

Abstract
APEX2, an engineered ascorbate peroxidase for high activity, is a powerful tool for proximity labeling applications. Owing to its lack of disulfides and the calcium‐independent activity, APEX2 can be applied intracellularly for targeted electron microscopy imaging or interactome mapping when fusing to a protein of interest. However, APEX2 fusion is often deleterious to the protein expression, which seriously hampers its wide utility. This problem is especially compelling when APEX2 is fused to structurally delicate proteins, such as multi‐pass membrane proteins. In this study, we found that a cysteine‐free single mutant C32S of APEX2 dramatically improved the expression of fusion proteins in mammalian cells without compromising the enzyme activity. We fused APEX2 and APEX2C32S to four multi‐transmembrane solute carriers (SLCs), SLC1A5, SLC6A5, SLC6A14, and SLC7A1, and compared their expressions in stable HEK293T cell lines. Except the SLC6A5 fusions expressing at decent levels for both APEX2 (70%) and APEX2C32S (73%), other three SLC proteins showed significantly better expression when fusing to APEX2C32S (69 ± 13%) than APEX2 (29 ± 15%). Immunofluorescence and western blot experiments showed correct plasma membrane localization and strong proximity labeling efficiency in all four SLC‐APEX2C32S cells. Enzyme kinetic experiments revealed that APEX2 and APEX2C32S have comparable activities in terms of oxidizing guaiacol. Overall, we believe APEX2C32S is a superior fusion tag to APEX2 for proximity labeling applications, especially when mismatched disulfide bonding or poor expression is a concern.
Keywords: APEX2, ascorbate peroxidase, proximity labeling, solute carriers
Abbreviations
- APX
ascorbate peroxidase
- BSA
bovine serum albumin
- EDTA
ethylenediaminetetraacetic acid
- EGFP
enhanced GFP
- GFP
green fluorescence protein
- HRP
horseradish peroxidase
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PMSF
phenylmethylsulfonyl fluoride
- RIPA
radioimmunoprecipitation assay
- SDS
sodium dodecyl sulfate
- SLC
solute carrier
1. INTRODUCTION
Heme peroxidases are very useful enzymes for biological applications owing to their great activity and broad substrate specificity.1 For instance, one of the most widely utilized heme peroxidases, horseradish peroxidase (HRP), is routinely applied as the reporters of chemiluminescence‐based western blot,2 immunohistochemistry,3 and enzyme‐linked immunosorbent assays (ELISA).4 Furthermore, HRP can activate various tyramide substrates, such as biotin5 or fluorophore derivatives,6 into short‐lived radical forms, which in turn covalently conjugate to neighboring proteins within 10–20 nm. This technique, known as proximity labeling, has been widely adopted in various applications, including tyramide signal amplification,7 electron microscopy tag,8 and extracellular proteomic mapping.9
A serious limitation of HRP is the loss of activity under a reducing environment, such as cytoplasm, because HRP requires four disulfide bonds and two calcium‐binding sites for its activity.10 On the other hand, ascorbate peroxidase (APX) is a 28‐kDa heme peroxidase that lacks disulfide bonds and calcium‐binding sites.11 Two generations of APX engineering, namely APEX12 and APEX2,13 have led to successful proximity labeling under the reducing environment in mammalian cells. By fusing to various proteins of interest, APEX2 has opened up avenues for spatially resolved proteomic mapping in live cell sub‐compartments, such as ciliary plasma membrane, mitochondrial matrix, endoplasmic reticulum membrane, and nucleus.14, 15, 16, 17, 18, 19
One prerequisite of a good tagging protein is to fold and express well when fusing with a protein of interest. We found that APEX2 fusions, however, often caused dramatically decreased expression in human cells. This problem is especially compelling when fusing to structurally delicate proteins, such as multi‐pass membrane proteins. For example, we observed that SLC1A5, a 10‐pass transmembrane solute carrier (SLC), was highly expressed at the plasma membrane when fusing with enhanced green fluorescence protein (EGFP) (92%), but poorly expressed when fusing with APEX2 (15%). We inspected the APEX2 sequence and found a single cysteine at position 32. It is known that disulfide bonds can form during the protein folding in endoplasmic reticulum20, 21 or even in the cytoplasm.22, 23, 24 We surmized that the APEX2 Cys‐32 residue may cause mismatched disulfides with other proteins, which leads to protein misfolding and degradation.
In this study, we showed that the APEX2C32S mutant is a superior fusion tag to the wild‐type APEX2 in terms of protein expression level in human cells. We constructed stable HEK293T cell lines that expressed APEX2C32S or APEX2 fused at the intracellular C‐terminus of four SLC members: SLC1A5 (10‐pass transmembrane), SLC6A5 (12‐pass transmembrane), SLC6A14 (12 pass‐transmembrane), and SLC7A1 (14‐pass transmembrane). Except SLC6A5 expressing well for both APEX2C32S and APEX2 fusions (73% vs. 70%), other three SLC fusions expressed significantly better when fusing to APEX2C32S (69 ± 13%) than APEX2 (29 ± 15%). Immunofluorescence and western blot experiments showed that all SLC‐APEX2C32S fusions correctly localized at the plasma membrane and performed efficient proximity labeling using biotin tyramide. The kinetic measurements revealed that APEX2C32S and APEX2 are equally active peroxidases for guaiacol oxidation.
2. RESULTS AND DISCUSSION
2.1. Previous reports suggested that the C32S mutation of APEX2 may not affect the peroxidase activity of small aromatic substrates
APX is known as a bi‐specific heme peroxidase that can oxidize either ascorbate or various aromatic small molecules in an H2O2‐dependent manner.25, 26 Sequence alignments revealed that Cys‐32 is the only conserved cysteine residue across all plant cytosolic APXs.27 Previous studies showed that the chemical modification of Cys‐32 by Ellman's reagent abolished the APX activity toward ascorbate but not other small aromatic substrates.25 Similarly, the APX C32S mutant decreased the ascorbate‐oxidizing activity by 70% but had no effects on guaiacol oxidation.25 These observations are in line with the APX crystal structures, which reveal two distinct substrate‐binding sites: (a) the ascorbate‐binding site at the edge of the heme plane that is in close vicinity to Cys‐32,28 and (b) the small aromatic molecule‐binding site on the top of the heme plane that does not interact with Cys‐32 (Figure 1).29 As most APEX2 applications utilize only small aromatic molecules, such as biotin tyramide for proteomic mapping, we reasoned that the C32S mutation of APEX2 would not compromise the activity in these applications.
Figure 1.

Crystal structures of ascorbate peroxidase in complex with (a) ascorbate (PDB: 1OAF) and (b) salicylhydroxamic acid (PDB: 1V0H) showed distinct binding sites of ascorbate and small aromatic molecules. Spheres: Cys‐32; magenta: heme; cyan: ascorbate in (a) or salicylhydroxamic acid in (b)
2.2. APEX2 and APEX2C32S exhibited comparable enzyme activity of guaiacol oxidation
To experimentally prove that the APEX2C32S activity is not altered for small aromatic substrates, we compared the guaiacol‐oxidizing kinetics of APEX2 and APEX2C32S. APEX2 and APEX2C32S proteins were recombinantly expressed, purified, and supplemented with hemin for the kinetic assays. Previous studies have shown that exorbitant H2O2 concentrations would inhibit the heme peroxidase activity. APEX2 has the maximal activity at ∼1 mM H2O2 and maintains >50% activity up to 10 mM H2O2, which provides an ideal range for proteomic and electron microscope labeling applications.13 We first compared the H2O2 susceptibility of APEX2 and APEX2C32S (Figure 2a). Across the H2O2 concentrations tested (0.125–8 mM), APEX2 and APEX2C32S showed similar activities with peaked values around 0.5–1 mM H2O2. Next, we determined the Michaelis–Menten kinetic parameters at a fixed concentration of H2O2 at 1 mM (Figure 2b). Similar to the wild‐type APX, the C32S mutation of APEX2 has no effects on k cat or K m (Figure 2c,d). Both APEX2 and APEX2C32S have k cat/K m ∼ 1.1 × 105 (M −1 s−1), which is similar to the previous report.13 We also examined the proximity labeling activity of APEX2 and APEX2C32S at the intracellular or extracellular sides of the plasma membrane using biotin tyramide. Regardless of the intracellular or extracellular environment, APEX2 and APEX2C32S showed similar activities when their expression levels were comparable (Figure S1).
Figure 2.
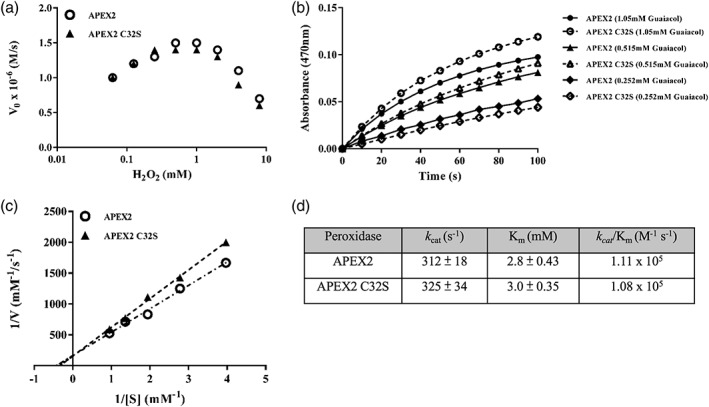
APEX2 and APEX2C32S showed comparable activity of guaiacol oxidation. (a) Initial rates of guaiacol oxidation (Vo) were measured in a range of H2O2 concentrations for APEX2 and APEX2C32S. A fixed concentration of guaiacol at 0.735 mM was used. (b) Kinetic measurements of guaiacol oxidation using APEX2 and APEX2C32S. A fixed concentration of H2O2 at 1 mM was used. For clarity, only three guaiacol concentrations are shown. (c) The Lineweaver–Burk (double reciprocal) plot of APEX2 and APEX2C32S enzyme kinetics. (d) The Michaelis–Menton parameters of APEX2 and APEX2C32S
2.3. Solute carrier family members are difficult targets for fusion tags
We chose SLC transporters as the proteins of interest. The SLC family, which comprises 52 subfamilies that contain more than 450 members, is the second largest family of human membrane proteins next to the G protein‐coupled receptors family.30, 31 They are transporters that control a wide range of solutes, including amino acids, nucleosides, sugars, ions, drugs, and so forth. In spite of the essential roles of SLCs in human health, however, SLCs are extremely under‐explored due to technical difficulties.32 Most SLCs are multi‐pass membrane proteins, which are structurally delicate and may be easily affected by a fusion protein. We tested three plasma membrane SLC proteins (SLC1A5, SLC6A5, and SLC6A14) with EGFP fused at the intracellular C‐terminus. Indeed, although all three SLC‐EGFP fusions glowed fluorescence in the transduced HEK293T cells (Figure 3a), only SLC1A5‐EGFP and SLC6A14‐EGFP were correctly trafficked to the plasma membrane. SLC6A5‐EGFP was retained in cytoplasm possibly due to the SLC protein misfolding (Figure 3b).
Figure 3.
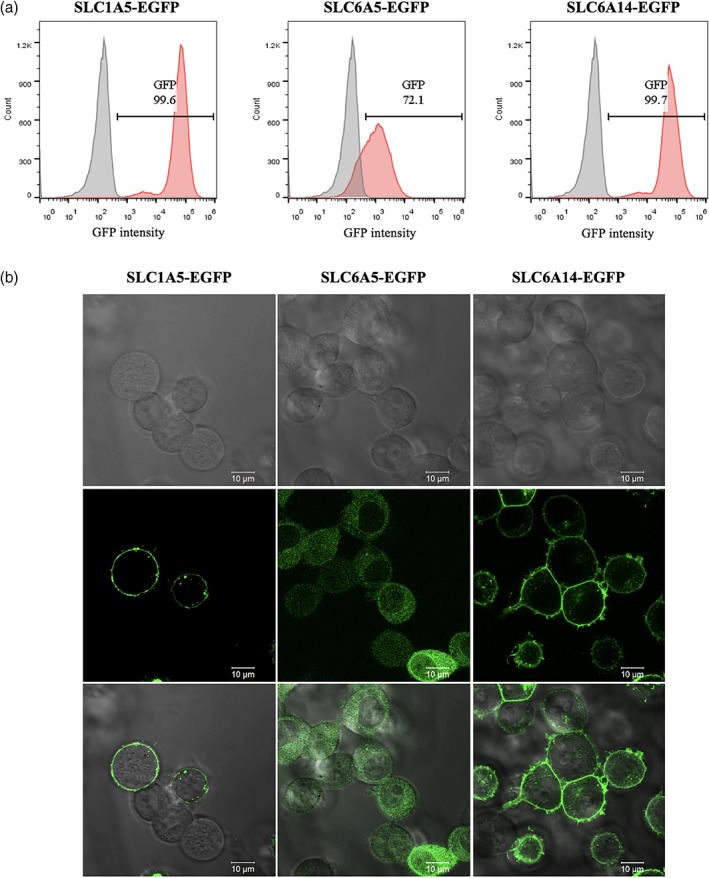
Expression level and subcellular localization of SLC‐EGFP fusions in HEK293T cells. (a) Expression levels of SLC‐EGFP fusions analyzed by flow cytometry. Red: SLC‐fusion; gray: parental HEK293T. (b) Sub‐localization of SLC‐EGFP fusions analyzed by confocal microscope. All SLC‐EGFP fusions were detected by EGFP fluorescence. EGFP, enhanced green fluorescence protein; SLC, solute carriers
2.4. APEX2C32S expression is superior to APEX2 when fusing to SLC proteins
Next, we sought to fuse APEX2 or APEX2C32S to four SLC proteins (SLC1A5, SLC6A5, SLC6A14, and SLC7A1) and compare their expressions in HEK293T cells. Unlike the EGFP fusions, all four SLCs were trafficked correctly to the plasma membrane when fusing to APEX2 or APEX2C32S (Figure 4b). However, they showed a binary expression pattern with a partial population not expressing the SLC fusion Figure 4a). Notably, only SLC6A5‐APEX2 exhibited a decent expression (69.8%), whereas other three SLC‐APEX2 fusions (SLC1A5, SLC6A14, and SLC7A1) showed poor expression levels (29 ± 15%), especially SLC1A5‐APEX2 at only 14.5% (Figure 4a). Strikingly, these three SLC expressions were significantly increased when fusing to APEX2C32S (69 ± 13%) Figure 4a). The immunofluorescence experiments confirmed that all SLC fusions were correctly localized at the plasma membrane Figure 4b). Given that these SLC proteins have multiple cysteine residues in the sequences, we conjectured that the APEX2C32S mutant might ameliorate the SLC fusion expression by eliminating mismatched disulfide bond formation between SLC and APEX2C32S.
Figure 4.
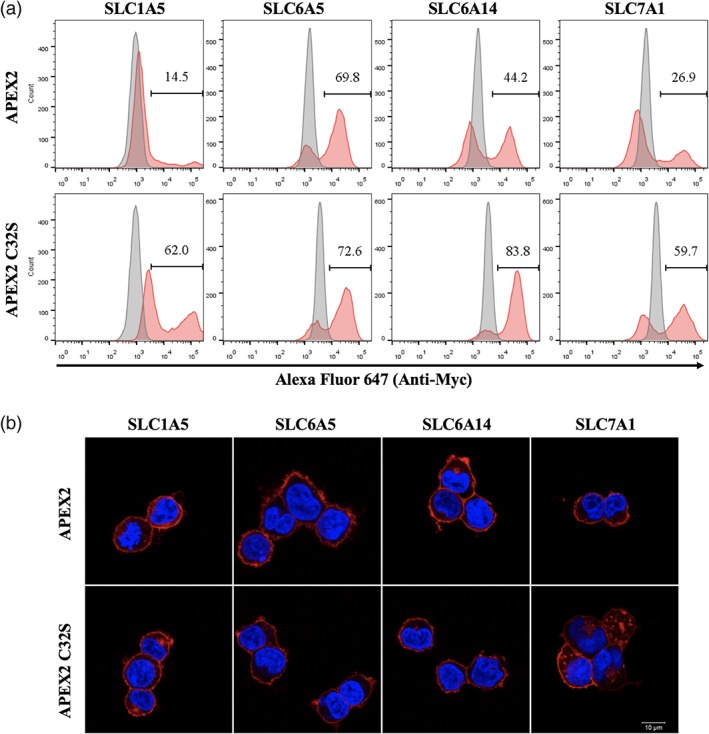
The expression levels of SLC‐APEX2C32S are generally better than SLC‐APEX2 in HEK293T cells. (a) Flow cytometry experiments compare the expressions levels of four SLC‐APEX2C32S and SLC‐APEX2 constructs. Red: SLC‐fusion; gray: parental HEK293T. (b) Immunofluorescence experiments show the correct plasma membrane localization of SLC‐APEX2C32S and SLC‐APEX2 proteins. All SLC‐APEX2 and SLC‐APEX2C32S fusions were detected by Alexa647 Myc‐tag staining. SLC, solute carriers
2.5. SLC‐APEX2C32S fusions showed efficient proximity labeling in human cells using biotin tyramide
Encouraged by the significant expressions of all four SLC‐APEX2C32S fusions, we further examined their peroxidase activity by performing proximity labeling using biotin tyramide in cells. The flow cytometry experiments showed 60, 71, 83, and 61% biotinylation levels of SLC1A5‐APEX2C32S, SLC6A5‐APEX2C32S, SLC6A14‐APEX2C32S, and SLC7A1‐APEX2C32S cells, respectively Figure 5a). The biotinylation levels were consistent with the SLC‐APEX2C32S expression levels in these four cell lines (Figure 4), indicating that most expressed SLC‐APEX2C32S proteins were functional. To acquire the spatial resolution, we further investigated the proximity labeling by immunofluorescence. While all four SLC‐APEX2C32S were correctly localized at the plasma membrane, the biotinylation signals were present at both the plasma membrane and cytoplasm Figure 5b). This is a common observation for APEX2‐membrane protein fusion. Owing to APEX2's strong activity, the proximity labeling can react on transient interactors at the plasma membrane,33, 34 which later diffuse to the cytoplasm. Overall, the four SLC‐APEX2C32S fusions tested exhibited strong proximity labeling activity and correct subcellular localization, which are two critical factors for successful proteomic mapping.
Figure 5.
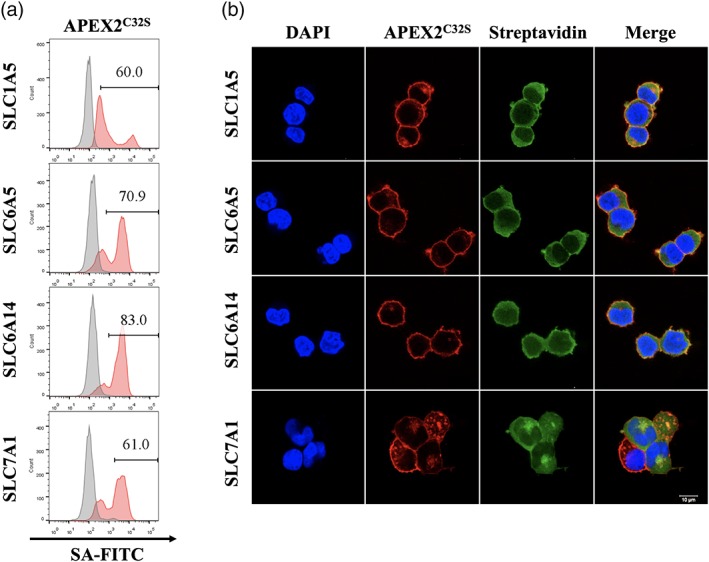
SLC‐APEX2C32S exhibited significant proximity labeling efficiency using biotin tyramide. (a) The proximity labeling efficiency was analyzed by flow cytometry for four SLC‐APEX2C32S constructs in HEK293T cells. Red: both biotin tyramide and H2O2 were added; gray: only H2O2 was added. (b) Immunofluorescence analyses of SLC‐APEX2C32S proximity labeling. APEX2C32S was detected by Myc‐tag staining (red). The biotin tyramide labeling was detected by streptavidin (green). SLC, solute carriers
2.6. SLC1A5‐APEX2C32S proximity labeling is more effective than SLC1A5‐APEX2 owing to the differential expression levels
Finally, we applied proximity labeling to SLC1A5‐APEX2 and SLC1A5‐APEX2C32S as a side‐by‐side comparison. Both constructs were made into HEK293T stable cell lines, and similar passage numbers of cells were used. The flow cytometry experiments showed that the biotinylated populations were 10 and 60% for SLC1A5‐APEX2 and SLC1A5‐APEX2C32S cells, respectively, which reflected the SLC1A5‐fusion expression levels (Figure 6a). As expected, SLC1A5‐APEX2C32S also showed much stronger signals than SLC1A5‐APEX2 on the western blot stained by streptavidin‐HRP (Figure 6b). Notably, several bands are very vague in SLC1A5‐APEX2 but clearly observed in SLC1A5‐APEX2C32S (arrows in Fig. 6b), which provides useful handles for downstream analyses, such as in‐gel digestion followed by liquid chromatography–mass spectrometry identification.
Figure 6.
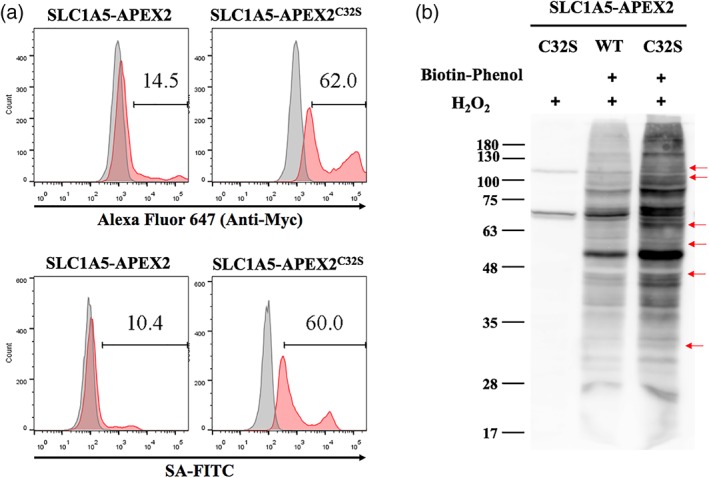
SLC1A5‐APEX2 proximity labeling is superior to SLC1A5‐APEX2C32S in HEK293T cells. (a) The expression level (upper) and the proximity labeling efficiency (bottom) of SLC1A5‐APEX2 and SLC1A5‐APEX2C32S were analyzed by flow cytometry. Upper: Myc‐tag staining for expression level (red: SLC1A5‐APEX2 or SLC1A5‐APEX2C32S cells; gray: parental HEK293T). Bottom: streptavidin staining for proximity labeling efficiency (red: both biotin tyramide and H2O2 were added; gray: only H2O2 was added). (b) The proximity labeling efficiency was analyzed by western blots. The red arrows denote the bands that are clearly observed in SLC1A5‐APEX2C32S, but very vague in SLC1A5‐APEX2. SLC, solute carriers
3. CONCLUSIONS
While a fusion tag is a powerful tool for biological studies, it is critical that the protein of interest's function, subcellular location, or expression is not affected by the fusion tag. For example, it is known that green fluorescence protein (GFP) could cause misfolding when fusing with other proteins.35, 36 This problem was ameliorated by engineering a robustly folded version of GFP called superfolder GFP (spGFP).37 In this study, we found that APEX2, a widely utilized fusion tag for proximity labeling applications, often expressed poorly when fusing with multi‐span membrane proteins, such as SLC family members. We engineered a cysteine‐free mutant APEX2C32S, which showed the same peroxidase activity toward small aromatic molecules as the wild‐type APEX2. We examined that APEX2C32S fused to four SLCs (SLC1A5, SLC6A5, SLC6A14, and SLC7A1) and showed that they all expressed significantly better than the wild‐type SLC‐APEX2 fusions in human cells. Furthermore, we showed that all these four SLC‐APEX2C32S proteins were correctly trafficked to the plasma membrane and performed efficient proximity labeling using biotin tyramide. The SLC proteins are very important transporters but extremely under‐investigated due to the lack of proper tools. The SLC‐APEXC32S fusions developed in this study provide a good handle for interactome mapping. Overall, we believe APEX2C32S is a good surrogate of APEX2 as a fusion tag, especially when there are concerns about protein misfolding or poor expression.
4. MATERIALS AND METHODS
4.1. Chemicals and reagents
The chemicals used in this work are commercially available and listed as follows: versene buffer: 0.5 mM ethylenediaminetetraacetic acid (EDTA) (Invitrogen, 15575‐038) in cell culture phosphate buffered saline (PBS). Radioimmunoprecipitation assay (RIPA) lysis buffer: 10 mM tris–HCl pH = 8.0 (Amresco, 1185‐53‐1), 1 mM EDTA, 1% triton‐X100 (Merck Millipore, 9002‐93‐1), 0.1% sodium deoxycholate (Sigma, 302‐95‐4), 0.1% sodium dodecyl sulfate (SDS, Merck Millipore, 1.13760.1000), 140 mM sodium chloride (Merck Millipore, 1.06404.5000), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Merck Millipore, P7626); protein purification buffer: 20 mM tris–HCl pH = 8.0 and 300 mM NaCl with 5 mM (resuspension), 20 mM (wash), or 250 mM (elution) imidazole; polyethyleneimine (Polyscience, 23966‐1); biotin‐phenol (IRIS Biotech, 41994‐02‐9).
4.2. Protein expression and purification
The N‐terminal His6‐tagged APEX2 and APEX2C32S were cloned into pET11a expression vector and expressed in Escherichia coli strain BL21‐DE3. These clones were cultured in 500 mL 2× YT broth with 100 μg/mL ampicillin at 37°C. Once the OD600 reached 0.6–0.8, the protein expression was induced with 420 μM isopropyl β‐D‐1‐thiogalactopyranoside (Sangon Biotech, 367‐93‐1) overnight at room temperature. Escherichia coli cells were centrifuged at 5000g at 4°C for 20 min and the pellet were resuspended with 20 mL resuspension buffer supplemented with 1 mM PMSF and 10 μg/mL DNaseI (Cyrusbioscience, 101.9003.98.9). Microfluidizer (M‐110S, Microfluidics) was used for the cell lysis. The lysates were centrifuged at 20,000g at 4°C for 20 min and the supernatant was incubated with 1 mL His‐tag binding agarose at 4°C for 1 hr. After protein binding, the His‐tag binding agarose was washed twice with 50 mL wash buffer. APEX2 and APEX2C32S were eluted with the elution buffer. The protein was concentrated and buffer‐exchanged to PBS using Amicon Ultra‐15 filter with Ultracel‐10K membrane (Merck Millipore). The purified protein was flash‐frozen in liquid N2 and stored at −80°C for storage.
4.3. Construct cloning
The APEX2 plasmid was received from Addgene (#49386) and APEX2C32S was constructed by mutagenesis. The cDNA plasmids of human SLC1A5 (HsCD00512455), SLC6A5 (HsCD00505746), SLC6A14 (HsCD00515993), and SLC7A1 (HsCD00514284) were received from DNASU. Standard polymerase chain reactions (PCRs) were performed to generate the SLC‐APEX2 fusion genes. PCR products were analyzed by 1% agarose and gel‐extracted for cloning. The gel‐extracted products were cloned into the puromycin‐resistance lentiviral vector using Gibson assembly followed by transforming into XL10‐Gold chemical competent cells. Plasmids were extracted and validated by Sanger sequencing before subsequent experiments.
4.4. Cell line, transfection, and lentivirus transduction
HEK293T cells were cultured in Dulbecco's Modified Eagle Medium with 10% fetal bovine serum and sodium bicarbonate at 37°C with 5% CO2. On the day prior to transfections, 6 × 105 HEK293T cells were seeded in a 6‐well culture plate. Subsequently, the gene transfer lentiviral plasmid was mixed with packaging vector pCMV‐dR8.91 and pMD2‐G in Opti‐MEM with polyethyleneimine for cell transfection. The culture medium was changed after 6 hr. After 48 hr of transfection, the lentiviral medium was filtered through a 0.45‐μm filter and mixed with 6 × 105 HEK293T cells with 4 μg/mL polybrene (Merck Millipore, TR‐1003‐G) for lentivirus transduction. The culture medium was changed after 4 hr. After 48 to 72 hr of transduction, the cells were selected with 2 μg/mL puromycin for 1 week to generate the stable clones.
4.5. Proximity labeling
SLC‐APEX2 or APEX2C32S stable cell lines were incubated in complete medium with 500 μM biotin‐phenol at 37°C for 30 min. The cells were subsequently mixed with 1 mM H2O2 for 1 min and quickly washed with PBS several times. After proximity labeling, the cells were fixed for immunofluorescence or flow cytometry experiments.
4.6. Flow cytometry analysis
After the proximity labeling reaction, the cells were lifted by versene buffer. Subsequently, the cells were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min followed by permeabilization with 0.1% triton‐X100 in PBS at room temperature for 10 min. The cells were then blocked at 4°C for 30 min with 1% bovine serum albumin (BSA) in PBS. The cells were stained at 4°C for 30 min with streptavidin fluorescein conjugated (Rockland, S000‐02) diluted 1:1000 and anti‐Myc Alexa 647 (Cell Signalling, 2233S) diluted 1:50 followed by PBS washes. The samples were analyzed by flow cytometry (LSRII, BD) and data were analyzed by FlowJo.
4.7. Immunofluorescence imaging
The cells were seeded on a 1.8 × 1.8 mm2 cover slip in a 6‐well culture plate on the day prior to the experiment. After proximity labeling, these cells were washed with PBS. The cells were fixed with 4% paraformaldehyde in PBS at room temperature for 15 min followed by permeabilization with 0.1% triton‐X100 in PBS at room temperature for 10 min. Subsequently, the cells were blocked at 4°C for 30 min with 1% BSA in PBS. The cells were stained at 4°C for 30 min with 4',6‐diamidino‐2‐phenylindole (BD Pharmingen) diluted 1:1000, streptavidin fluorescein conjugated diluted 1:1000, and anti‐Myc Alexa 647 diluted 1:100. Immunofluorescence images were acquired from confocal microscopy (Zeiss LSM 700) and analyzed by Zen.
4.8. Western blot
After proximity labeling, the cells were washed with PBS and lysed by RIPA lysis buffer. The protein concentration was quantified by bicinchoninic acid assay protein assay kit (Thermo‐Fisher, 23225). The lysate was mixed with the sample buffer and boiled for 10 min. The proteins were separated by gradient SDS‐polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Merck Millipore, IPVH00010) at 100 V for 2 hr. The membrane was blocked with 5% BSA in PBS overnight at 4°C and then incubated with streptavidin‐HRP conjugated (Rockland, S000‐03) diluted 1:20000 at room temperature for 1 hr. Subsequently, chemiluminescent luminescent HRP substrate (Merck Millipore, P90720) was added and detected by the luminescent analyzer (LAS‐4000 mini).
4.9. Guaiacol steady‐state kinetic assays
The purified APEX2 or APEX2C32S was mixed with hemin (Sigma, 51280, mole ratio: 1.5:1 to protein). The absorbance ratio of A405/A280 was greater than 2.0, which represented a full hemin occupancy. The working concentration of APEX2 and APEX2C32S in the kinetic experiments was 20 nM. The guaiacol (Cayman, 90‐05‐1) was diluted in PBS to the concentrations ranging from 0.25 to 1.5 mM. Guaiacol, H2O2, and the enzyme were mixed in 96‐well plates immediately followed by measuring the absorbance at 470 nm every 10 s using a SpectraMax 190 Microplate Reader (Molecular Devices) at room temperature.
Supporting information
Figure S1. Flow cytometry experiments showed that APEX2 and APEX2C32S have similar proximity labeling activity using biotin tyramide at either the intracellular or extracellular side of the plasma membrane. (a) APEX2 or APEX2C32S is fused to the intracellular C‐terminus of SLC6A5. (b) APEX2 or APEX2C32S is fused to the extracellular N‐terminus of a single‐pass transmembrane domain from the platelet‐derived growth factor receptor. All APEX2 and APEX2C32S constructs were detected by anti‐Myc staining (x‐axis). The biotin labeling was detected by streptavidin (y‐axis)
ACKNOWLEDGMENTS
We thank the Light Microscopy and Flow Cytometry Cores of Institute of Biomedical Sciences for their technical supports. We thank the Academia Sinica DNA Sequencing Core Facility (AS‐CFII‐108‐115). This work is supported by the Academia Sinica Career Development Award (AS‐CDA‐108‐L07) and the Ministry of Science and Technology, Taiwan (107‐2113‐M‐001‐013).
Huang M‐S, Lin W‐C, Chang J‐H, Cheng C‐H, Wang HY, Mou KY. The cysteine‐free single mutant C32S of APEX2 is a highly expressed and active fusion tag for proximity labeling applications. Protein Science. 2019;28:1703–1712. 10.1002/pro.3685
Funding information Academia Sinica, Grant/Award Number: AS‐CDA‐108‐L07; Ministry of Science and Technology, Grant/Award Number: 107‐2113‐M‐001‐013
REFERENCES
- 1. Poulos TL. Heme enzyme structure and function. Chem Rev. 2014;114:3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mruk DD, Cheng CY. Enhanced chemiluminescence (ECL) for routine immunoblotting: an inexpensive alternative to commercially available kits. Spermatogenesis. 2011;1:121–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchwalow I, Boecker W, Wolf E, Samoilova V, Tiemann M. Signal amplification in immunohistochemistry: loose‐jointed deformable heteropolymeric HRP conjugates vs. linear polymer backbone HRP conjugates. Acta Histochem. 2013;115:587–594. [DOI] [PubMed] [Google Scholar]
- 4. Sheng YM, Wang K, Lu QZ, et al. Nanobody‐horseradish peroxidase fusion protein as an ultrasensitive probe to detect antibodies against Newcastle disease virus in the immunoassay. J Nanobiotechnology. 2019;17:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toda Y, Kono K, Abiru H, et al. Application of tyramide signal amplification system to immunohistochemistry: a potent method to localize antigens that are not detectable by ordinary method. Pathol Int. 1999;49:479–483. [DOI] [PubMed] [Google Scholar]
- 6. Faget L, Hnasko TS. Tyramide signal amplification for immunofluorescent enhancement. Methods Mol Biol. 2015;1318:161–172. [DOI] [PubMed] [Google Scholar]
- 7. Schonhuber W, Fuchs B, Juretschko S, Amann R. Improved sensitivity of whole‐cell hybridization by the combination of horseradish peroxidase‐labeled oligonucleotides and tyramide signal amplification. Appl Environ Microbiol. 1997;63:3268–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cruz‐Lopez D, Ramos D, Castilloveitia G, Schikorski T. Quintuple labeling in the electron microscope with genetically encoded enhanced horseradish peroxidase. PLoS One. 2018;13:e0200693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li XW, Rees JS, Xue P, et al. New insights into the DT40 B cell receptor cluster using a proteomic proximity labeling assay. J Biol Chem. 2014;289:14434–14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howes BD, Feis A, Raimondi L, Indiani C, Smulevich G. The critical role of the proximal calcium ion in the structural properties of horseradish peroxidase. J Biol Chem. 2001;276:40704–40711. [DOI] [PubMed] [Google Scholar]
- 11. Patterson WR, Poulos TL. Crystal structure of recombinant pea cytosolic ascorbate peroxidase. Biochemistry. 1995;34:4331–4341. [DOI] [PubMed] [Google Scholar]
- 12. Martell JD, Deerinck TJ, Sancak Y, et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;30:1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lam SS, Martell JD, Kamer KJ, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hung V, Udeshi ND, Lam SS, et al. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc. 2016;11:456–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hung V, Lam SS, Udeshi ND, et al. Proteomic mapping of cytosol‐facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife. 2017;6:e24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rhee HW, Zou P, Udeshi ND, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han S, Udeshi ND, Deerinck TJ, et al. Proximity biotinylation as a method for mapping proteins associated with mtDNA in living cells. Cell Chem Biol. 2017;24:404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benhalevy D, Anastasakis DG, Hafner M. Proximity‐CLIP provides a snapshot of protein‐occupied RNA elements in subcellular compartments. Nat Methods. 2018;15:1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohli P, Hoxhne M, Jungst C, et al. The ciliary membrane‐associated proteome reveals actin‐binding proteins as key components of cilia. EMBO Rep. 2017;18:1521–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oka OB, Bulleid NJ. Forming disulfides in the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2425–2429. [DOI] [PubMed] [Google Scholar]
- 21. Zapun A, Jakob CA, Thomas DY, Bergeron JJ. Protein folding in a specialized compartment: the endoplasmic reticulum. Structure. 1999;7:R173–R182. [DOI] [PubMed] [Google Scholar]
- 22. Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–21758. [DOI] [PubMed] [Google Scholar]
- 23. Chakravarthi S, Jessop CE, Willer M, Stirling CJ, Bulleid NJ. Intracellular catalysis of disulfide bond formation by the human sulfhydryl oxidase, QSOX1. Biochem J. 2007;404:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saaranen MJ, Ruddock LW. Disulfide bond formation in the cytoplasm. Antioxid Redox Signal. 2013;19:36–43. [DOI] [PubMed] [Google Scholar]
- 25. Mandelman D, Jamal J, Poulos TL. Identification of two electron‐transfer sites in ascorbate peroxidase using chemical modification, enzyme kinetics, and crystallography. Biochemistry. 1998;37:17610–17617. [DOI] [PubMed] [Google Scholar]
- 26. Gumiero A, Murphy EJ, Metcalfe CL, Moody PC, Raven EL. An analysis of substrate binding interactions in the heme peroxidase enzymes: a structural perspective. Arch Biochem Biophys. 2010;500:13–20. [DOI] [PubMed] [Google Scholar]
- 27. Gelhaye E, Navrot N, Macdonald IK, Rouhier N, Raven EL, Jacquot JP. Ascorbate peroxidase‐thioredoxin interaction. Photosynth Res. 2006;89:193–200. [DOI] [PubMed] [Google Scholar]
- 28. Sharp KH, Mewies M, Moody PC, Raven EL. Crystal structure of the ascorbate peroxidase‐ascorbate complex. Nat Struct Biol. 2003;10:303–307. [DOI] [PubMed] [Google Scholar]
- 29. Sharp KH, Moody PC, Brown KA, Raven EL. Crystal structure of the ascorbate peroxidase‐salicylhydroxamic acid complex. Biochemistry. 2004;43:8644–8651. [DOI] [PubMed] [Google Scholar]
- 30. Fredriksson R, Nordstrom KJ, Stephansson O, Hagglund MG, Schioth HB. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett. 2008;582:3811–3816. [DOI] [PubMed] [Google Scholar]
- 31. Hediger MA, Clemencon B, Burrier RE, Bruford EA. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol Aspects Med. 2013;34:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cesar‐Razquin A, Snijder B, Frappier‐Brinton T, et al. A call for systematic research on solute carriers. Cell. 2015;162:478–487. [DOI] [PubMed] [Google Scholar]
- 33. Chen CL, Perrimon N. Proximity‐dependent labeling methods for proteomic profiling in living cells. Wiley Interdiscip Rev Dev Biol. 2017;6:e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paek J, Kalocsay M, Staus DP, et al. Multidimensional tracking of GPCR signaling via peroxidase‐catalyzed proximity labeling. Cell. 2017;169:338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. [DOI] [PubMed] [Google Scholar]
- 36. Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nat Biotechnol. 2005;23:102–107. [DOI] [PubMed] [Google Scholar]
- 37. Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:1170–1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow cytometry experiments showed that APEX2 and APEX2C32S have similar proximity labeling activity using biotin tyramide at either the intracellular or extracellular side of the plasma membrane. (a) APEX2 or APEX2C32S is fused to the intracellular C‐terminus of SLC6A5. (b) APEX2 or APEX2C32S is fused to the extracellular N‐terminus of a single‐pass transmembrane domain from the platelet‐derived growth factor receptor. All APEX2 and APEX2C32S constructs were detected by anti‐Myc staining (x‐axis). The biotin labeling was detected by streptavidin (y‐axis)