genetic data are reused, that is, for the second, third, and so on, PGx-guided therapy.4
Given the large and growing number of PGx opportunities, strategies to identify and test populations to maximize the potential value of genomic testing are needed. As of now, it is cost-prohibitive to genotype all patients in a population of a health system, regardless of clinical need. We recently described an approach to identify patients for panel-based, preemptive genotyping that was founded on the clinical risk of developing an indication for one of the targeted medications.5 Briefly, we identified eligible patients by estimating risk of being prescribed any of three medications with known PGx effects (statin, warfarin, or an antiplatelet) within 3 years of patients’“medical home” date.5 We constructed a single risk prediction model, and we made genotyping decisions based on whether or not the risk estimate exceeded a predefined cutoff value. This decision approach was then implemented within the Pharmacogenomic Resource for Enhanced Decisions in Care and Treatment (PREDICT) program, a clinical quality improvement initiative at Vanderbilt University Medical Center (VUMC).6 The program incorporated genetic information into the electronic health record (EHR) and, when appropriate, permitted genotype-guided prescribing.6–8 Thus far, almost 15,000 patients have been genotyped since 2010 as a part of routine care.
In this article, we extend this previous work to formalize a value-based approach that incorporates the magnitude, severity, and cost of the adverse drug reaction averted by pharmacogenetics. Specifically, we detail 1) predictive modeling approaches using readily available clinical data from an EHR to capture patient-level risk of being prescribed each PGx medication separately; 2) a discrete event simulation that uses literature-based estimates of variant allele frequencies, adverse event rates, and secular death rates to estimate the cost and quality-adjusted life year (QALY) benefit of genotype-guided therapy after an indication occurs; and 3) a decision-theoretic approach to combine points (1) and (2) to develop genotyping rules that optimizes preemptive genotyping value based on user defined utility functions.
Methods
The data for this value-based, preemptive genotyping program proof of concept were derived from EHRs from VUMC.
Predicting PGx Medication Prescriptions
In the next several subsections, we detail an approach to using readily available clinical data from the VUMC EHR to estimate risk of being prescribed an antiplatelet medication following a percutaneous coronary intervention (PCI), a statin, and warfarin, and to validate the models. We focus on antiplatelet medication following a PCI (e.g., a coronary stent) due to the availability of compelling evidence of genetic testing efficacy as compared to other indications for antiplatelets where the benefit is less clear.9,10
Study Cohorts
We included patients with records in the Vanderbilt Synthetic Derivative (SD)11 and with VUMC established as their “medical home” (MH). We considered patients to have a VUMC MH when we observed, for the first time, the third distinct visit within a 2-year timeframe to primary care and specialty medical and surgical clinics delivering non-acute care. Patients were required to be 18 years old on the MH date, and for each medication considered, they must have had no observed history of receiving the medication (medication naïve on the MH date). Those who satisfy MH criteria for the first time between July 2010 and December 2012 comprise the training cohort, and those who satisfy the criteria between January 2013 and December 2015 comprise the validation cohort. For the purpose of modeling, baseline characteristics were established on the MH date and were updated longitudinally on each patient as clinical data accumulated.
Data Preparation
For the purpose of modeling, we included independent variables that were readily available in the EHR including patient demographics (age, sex, race, body mass index), vitals (diastolic and systolic blood pressure), history of chronic medical conditions defined based on ICD-9 codes (hypertension, type 2 diabetes, coronary artery disease, atrial fibrillation, atherosclerosis, congestive heart failure, dialysis, and cerebrovascular disease), other medication history (e.g., if modeling statin prescriptions, then include history of antiplatelet and warfarin prescriptions), history of acute cardiovascular events (acute coronary syndrome, coronary catheterization with revascularization, or coronary artery bypass grafting), number of clinic visits in the past 6 months (number of cardiology clinic and non-cardiology clinic visits), and laboratory data (low-density lipoprotein, high-density lipoprotein, triglycerides, HgbA1C, and creatinine).
Since physician-based prescribing decisions rely on patient observation and what is available in the EHR, we used the most recently observed weight and lab data at each visit when not observed at the current visit. When, according to the EHR, lab values (e.g., HgbA1C) had yet to be observed in an individual, we assigned a value within the normal range reflecting our belief that if no lab test was ordered, the physician was effectively assuming a normal value or was not concerned about its value. In addition to observed or imputed lab values, we included indicators (yes/no) of whether the lab test was performed. As the goal of our preemptive genotyping program was to focus on outpatient visit opportunities to identify patients suitable for preemptive genotyping, we updated patient data, at most, once every 6 months.
Modeling Strategy
Since patients were observed for variable amounts of time and were administratively censored due to the timing of data acquisition, we used survival analysis approaches to model, separately, time to antiplatelet therapy following placement of a coronary stent, statin drug initiation, and warfarin prescription. Because we had multiple measurements per subject and we sought to predict future prescriptions from the time of each clinic visit, we employed a partly conditional survival model12 for longitudinal data and we used a residual survival time scale outcome.12,13 For estimation we implemented time-varying, Cox proportional hazard models14 with robust, sandwich standard error estimates15 and random survival forests.16
Cox proportional hazard model
We developed time-dependent Cox proportional hazard models12,15 using two strategies: one that does and one that does not include independent variable interactions. First, we built a Cox model with all 28 predictors described previously. For all continuous variables, we used restricted cubic splines with 5 knots specified at the 5th, 25th, 50th, 75th, and 95th percentiles to allow for nonlinear associations. In total, this modeling approach estimated 53 parameters for each of the three outcome models. Next, we sought to add two-way interactions. Since it is difficult to detect interactions between correlated variables and with very rare variables, we added interactions with the following approach. First, we conducted a hierarchical cluster analysis17,18 to identify uncorrelated clusters of variables after excluding binary variables with prevalence less than 0.05. We then performed principle component analysis, computed the first principle component for each cluster, and added all pairwise interactions among the principle components to the Cox models.19–21 For the antiplatelet, statin, and warfarin models we estimated 182, 166, and 124 parameters, respectively, across all main effects and interactions. The difference in the number of parameters estimated for each outcome is due to the difference in the patients included in the analyses. Subjects included in each medication model were required to be naïve to the medication at the MH date, and so sample sizes for the three models differed (though there is much overlap). In total, for the 2010 to 2012 training cohort, 74,607, 45,532, and 75,795 unique patients contributed longitudinal data to the antiplatelet, statin, and warfarin models, respectively. For the 2013 to 2015 validation cohort, 52,588, 34,373, and 54,495 patients contributed data.
Random survival forests
Random survival forests (RSF)16 are a popular, ensemble-based, machine-learning algorithm for variable selection and risk prediction that naturally includes high dimensional interactions, and that reduces model overfitting and prediction bias by averaging across a number of individual trees. When constructing our final models, we conducted a thorough grid search for all common tuning parameters, and in Appendix I, we show the extent to which predictive model summaries depend on the tuning parameters: number of trees in the forest (ntree) and the average number of observations in terminal nodes (nodesize). Based on a grid search, we found that tuning parameters, number of candidate predictors randomly selected at each split (mtry) of a tree, and the maximum number of splits for continuous variables (nsplit) had a smaller impact on predictive model performance. We set mtry = 5 (i.e., approximately equal to the commonly used square root of the number of independent variables) and nsplit = 10. In Appendix I (Tables A1.1 and A1.2), we show C-Indices for the training data, the out-of-bag samples, and the validation data, and in Figure A1.1, we include calibration curves based on applying the RSFs constructed with the training data but applied to the validation data. When evaluating the performance of RSFs we chose ntree = 500 for all medications, and nodesize = 5 for statins and nodesize = 25 for antiplatelets and warfarin, respectively. When making these decisions, we balanced model calibration, the standard deviation of the predictive distribution (with larger values being better), and the C-Index.
Model Evaluation
We constructed the Cox models and RSFs using nonparametric bootstrap samples from the 2010–2012 cohort. We then evaluated each of the models using the out of bag samples from the 2010–2012 cohort and the independent 2013–2015 cohort. In this article, we focus exclusively on evaluation of performance on the 2013–2015 cohort. We examined model calibration and predictive distributions graphically and captured discrimination with the C-index21 at 2 years following each clinic visit. All analyses were performed using the R programming language 3.3.0 (R Foundation for Statistical Computing, Vienna, Austria) with rms,22 randomForestSRC,16,23,24 and survivalROC25 libraries.
Calculating the Value of Genotyping Using Discrete Event Simulation
To estimate the incremental value of genotype tailored therapy over standard prescribing, we defined four distinct utility functions that represent different approaches to summarizing adverse outcomes, costs of treatment for adverse outcomes, and costs of testing. Let be the expected utility associated with therapeutic approach b once an indication for medication med has occurred, and let b = std for standard, non-tailored therapy and b = gtt for genotype tailored therapy. To examine the impact of genotype tailored therapy, we calculate , , and , where , a function of , and , captures the expected benefit of genotype tailored therapy after an indication arises. The value depends on variant allele frequencies, adverse event/failure rates, relative rate reductions due to genotyping, secular death rates, costs, the follow-up timeframe, and so on. In this article, we consider the utility functions: all adverse events (AEs), severe adverse events (SAEs), quality-adjusted life years (QALY), and net health benefit (NHB). For AE, SAE, and QALY, is a simple difference (i.e., the difference in the expected number of AEs). In contrast, for NHB, where and are the difference in QALYs and costs, respectively, for genotype tailored therapy compared to standard therapy, and is a willingness to pay threshold that represents the amount society is willing to pay per quality-adjusted life year gained. In this article, we used a willingness to pay threshold equal to $100,000 ($50,000 is another commonly used value).26
To calculate expected benefits for each utility function by medication combination, we appeal to discrete event simulation (DES) methods.27–30 As described in detail in previously published work31 and using parameters shown in Appendix II, the underlying DES jointly models the prescription incidence, health, and mortality outcomes among patients prescribed antiplatelet therapy, a statin, or warfarin. The demographic profile of the simulated patient population was matched to the VUMC MH population based on age and gender. The model assumes the health sector perspective and incorporates medical costs, pharmaceutical costs, and clinical outcomes based on prior randomized trials and on previous cost-effectiveness studies of antiplatelet therapy, simvastatin, and warfarin. The DES compared a base case scenario under which no genetic testing is used to guide therapeutic decisions to a genotype-tailored scenario under which a genetic test is ordered and acted upon when appropriate. For our DES model outcomes, we consider a 1-year timeframe after medication prescriptions, as the differential impact of genotype-tailored therapy versus standard therapy on AEs is concentrated within the year after initiating therapy.
Decision Analysis for Genotyping
The benefits of genotyping differ according to medications and indications, with genotype-tailored therapy for certain “high-yield” drug-gene pairs shown to have a higher net monetary benefit as compared with other drug-gene pairs.31 Thus, a naïve approach to the preemptive genotyping decision (i.e., whom to genotype) that simply averages predicted risks across PGx medications into a single summary risk score will assign equal weight to higher and lower value PGx scenarios and will weigh the overall risk score toward the drug with the largest indication frequency in the sample. Ideally, a summary risk score used to make clinical decisions to preemptively genotype would be based on a value-weighted combination of predicted risks for each drug type. That is, a preemptive genotyping decision should ideally combine 1) medication prescription risk with 2) the estimated benefit of genotype tailored therapy once the indication occurs.
Let be the 2-year prescription risk for medication med. For the present analyses, we calculated , , and at all patient-clinic visits in the 2013–2015 cohort using results from the Cox model and RSF fits that were constructed using the 2010–2012 cohort. After running the discrete event simulations, we calculated expected benefit of genotyping once a medication is indicated, , , and along a variety of dimensions (e.g., number of SEAs avoided, NHB, etc.) under the recognition that some health systems and decision makers may prefer one definition of value over another. Finally, for each med, is the value of preemptively genotyping for medication med.
A decision rule that optimizes expected benefit for panel-based, preemptive genotyping genotypes patients with the highest total value across all available medications: This total value quantity has intuitive interpretations. For example, if the utility function represents a sum of all AEs, then is the anticipated number of AEs that would be avoided under preemptive genotyping. Notably, a patient who has already been exposed to one or two of the medications may still benefit from preemptive genotyping. For example, if a patient had already been exposed to, say, statins then the total value is calculated with the remaining medications, , as it is still a valid measure of expected benefit. We also recommend setting when it is negative, since genotype-tailored therapy would not be used for the medication.
Results
Patient data used to calculate medication prescription risk are summarized in Table 1. We study 85,837 patients (534,962 clinic visits) in the 2010–2012 training cohort, and 59,953 patients (194,187 clinic visits) in the 2013–2015 validation cohort. The training cohort tended to be older (median age 58 v. 55), to have higher rate of type 2 diabetes (21% v. 13.8%), hypertension (60.1% v. 46.1%), congestive heart failure (9.7% v. 6.8%), and atherosclerosis (24.7% v. 18.8%) than the validation cohort. However, it had a lower prevalence of cardiac clinic visits (22.6% v. 25.1%). Figure 1 displays medication-free, Kaplan-Meier curves14 using the longitudinal training and validation data for the three medications. Compared with the validation cohort, the training cohort had a somewhat higher rate of being prescribed the medications at 2 years. The 2-year risk estimates [95% confidence intervals] for the training and validation cohorts were, respectively, 0.022 [0.021, 0.023] and 0.018 [0.017, 0.019] for stent-indicated antiplatelet prescriptions, 0.146 [0.145, 0.148] and 0.126 [0.123, 0.129] for statin prescriptions, and 0.042 [0.041, 0.043] and 0.035 [0.034, 0.036] for warfarin prescriptions.
Table 1.
Patient Demographics, Vitals, Labs, Medical History, Chronic Medical Conditions, and 2-Year Prescription Event Rates for the 2010–2012 (Training) and 2013–2015 (Validation) Cohortsa
| Train Cohort | Validation Cohort | |||
|---|---|---|---|---|
| Baseline | Longitudinal | Baseline | Longitudinal | |
| N | 85,837 | 534,962 | 59,953 | 194,187 |
| Demographics | ||||
| Age (years) | 55 [29, 75] | 58 [33, 77] | 52 [26, 73] | 55 [28, 75] |
| Female | 58 | 58 | 58 | 57 |
| Race | ||||
| White | 85 | 85 | 86 | 86 |
| Black | 12 | 12 | 11 | 11 |
| Other | 3 | 3 | 3 | 3 |
| Vitals and labs | ||||
| BMI (kg/m2) | 28 [21, 38] | 28 [22, 39] | 28 [21, 38] | 28 [22, 39] |
| BP available? (%) | 97.4 | 90.3 | 97.6 | 89.7 |
| Diastolic (mm Hg) | 74 [60, 90] | 74 [60, 89] | 74 [60, 90] | 74 [60, 89] |
| Systolic (mm Hg) | 118 [ 67, 144] | 120 [ 67, 146] | 122 [ 82, 147] | 123 [ 88, 148] |
| Lipids available? (%) | 17.4 | 16.2 | 8.6 | 9 |
| LDL (mg/dL) | 100 [ 72, 132] | 100 [ 66, 136] | 100 [ 84, 122] | 100 [ 77, 127] |
| HDL (mg/dL) | 49 [35, 66] | 49 [34, 69] | 50 [39, 60] | 50 [37, 63] |
| Triglycerides (mg/dL) | 110 [ 66, 197] | 111 [ 64, 221] | 110 [ 79, 159] | 110 [ 73, 182] |
| HgbA1C available? (%) | 5.6 | 8 | 5.1 | 5.3 |
| HgbA1C value (%) | 5.2 [4.4, 6.4] | 5.3 [4.5, 6.9] | 5.1 [4.4, 6.0] | 5.2 [4.4, 6.2] |
| Creatinine available? (%) | 42.7 | 38.6 | 32.1 | 30.6 |
| Creatinine (mg/dL) | 0.8 [0.6, 1.3] | 0.9 [0.6, 1.4] | 0.8 [0.6, 1.2] | 0.8 [0.6, 1.2] |
| Medical history and chronic conditions (%) | ||||
| Type 2 diabetes | 14.8 | 21 | 10.1 | 13.8 |
| CAD | 4 | 6 | 3.1 | 3.9 |
| Atrial fibrillation | 3.2 | 5 | 2.2 | 3.2 |
| Hypertension | 49 | 60.1 | 37.8 | 46.1 |
| Congestive heart failure | 6.8 | 9.7 | 4.9 | 6.8 |
| Atherosclerosis | 18.9 | 24.7 | 14.9 | 18.8 |
| Cerebrovascular event | 4.9 | 7.6 | 3.1 | 4.5 |
| Dialysis | 10.4 | 14.1 | 7.7 | 10.1 |
| Any acute event | 7.5 | 11.3 | 6.4 | 8.6 |
| Cardiac clinic | 24.4 | 22.6 | 26.9 | 25.1 |
| Antiplatelet | 6.3 | 9.9 | 5 | 6.9 |
| Statins | 47 | 58.6 | 42.7 | 50.6 |
| Warfarin | 11.7 | 16.5 | 9.1 | 12.2 |
| 2-Year event rateb (number of events/Nc) | ||||
| Antiplatelet | 2.3 (1,699/74,607) | 2.2 (8,092/426,409) | 1.9 (835/52,588) | 1.8 (1,883/149,985) |
| Statin | 11.3 (5,041/45,532) | 14.7 (26,656/210,254) | 14.7 (4,130/34,373) | 12.6 (7,584/86,418) |
| Warfarin | 3.7 (2,755/75,795) | 4.2 (15,395/424,812) | 4.3 (1,949/54,495) | 3.5 (3,759/153,105) |
BMI, body mass index; BP, blood pressure; CAD, coronary artery disease; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Continuous variables are summarized with the 50th [10th, 90th] percentiles and categorical variables with percentages.
A Kaplan-Meier estimate was used for prescription rate.
N corresponds to the sample size that is naïve to the medication at the medical home date.
Figure 1.
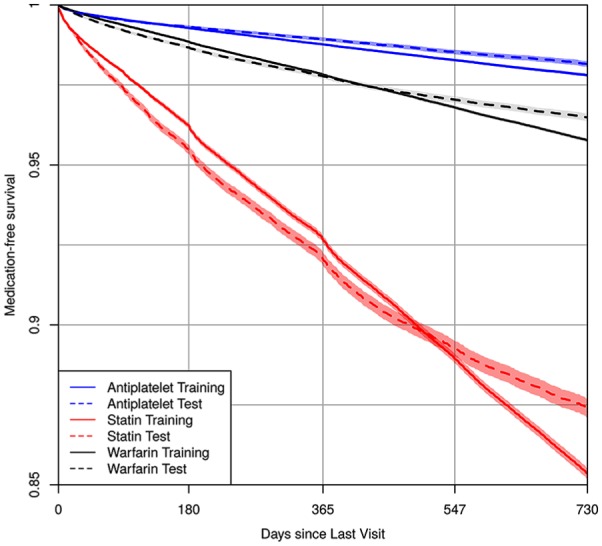
Kaplan-Meier estimates of medication-free survival probability using the longitudinal train and validation cohort data for antiplatelet, statin, and warfarin. The x-axis is the time since last clinic visit using the residual time scale.
Medication Prescription Risk Modeling
Figure 2 displays calibration curves, predictive distribution summaries, and C-Indices for 2-year risks when applying models developed using the training cohort data (2010–2012) to the validation cohort data (2013–2015). We display summaries for the three medications and the three modeling approaches (Cox without interactions, Cox with interactions, and RSF). None of the modeling approaches clearly outperformed the others across the three medications. The models were quite well calibrated for the statin and antiplatelet models although all models, and particularly the Cox without interaction model, appeared to overestimate warfarin risk at the upper tail of the distribution. All modeling approaches also yielded quite similar C-Indices (0.80–0.81 for antiplatelets, 0.69–0.70 for statins, and 0.69 for warfarin). In online appendices, we display parameter estimates, confidence intervals, and nomograms using the Cox without interactions model to enhance readers’ understanding of independent variable contributions to the risk estimates (see Appendix I, Figures A1.2–A1.7).
Figure 2.

Calibration and distribution for predicted 2-year risks. Risk estimates were calculated by applying Cox main effect model, Cox interaction model, and RSF to the validation cohort dataset. Each dot stands for the average risk over 300, 173, and 306 observations, which provides 500 dots (bins) for antiplatelet, statin, and warfarin. The “syringe” plot or extended boxplot shows the 1st, 10th, 25th, 50th, 75th, 90th, 99th percentiles, and the standard deviation of the predicted risk distribution is displayed numerically. The smooth lines and the 95% confidence band were calculated using the locally estimated scatterplot smoothing.
Discrete Event Simulation
For each medication, we conducted a DES using literature-based values for secular death rates, variant allele frequencies, AE rates, and costs to estimate the expected benefit of genotyping in the 1 year following each medication prescription. We calculated , , and using benefit measures: number of SEAs avoided, total number of AEs avoided, disutility-weighted QALY gained, and NHB with a willingness to pay threshold of $100,000. We assumed that genotype tailored therapy affected outcomes only by altering the medication choice (i.e., switching from clopidgrel to ticagrelor or altering the warfarin dose) in those with actionable variants. For each of the six scenarios (antiplatelet, statin, and warfarin indications under standard therapy and genotype-tailored therapy) we simulated a population of 10 million patients to estimate expected costs and QALY. Then, for each medication, we calculated the expected benefit using the difference in expected utility between the genotype-tailored therapy and the standard therapy populations. Since metabolizer status is known to vary by race for CYP2C19 and SLCO1B1, we calculated and for white, black, and other races separately. We did not conduct a race-based calculation for warfarin therapy due to inadequate data on specific impact of variants VKORC1 and CYP2C9 on the time to controlled international normalization ratio (INR). Instead we used the results from Pirmohamed et al32 that directly modeled the impact of genotype-tailored therapy versus standard therapy on time to INR control. For the purpose of this proof of concept, we assumed that metabolizer status for those in the “other” race category was similar to Asian populations and we use values based on Scott et al9 for CYP2C19 and School of Medicine, Indiana University,33 for SLCO1B1.
Table 2 shows the results of the discrete event simulation for combinations of the utility function, 2-year medication prescription risks, and race. When using SEAs and quality-adjusted life day utilities, the expected benefit of genotype-tailored therapy is far greater for those prescribed antiplatelets than those prescribed statins or warfarin. For example, while with antiplatelet therapy, we estimated avoiding 2.6 (white), 3.2 (black), and 5.1 (other race) SEAs per 1,000 patients genotyped (see ΔUmed column), with statin and warfarin therapy, we estimate avoiding less than 0.15 SEAs in all subgroups. In contrast, if considering all AEs together, including nonsevere events, we estimate that genotype-tailored therapy avoids far more statin-related AEs in white and other races (e.g., compare 19.7 statin-related AEs avoided in the other race compared to 4.8 and 3.0 antiplatelet-related and warfarin-related AEs). Table 2 also shows the 95th, 97.5th, and 99th percentiles of 2-year medication prescription risk () and the associated expected benefit of preemptive genotyping () for each medication, utility function, and race. Clearly, anticipated benefit of genotyping () and the 2-year risk of being prescribed the medication () both factor into the anticipated benefit of preemptive genotyping . The NHB was observed to be negative for all three medications, and so for 1-year timeframe that we used here and for the medications we studied, we would not choose to genotype patients based exclusively on the NHB. Such a conclusion could change with longer follow-up periods and with different medications. Notably, for each medication and utility function, the differences in post-medication, expected benefit () among the races are explained by the difference in the low metabolizer rates (e.g., 0.146 v. 0.290 poor metabolizer prevalence in white versus other race). This is due to the fact that the discrete event simulation did not factor race into the calculation beyond metabolizer status.
Table 2.
Discrete Event Simulationa
| ΔUmed | rmed | Vmed | ||||||
|---|---|---|---|---|---|---|---|---|
| Medication | Race | SAEb | AEc | QALD | Two-Year Prescription Risk in the 2013–2015 VUMC Cohorts | SAEb | AEc | QALD |
| Antiplatelets | White (CYP2C19 poor metabolizer prevalence = 0.146)9 | 2.554 | 2.427 | 185.325 | 0.059 (95th) | 0.150 | 0.143 | 10.897 |
| 0.080 (97.5th) | 0.204 | 0.194 | 14.836 | |||||
| 0.113 (99th) | 0.288 | 0.273 | 20.876 | |||||
| Black (CYP2C19 poor metabolizer prevalence = 0.183)9 | 3.201 | 3.043 | 230.309 | 0.059 (95th) | 0.190 | 0.180 | 13.635 | |
| 0.090 (97.5th) | 0.288 | 0.274 | 20.755 | |||||
| 0.144 (99th) | 0.460 | 0.437 | 33.098 | |||||
| Other (CYP2C19 poor metabolizer prevalence = 0.290)9 | 5.073 | 4.821 | 360.400 | 0.023 (95th) | 0.115 | 0.109 | 8.155 | |
| 0.036 (97.5th) | 0.183 | 0.174 | 13.010 | |||||
| 0.055 (99th) | 0.279 | 0.265 | 19.807 | |||||
| Statin | White (SLCO1B1 poor/medium metabolizer prevalence = 0.029/0.282)33 | 0.144 | 26.252 | 8.925 | 0.233 (95th) | 0.034 | 6.128 | 2.083 |
| 0.284 (97.5th) | 0.041 | 7.448 | 2.532 | |||||
| 0.357 (99th) | 0.051 | 9.369 | 3.185 | |||||
| Black (SLCO1B1 poor/medium metabolizer prevalence = 0.000/0.039)33 | 0.017 | 2.896 | 1.015 | 0.283 (95th) | 0.005 | 0.820 | 0.288 | |
| 0.348 (97.5th) | 0.006 | 1.008 | 0.353 | |||||
| 0.437 (99th) | 0.007 | 1.265 | 0.444 | |||||
| Other (SLCO1B1 poor/medium metabolizer prevalence = 0.017/0.226)33 | 0.110 | 19.742 | 6.765 | 0.188 (95th) | 0.021 | 3.709 | 1.271 | |
| 0.239 (97.5th) | 0.026 | 4.726 | 1.619 | |||||
| 0.314 (99th) | 0.034 | 6.191 | 2.121 | |||||
| Warfarin | Overall | 0.007 | 2.969 | 10.843 | 0.088 (95th) | 0.001 | 0.261 | 0.953 |
| 0.108 (97.5th) | 0.001 | 0.321 | 1.173 | |||||
| 0.138 (99th) | 0.001 | 0.409 | 1.495 | |||||
We report the expected benefit of genotype-tailored therapy in the year following a prescription per 1,000 patients genotyped () by race, the 95th, 97.5th, and 99th percentiles of 2-year medication prescription risk (rmed) among patients in the 2013–2015 cohort, and the expected benefit of preemptively genotyping 1,000 patients () by medication prescription risk and race. We consider utility functions: severe adverse events (SAEs), all adverse events (AEs), and quality-adjusted life days (QALDs). Net Health Benefit (willingness to pay threshold set to $100,000) was negative in all scenarios and is not included. For each medication, we simulated 10,000,000 patients with age and gender distributions that resemble the 2013–2015 validation cohort. We rescaled results so that and are both on a “per 1,000 patients genotyped” scale.
For antiplatelets, SAE includes ST event, MI (myocardial infarction), revascularization, major bleed. For statin, SAE includes moderate or severe myopathy, cerebrovascular event. For warfarin, SAE includes major bleed, stroke, DVT (deep vein thrombosis) event, pulmonary embolism.
For antiplatelets, AE includes serious adverse events and non-serious events including minor bleed. For statin, AE includes serious adverse events and non-serious mild myopathy events. For warfarin, AE includes serious adverse events and non-serious minor bleeds.
Application of the Decision Rule to Identify Patients to Genotype
Since genotyping is still too expensive to perform on all patients, for this demonstration, we assume we can only genotype the 1,500 patients in each scenario, and Table 3 shows the extent to which this preemptive genotyping program could be used to improve outcomes if applied in the validation cohort. Examining the quality-adjusted life day utility function, we can see that with the decision rule to genotype when , we enrich the sample with black patients compared to the original cohort (19% v. 11%), and number needed to genotype to gain one quality adjusted life day is 34. In contrast, the number needed to genotype under a random sampling scheme is 208.
Table 3.
Extent of Enrichment of the Genotyped Cohorts Using Rules That Genotype When the Total Expected Benefit of Preemptive Genotyping, , Exceeds the Thresholds Showna
| White | Black | Other | Overall | |
|---|---|---|---|---|
| Proportion in the 2013–2015 VUMC cohort of 58,499 patients | 85.9 | 11.2 | 2.9 | |
| SAE genotyping rule: Genotype if | ||||
| Proportion in a genotyped cohort of 1,500 patients | 78.9 | 19.2 | 1.9 | |
| NNT in a genotyped cohort to prevent one SAE | 2,661 | 2,073 | 2,130 | 2,512 |
| NNT in a randomly genotyped cohort to prevent one SAE | 16,061 | 15,207 | 19,425 | 16,045 |
| AE genotyping rule: Genotype if | ||||
| Proportion in a genotyped cohort of 1,500 patients | 99.3 | 0 | 0.7 | |
| NNT in a genotyped cohort to prevent one AE | 91 | — | 95 | 91 |
| NNT in a randomly genotyped cohort to prevent one AE | 421 | 2,200 | 597 | 468 |
| QALD genotyping rule: Genotype if | ||||
| Proportion in a genotyped cohort of 1,500 patients | 79.3 | 19.0 | 1.7 | |
| NNT in a genotyped cohort for one additional QALD | 36 | 28 | 28 | 34 |
| NNT in a randomly genotyped cohort for one additional QALD | 207 | 198 | 264 | 208 |
AE, all adverse event derived genotyping rule; QALD, quality adjusted life day derived genotyping rule; SAE, serious adverse event based genotyping rule.
We chose the thresholds for this demonstration project in a manner that genotypes 1,500 patients. For each med, , where is the estimated 2-year medication prescription risk in the 2013–2015 cohort and is estimated from the discrete event simulation. We show the racial makeup of the genotyped pool under each rule and the number needed to genotype (NNT) under each rule and under random sampling.
Figure 3 displays cumulative incidence of being prescribed each medication among the subset of 1,500 participants genotyped under each decision rule. By comparing these values to the cumulative incidence under a random sampling design, we observe the extent to which the pool of genotyped patients was enriched with those who were prescribed medications within 2 years of each clinic visit. The figure highlights the tradeoffs that are made when choosing a utility function. Genotyping rules that seek to minimize SAEs and increase the number of QALYs both enrich the sample with those prescribed antiplatelets (top left panel). In contrast, genotyping rules that seek to reduce all AEs enrich the genotyped pool primarily with those prescribed statins (top right panel).
Figure 3.

Cumulative incidence of medication prescriptions under four genotyping rules. We selected for genotyping the 1,500 patients in the 2013–2015 dataset who crossed the genotyping threshold for for utility functions: serious adverse events only (red), all adverse events (orange), quality-adjusted life days (blue), and a random selection of 1,500 patients (gray). Once patients were selected into the genotyping pool, we estimated their cumulative risk of being prescribed each medication over time with Kaplan-Meier estimates. To be included in the risk set for each figure, patients must have been naïve to the medication at the medical home date. To be included in the lower right panel, patients must have been naïve to at least one of the medications at the medical home date. Compared to random selection, all genotyping approaches enriched the pool of genotyped patients with those who are likely to be prescribed one or more of the medications.
Discussion
This article provides a framework for institutions and health systems to develop a preemptive genotyping program that maximizes societal and institutional value by integrating the advantages of genotype-tailored therapy with selection of a health care population that is most likely to benefit from testing. The framework is applied retrospectively using data from a single institution to demonstrate the feasibility and potential utility of the method. As a three-step process, the framework is more complex than what is currently implemented at several institutions including our own.34–39 However, if implemented, it could potentially balance the investment in testing with the downstream value of improving prescription-related outcomes.
We evaluated two different predictive modeling strategies—a Cox survival model and a machine learning–based RSF—and performance was comparable. This is consistent with a lack of high-order interaction terms that are naturally captured with RSFs and can be explained by noting that medication prescriptions represent a clinician-made decision. Thus, the predictive models can only be as complex as clinicians’ ability to assimilate patients’ clinical profiles. In addition, we implemented a discrete event simulation to estimate the expected benefit of genotype-tailored therapy once medications are prescribed, and we combined the prescription risk estimates with the expected benefit after prescriptions to derive expected benefit associated with preemptive genotyping. Under this program, for a chosen utility function, patients with the highest expected benefit across all medications should be genotyped.
In this demonstration, improvements may be perceived as modest. Observed results will improve with a larger number of medications with PGx effects, with a greater focus on high-yield indications for which genotype-tailored therapy has been shown to be beneficial, with increased targeting of those at highest risk of medication prescriptions, with longer follow-up times (both for prescribing and for observing downstream events), and with a more focused targeting of subgroups of patients known to have relatively high rates of risk conferring alleles. Notably, the strategy we propose is highly adaptable and can be easily modified as new evidence emerges (i.e., medications and medication indications can be added and removed from all calculations). Whether health systems can justify implementing preemptive, panel-based pharmacogenomic testing, or payors agree to fund such testing, may depend on the use of both predictive modeling and careful consideration of the economic and clinical benefits of tailored prescribing.
Supplemental Material
Supplemental material, Appendix_online_supp for A Decision-Theoretic Approach to Panel-Based, Preemptive Genotyping by Yaping Shi, John A. Graves, Shawn P. Garbett, Zilu Zhou, Ramya Marathi, Xiaoming Wang, Frank E. Harrell, Thomas A. Lasko, Joshua C. Denny, Dan M. Roden, Josh F. Peterson and Jonathan S. Schildcrout in MDM Policy & Practice
Footnotes
ORCID iD: Jonathan S. Schildcrout  https://orcid.org/0000-0003-2276-2551
https://orcid.org/0000-0003-2276-2551
Supplemental Material: Supplementary material for this article is available on the Medical Decision Making Policy & Practice website at https://journals.sagepub.com/home/mpp.
Contributor Information
Yaping Shi, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee.
John A. Graves, Department of Health Policy, Vanderbilt University Medical Center, Nashville, Tennessee Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Vanderbilt University School of Medicine, Nashville, Tennessee.
Shawn P. Garbett, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee
Zilu Zhou, Department of Health Policy, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee.
Ramya Marathi, Department of Health Policy, Vanderbilt University Medical Center, Nashville, Tennessee.
Xiaoming Wang, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, Tennessee.
Frank E. Harrell, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee Vanderbilt University School of Medicine, Nashville, Tennessee; Vanderbilt Kennedy Center, Nashville, Tennessee.
Thomas A. Lasko, Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee
Joshua C. Denny, Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Vanderbilt University School of Medicine, Nashville, Tennessee.
Dan M. Roden, Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Pharmacology, Vanderbilt University Medical Center, Nashville, Tennessee; Vanderbilt University School of Medicine, Nashville, Tennessee.
Josh F. Peterson, Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, Tennessee Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; Vanderbilt University School of Medicine, Nashville, Tennessee.
Jonathan S. Schildcrout, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, Tennessee; Department of Anesthesiology, Vanderbilt University Medical Center, Nashville, Tennessee; Vanderbilt University School of Medicine, Nashville, Tennessee.
References
- 1. Roden DM, Wilke RA, Kroemer HK, Stein CM. Pharmacogenomics: the genetics of variable drug responses. Circulation. 2011;123(15):1661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. US Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. Available from: http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm
- 3. Van Driest S, Shi Y, Bowton Eet al. Clinically actionable genotypes among 10 000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 2014;95(4):423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schildcrout JS, Denny JC, Bowton Eet al. Optimizing drug outcomes through pharmacogenetics: a case for preemptive genotyping. Clin Pharmacol Ther. 2012;92(2):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schildcrout JS, Shi Y, Danciu Iet al. A prognostic model based on readily available clinical data enriched a pre-emptive pharmacogenetic testing program. J Clin Epidemiol. 2016;72:107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pulley JM, Denny JC, Peterson JFet al. Operational implementation of prospective genotyping for personalized medicine: the design of the Vanderbilt PREDICT Project. Clin Pharmacol Ther. 2012;92(1):87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peterson JF, Field JR, Shi Yet al. Attitudes of clinicians following large-scale pharmacogenomics implementation. Pharmacogenomics J. 2016;16(4):393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peterson J, Field J, Unertl Ket al. Physician response to implementation of genotype-tailored antiplatelet therapy. Clin Pharmacol Ther. 2016;100(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scott SA, Sangkuhl K, Stein CMet al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorich MJ, Rowland A, McKinnon RA, Wiese MD. CYP2C19 genotype has a greater effect on adverse cardiovascular outcomes following percutaneous coronary intervention and in Asian populations treated with clopidogrel: a meta-analysis. Circ Cardiovasc Genet. 2014;7(6):895–902. [DOI] [PubMed] [Google Scholar]
- 11. Roden D, Pulley J, Basford Met al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84(3):362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zheng Y, Heagerty PJ. Partly conditional survival models for longitudinal data. Biometrics. 2005;61(2):379–91. [DOI] [PubMed] [Google Scholar]
- 13. Shi M, Taylor JM, Currier RJet al. Replacing time since human immunodeficiency virus infection by marker values in predicting residual time to acquired immunodeficiency syndrome diagnosis. Multicenter AIDS Cohort Study. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12(3):309–16. [DOI] [PubMed] [Google Scholar]
- 14. Cox DR. Regression models and life-tables. In: Kotz S, Johnson NL, eds. Breakthroughs in Statistics. New York: Springer; 1992. p 527–41. [Google Scholar]
- 15. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model: Series: Statistics for Biology and Health. New York: Springer; 2000. p 350. [Google Scholar]
- 16. Ishwaran H, Kogalur UB, Blackstone EH, Lauer MS. Random survival forests. Ann Appl Stat. 2008;2(3):841–60. [Google Scholar]
- 17. Anderberg MR. Cluster Analysis for Applications. New York: Academic Press; 1973. [Google Scholar]
- 18. Hoeffding W. A non-parametric test of independence. Ann Math Stat. 1948;19:546–57. [Google Scholar]
- 19. Harrell FE, Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for improved prognostic prediction. Stat Med. 1984;3(2):143–52. [DOI] [PubMed] [Google Scholar]
- 20. Harrell FE., Jr. Regression Modeling Strategies With Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis. Cham, Switzerland: Springer; 2015. [Google Scholar]
- 21. Harrell FE, Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15(4):361–87. [DOI] [PubMed] [Google Scholar]
- 22. Harrell FE., Jr rms: Regression modeling strategies. Available from: https://CRAN.R-project.org/package=rms
- 23. Ishwaran H, Kogalur UB. Random forests for survival, regression and classification (RF-SRC). Available from: https://mran.microsoft.com/snapshot/2016-04-02/web/packages/randomForestSRC/randomForestSRC.pdf
- 24. Ishwaran H, Kogalur UB. Random survival forests for R. R News. 2007;7(2):25–31. [Google Scholar]
- 25. Heagerty PJ, Saha-Chaudhuri P. survivalROC: Time-dependent ROC curve estimation from censored survival data. Available from: https://CRAN.R-project.org/package=survivalROC
- 26. Cameron D, Ubels J, Norström F. On what basis are medical cost-effectiveness thresholds set? Clashing opinions and an absence of data: a systematic review. Glob Health Action. 2018;11(1):1447828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Standfield L, Comans T, Scuffham P. Markov modeling and discrete event simulation in health care: a systematic comparison. Int J Technol Assess Health Care. 2014;30(2):165–72. [DOI] [PubMed] [Google Scholar]
- 28. Karnon J, Stahl J, Brennan A, Caro JJ, Mar J, Möller J. Modeling using discrete event simulation: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force-4. Med Decis Mak. 2012;32(5):701–11. [DOI] [PubMed] [Google Scholar]
- 29. Caro JJ, Møller J, Karnon J, Stahl J, Ishak J. Discrete Event Simulation for Health Technology Assessment. Boca Raton: CRC Press; 2016. [Google Scholar]
- 30. Jacobson SH, Hall SN, Swisher JR. Discrete-event simulation of health care systems. In: Hall RW, ed. Patient Flow: Reducing Delay in Healthcare Delivery. Boston: Springer; 2006. p 211–52. [Google Scholar]
- 31. Graves J, Garbett S, Zhou Z, Peterson J. The Value of Pharmacogenomic Information (Working Paper No. W241343). Cambridge: National Bureau of Economic Research; 2017. Available from: http://www.nber.org/papers/w24134.pdf [Google Scholar]
- 32. Pirmohamed M, Burnside G, Eriksson Net al. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369(24):2294–303. [DOI] [PubMed] [Google Scholar]
- 33. School of Medicine, Indiana University. PGX SLCO1B1 Genotyping For detection of SLCO1B1 variants affecting drug metabolism. Available from: http://geneticslab.medicine.iu.edu/dgg_dbadmin/public/files/documents/2016/01/tech-SLCO1B1.pdf
- 34. Dunnenberger HM, Crews KR, Hoffman JMet al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu Rev Pharmacol Toxicol. 2015;55:89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bielinski SJ, Olson JE, Pathak Jet al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time—using genomic data to individualize treatment protocol. Mayo Clin Proc. 2014;89(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoffman JM, Haidar CE, Wilkinson MRet al. PG4KDS: a model for the clinical implementation of pre-emptive pharmacogenetics. Am J Med Genet C Semin Med Genet. 2014;166(1):45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gottesman O, Scott SA, Ellis SBet al. The CLIPMERGE PGx program: clinical implementation of personalized medicine through electronic health records and genomics–pharmacogenomics. Clin Pharmacol Ther. 2013;94(2):214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson JA, Elsey AR, Clare-Salzler MJ, Nessl D, Conlon M, Nelson DR. Institutional profile: University of Florida and Shands Hospital Personalized Medicine Program: clinical implementation of pharmacogenetics. Pharmacogenomics. 2013;14(7):723–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O’Donnell PH, Bush A, Spitz Jet al. The 1200 patients project: creating a new medical model system for clinical implementation of pharmacogenomics. Clin Pharmacol Ther. 2012;92(4):446–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Appendix_online_supp for A Decision-Theoretic Approach to Panel-Based, Preemptive Genotyping by Yaping Shi, John A. Graves, Shawn P. Garbett, Zilu Zhou, Ramya Marathi, Xiaoming Wang, Frank E. Harrell, Thomas A. Lasko, Joshua C. Denny, Dan M. Roden, Josh F. Peterson and Jonathan S. Schildcrout in MDM Policy & Practice