

Abstract
Computational tools for the analysis of protein data and the prediction of biological properties are essential in life sciences and biomedical research. Here, we introduce ProtDCal‐Suite, a web server comprising a set of machine learning‐based methods for studying proteins. The main module of ProtDCal‐Suite is the ProtDCal software. ProtDCal translates the structural information of proteins into numerical descriptors that serve as input to machine‐learning techniques. The ProtDCal‐Suite server also incorporates a post‐processing optional stage that allows ranking and filtering the obtained descriptors by computing their Shannon entropy values across the input set of proteins. ProtDCal's codification was used in the development of models for the prediction of specific protein properties. Thus, the other modules of ProtDCal‐Suite are protein analysis tools implemented using ProtDCal's descriptors. Among them are PPI‐Detect, for predicting the interaction likelihood of protein–protein and protein–peptide pairs, Enzyme Identifier, for identifying enzymes from amino acid sequences or 3D structures, and Pred‐NGlyco, for predicting N‐glycosylation sites. ProtDCal‐Suite is freely accessible at https://protdcal.zmb.uni‐due.de.
Keywords: descriptor, enzymes, machine‐learning, N‐glycosylation, protein–protein interactions, web server
1. INTRODUCTION
The analysis of protein data and the prediction of protein properties are of fundamental importance in modern Molecular Biology. Subjects such as the elucidation of protein–protein interaction networks, protein function prediction, and computational drug design, all benefit from massive computational analysis of the known protein data to extrapolate new knowledge of biological function.1, 2, 3, 4 The numerical encoding of raw protein sequences or structural data plays an important role for the development of robust prediction tools based on machine‐learning techniques.
In this context, ProtDCal is a software package that transforms protein sequences or 3D‐structures into general‐purpose numerical descriptors, accounting for both global and local information.5 Due to its complementary performance with respect to other well‐established tools in the field like PROFEAT6 and PseAcc7 (later extended to Pse‐in‐one8), ProtDCal has been used in a number of studies.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 Notable among them are the modeling of posttranslational modifications,14 the prediction of protein enzymatic function,15 the prediction of antimicrobial activity in peptides,16 the determination of residues critical for protein function,17 and the prediction of stability changes upon mutations.18 Very recently, ProtDCal was enhanced with a procedure for encoding protein pairs, which allows targeting the protein–protein interaction identification problem.19
Here, we present ProtDCal‐Suite, a versatile platform for granting web access to the wealth of encoding approaches implemented within ProtDCal, as well as to several protein analysis tools developed using ProtDCal's descriptors. Currently, ProtDCal‐Suite allows predicting the enzyme‐like character of proteins (Enzyme Identifier)15 and N‐glycosylation (Pred‐NGlyco) sites5, 14 as well as evaluating the likelihood of protein–protein interactions (PPI‐Detect).19 Recently, a tool for the prediction of methylation sites (MethylSight)20 was also incorporated by us in ProtDCal‐Suite. These applications of ProtDCal are useful on their own right, but also illustrate the capabilities of ProtDCal‐derived features for novel and diverse protein analysis tasks.
2. RESULTS
ProtDCal‐Suite consists of a main module (ProtDCal) and a set of secondary modules that provide access to machine learning‐based tools. These applications are used to predict specific protein functions and were created using ProtDCal descriptors. Next, we describe the generalities of the suite and the available tools.
2.1. The ProtDCal‐Suite
The graphical design of ProtDCal‐Suite is highly intuitive (Figure 1). Each tool has its own interface but shares a similar layout for quick familiarization by users. We documented all individual tools with help content and usage examples. Extended documentation and a tutorial, explaining the protein‐encoding features of ProtDCal, are also available. Template python scripts allow remotely accessing the web services and parsing the output data. This way, users can also submit jobs without using the web interface. This feature is valuable for remotely invoking the server services or for integrating the calculation of descriptors into custom third‐party workflows.
Figure 1.

Main interface of ProtDCal‐Suite
2.1.1. ProtDCal‐Suite input
All the predictive tools implemented in ProtDCal‐Suite accept input files containing the sequence information of proteins in FASTA format (Enzyme Identifier, PPI‐Detect, MethylSight and Pred‐NGlyco) and/or structural information in PDB format (Enzyme Identifier). In the main module (ProtDCal), the user can also specify options for the calculation of protein descriptors via the web interface. In the documentation of the interfaces for the different tools within ProtDCal‐Suite we provide information about the input formats and offer examples for the submission of jobs. Besides the input data, the user enters a job name and (optionally) an email address to receive information about the progress of the job. Using the identification code (ID) assigned to the job, the user can follow its status in the computing queue and subsequently retrieve the results of the calculations.
2.1.2. ProtDCal‐Suite output
Once a job is completed, there are two main output interfaces depending on whether the used tool was (1) ProtDCal or (2) any of the ProtDCal‐based applications. In the first case, the output is a download link to access the file containing the complete descriptor matrix. In addition, the output interface permits the user to post‐process the computed descriptors using an unsupervised feature selection approach based on Shannon Entropy (see section Analysis of ProtDCal's outcome). The use of Shannon Entropy allows for a preliminary reduction of the dimensionality of the descriptor matrix. For ProtDCal‐based applications, the predictions are visualized directly in the web, using a tabular form. All the results can be downloaded in CSV format.
2.2. ProtDCal
ProtDCal is a computational package5 for encoding the sequences and structures of proteins into numerical descriptors. These descriptors are the input to machine‐learning techniques (artificial neural networks,21 support vector machine,22 and random forest,23 among others) used for the development of novel predictors of protein functions and properties. ProtDCal splits the protein into different residue groups. Then, the contributions of the residues in each group are aggregated using diverse descriptive statistics (such as averages, variance, minimum or maximum values). This aggregation gives rise to a large variety of scalar descriptors, each of which represents local or global properties of the protein. The resulting vector is applicable to data mining problems such as protein classification, similarity analysis, and function prediction.
2.2.1. ProtDCal steps for calculating a protein descriptor
Figure 2 illustrates the process of obtaining the descriptor FD_AC2_GLY_Ar for the human prion protein fragment described by the PDB entry 1OEH 27 with sequence: HGGGTGQP. The notation used in ProtDCal to label the final descriptors directly refers to the options chosen by the user in the input step. A combinatorial algorithm composed of four steps (Figure 2, top), each with several options (that can be defined by the user) is implemented in ProtDCal. The program computes all the combinations of defined options, thus producing one individual descriptor from each combination. The combination of the selected indices (In), vicinity operators (VO), groups (Gp), and aggregation operators (AO) results in a large set of descriptors for each protein. All these descriptors are univocally identified following the convention: In_VO_Gp_AO. In the example shown in Figure 2, the options selected to generate the descriptor are highlighted in red.
Figure 2.
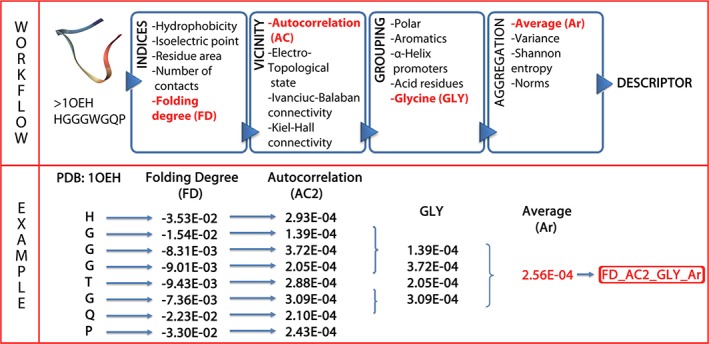
ProtDCal steps for calculating a protein descriptor. The fragment of a human prion protein (upper panel, far left) with Protein Data Bank24, 25, 26 identification code 1OEH 27 is used as an example of protein under codification
Next, we briefly describe, step by step, the general process of calculating the protein descriptors using ProtDCal, for the human prion protein fragment shown in Figure 2.
Step 1: Residue codification (indices). ProtDCal has implemented a list of indices (Tables S1–S4), mostly extracted from the AAindex database28 that represent several structural and chemical physical properties of amino acids. For each residue in the protein, according to the indices selected by the user, an array of numerical values is created. This list of indices is then used to encode the residues in the protein in order to obtain sequence‐based and 3D‐structure protein descriptors.
In the example shown in Figure 2, we use the folding degree (FD) as residue index. FD is a geometrical parameter,29 which significantly correlates with the folding rate constant and the average of the logarithm of the folding degree (lnFD) along all the residues in the protein.
where d is the spatial Euclidian distance, N the length of the protein, and x a parameter that takes value 2 for terminal residues and 3 for all the others. In the example, FD is selected as index to provide an initial numerical characterization of all residues in the protein. In addition to the folding degree, more than 30 geometrical and chemical–physical indices (e.g., hydrophobicity, number of contacts, molar weight, solvent accessible surface area) are implemented in ProtDCal, which results in a great variability of the information captured by different descriptors.
Step 2: Modification by vicinity. Here, the numeric values in each array of index values are modified according to the values of neighboring residues within the sequence. Different definitions of “neighborhood” result in several potential vicinity operators (Table S5). The application of vicinity‐modification operators to the values of a specific index array allows to include information in the final descriptor that reflects the ordering of the amino acids within the protein.
In the example of Figure 2, the autocorrelation operator of order 2 (AC2) is used to modify the initial FD values of each residue. This is achieved by incorporating information of the values from residues separated by two amino acids along the sequence. The operator is formulated as:
where i represents the i‐th residue in a protein and k corresponds to the order of the autocorrelation.
Step 3: Grouping. Subarrays of groups of residues are formed, according to a set of grouping criteria implemented in ProtDCal5 (Tables S6–S8). Among them, the entire protein forms the largest group, while the shortest group could contain a single type of residue. Such splitting of information in the amino acid sequence results in highly specific descriptors applicable to various protein analysis‐related problems. In the example shown in Figure 2, the group is formed by all glycine residues (GLY) in the protein.
Step 4: Aggregation operators. Finally, an aggregation operator is applied to the columns of each matrix obtained after grouping, to transform such matrix into a final numeric descriptor. Available aggregation operators include the p‐norms of orders p = 1 to p = 3,30 central‐tendency measures (geometric, average, and harmonic means, among others), dispersion and distribution parameters (kurtosis, variance, quartiles, skewness), and information‐theoretic measures based on Shannon entropy31 (Tables S9–S12). The different aggregation operators deliver distinct information about the property and the group used to generate the descriptors. In this way, descriptors derived from norms are most appropriate for modeling protein functions and classes that are dependent on protein size. On the contrary, for classes that are not related to the number of residues, descriptors obtained with dispersion and central tendency (means) aggregation operators may be preferable. In Figure 2, the arithmetic mean (Ar) is used to aggregate the values in the group into a single scalar value.
After following these four steps, the final descriptor resulting from the selected options (Figure 2, highlighted in red) is: FD_AC2_GLY_Ar. Hence, the structural information in this descriptor can be read as the average value (Ar) for all glycine amino acids (GLY), of the modified folding degree (FD) property, according to the autocorrelation (AC2) operator between neighboring residues.
2.2.2. Analysis of ProtDCal's outcome
PROFEAT,6 PROTEIN RECON,32 and PseAAC7, 8 are among the most notable available tools for calculating large numbers of sequence‐based physicochemical protein features. We used principal component analysis (PCA) to compare these methods to ProtDCal5 (Figure 3). PCA was applied on the matrix of all computed descriptors. Then, the contribution of each program was measured using the loading values to evaluate the correlation between the original descriptors and the principal components. A given component is said to be loaded by a descriptor arising from one program when the correlation between the descriptor and a component is higher than 0.7.
Figure 3.
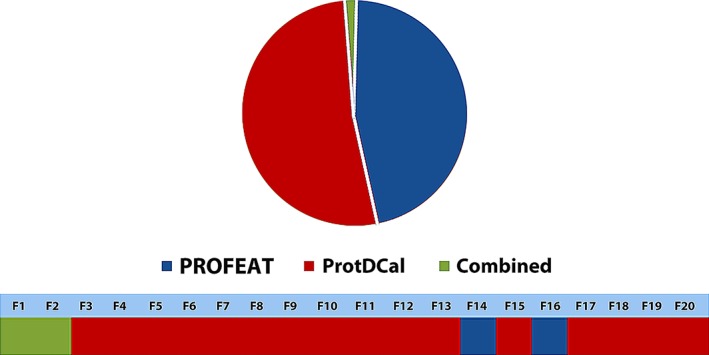
PCA test. Top: Pie chart showing all 191 principal components. Bottom: Bar diagram of the 20 top‐ranked composed components of the test. The descriptors from the RECON program were highly redundant, thus they are found only within the first two “combined components.”
The application of PCA resulted in 191 principal components, explaining 95% of the total variance in the descriptor data. Notably, while PROFEAT explains 45% of the variance (90 components loaded), ProtDCal descriptors are able to explain 52% of the variance (103 components loaded, Figure 3 top). Of the 20 top‐ranked components (Figure 3, bottom), 16 have high loadings uniquely from ProtDCal. This analysis indicates that the components of ProtDCal capture most of the data variance. Importantly, ProtDCal captures information that it is not contained in other descriptors such as those of PROFEAT and PROTEIN RECON.
The information content of the structural descriptors generated by ProtDCal makes them suitable for modeling various functions and properties of proteins. However, given the large number of descriptors that ProtDCal delivers, the application of feature selection methods is required as an intermediate step between generating a raw feature matrix and training the final model. Machine‐learning platforms, such as Weka33 offer several methods to perform feature selection based on both unsupervised and supervised approaches. Depending of the size of the data set and the number of initial features, this step can be computationally demanding. Importantly, the resulting subset of features can determine the quality of the final model. Thus, to offer users an initial processing of the feature matrix, our web server characterizes each descriptor using standardized Shannon Entropy (sSE).
where p i is the probability that a randomly selected instance (protein) belongs to the interval i and N is the number of intervals over which the range of descriptor values is split. We use uniform splitting to obtain all the intervals. The number of instances in the data set determines the number of bins. In this way, the range of the sSE values for each descriptor is within (0,1), ranging from zero, corresponding to a total absence of variability, to one, corresponding to a uniformly distributed data set along the descriptor range. Accordingly, plots of the frequency histogram per interval of sSE and of the cumulative frequency along the data set are provided to the user (Figure 4). Then, users can perform an initial reduction of the feature matrix by requesting a subset of descriptors within a custom interval of sSE. This preprocessing step presents, in a user‐friendly manner, the dispersion of the obtained descriptors along the data set of input proteins. In addition, it enables the elimination of invariant descriptors that do not provide useful information. This step also allows discarding highly variable features that may not be as effective to model discrete properties, such as in a binary classification problem (e.g., active vs. inactive peptide drugs), where we generally seek descriptors following a bimodal distribution.
Figure 4.
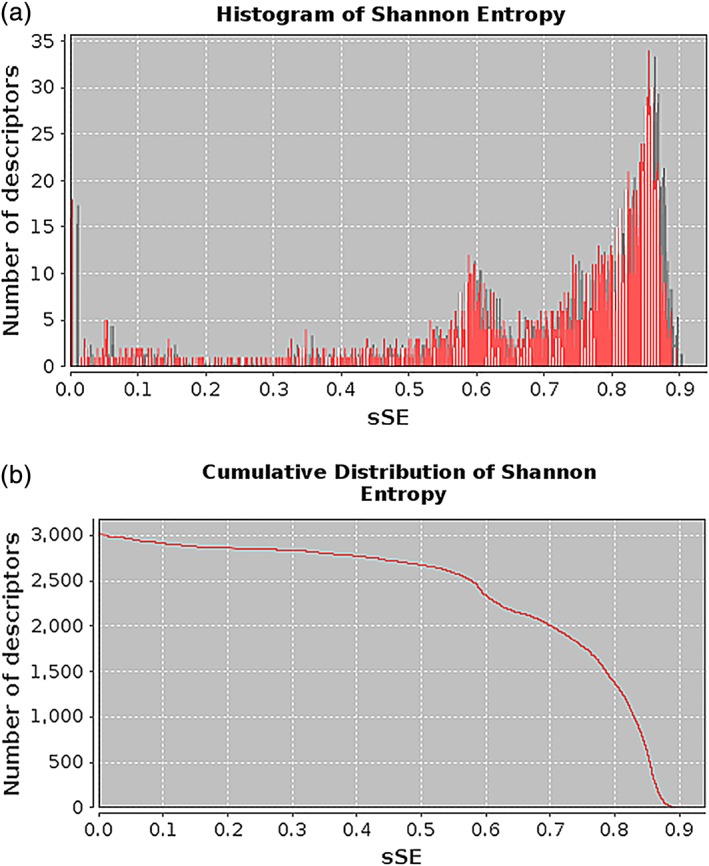
Illustration of the information content plots derived from a set of 3,000 descriptors calculated with ProtDCal. (a) A frequency histogram per interval of standardized Shannon entropy (sSE) is presented. (b) The cumulative frequency along the range of sSE is depicted
Independent tools, such as the IMMAN program,34 allow for the advanced use of SE and several other information theoretic measures for applying both unsupervised and supervised feature selection to a set of descriptors. Information gain35, 36 is another widely used measure for supervised feature selection in machine‐learning approaches. In future developments of our web server, we intend to implement these and other feature‐selection analysis tools, for post‐processing the descriptors generated by the ProtDCal server.
2.3. Protein analysis tools
ProtDCal's features have been used to develop predictors for protein analysis.9, 14, 15, 16, 17, 19, 20 In ProtDCal‐Suite we provide, for the first time, web access to some of these tools.
2.3.1. Performance measures
Next, we summarize the set of measures used to evaluate the predictors implemented in the different protein analysis tools.
where TP means true positive predictions, TN corresponds to true negative predictions, FP represents false positives, and FN indicates false negative predictions.
2.3.2. PPI‐detect
PPI‐Detect19 is a support vector machine (SVM) model that allows predicting the likelihood of interactions between two proteins based on their sequence information. The method is based on a new formalism that transforms pairs of amino acid sequences into general‐purpose‐numerical descriptors, which are used as input to an SVM classifier.
The benchmark employed for PPI‐Detect was created using the publicly available databases of protein domains interaction data: 3did37 and IPfam,38 containing pairs of domains reported as interacting, and Negatome 2.0,39 containing pairs of domains with no reported interactions. For each domain, the corresponding sequences were obtained from Pfam, a database with a large collection of protein families.40 The final dataset comprises 1,922 interacting pairs and 2,405 noninteracting pairs of domains. Then, the data set was split into training (3,491 pairs: 1,613 positive and 1,878 negatives) and test (836 pairs: 309 positives and 527 negatives).
The theoretical background of PPI‐Detect is described elsewhere.19 Shortly, we defined new pairwise protein descriptors as follows: Provided two amino acid sequences A and B, and the reaction:
where AB and BA are block copolymers formed by the sequences of A and B.
The pairwise descriptor D(A‐B) is calculated as: D(A‐B) = D(AB) + D(BA) − 2D(A) − 2D(B), where D(X) corresponds to the value of the single‐chain descriptor for a given sequence X (A, B, AB, or BA in this example). The value of D(A‐B) is related to the change in the topological information upon the dimerization process. We note that the contribution of the unaltered partners is removed, thus the descriptors are a numerical representation of the relation between the independent sequences. We obtained the individual descriptors using the electro‐topological state (E‐State) vicinity operator, which allows capturing the topological information of both the original and combined sequences.
The training was performed with the SVM package SMO22, 41 and the final model was selected with a linear kernel and a cost (C) for misclassified cases, C = 11.3. The results of an external test for PPI‐Detect and the tools PIPE,42 Pred‐PPI,43 and SPPS44 indicate that PPI‐Detect outperforms, in terms of accuracy, the other tools (Table 1).
Table 1.
Comparison of the accuracy values for PPI‐detect and other PPI predictors19
| PIPE | Pred‐PPI | SPPS | PPI‐detect | |
|---|---|---|---|---|
| Accuracy (%) | 63.9 | 43.5 | 61.7 | 66.1 |
Abbreviation: PPI, protein–protein interaction.
PPI‐Detect was successfully used to identify improved derivatives of EPI‐X4,45, 46 an endogenous peptide inhibitor of the G‐protein‐coupled receptor CXCR4.19
2.3.3. Enzyme identifier
Enzyme Identifier is a SVM predictor for identifying enzyme‐like proteins15 from sequence or structural data. Accordingly, two models are implemented in Enzyme Identifier: sequence‐based (using FASTA Files) and structure‐based (using PDB files).
The data set employed for training both models was taken from Dobson and Doig (D&D),47 comprising a total of 1178 structurally diverse proteins (691 enzymes and 487 nonenzymes), extracted from the PDB and Medline Abstracts databases. The Enzyme Identifier SVM models were generated and validated using 10 × 10‐fold CV. The accuracy values reported in Table 2 illustrate how this structure‐based model outperforms structure‐based predictors developed by other authors using the same data set.
Table 2.
Comparison of performance measures in 10‐fold cross‐validation for ProtDCal‐based models (enzyme identifier) and other methods15
| Reference | Accuracy (%) |
|---|---|
| Enzyme identifier (3D structures)15 | 82.0 ± 0.3 |
| Shervashidze48 | 81.5 ± 1.5 |
| Senelle49 | 80.3 |
| Dobson et al.47 | 80.2 ± 1.2 |
| Shervashidze et al.50 | 79.8 ± 0.4 |
| Neumann et al.51 | 79.0 ± 0.2 |
| Enzyme identifier (amino acid sequences)a | 78.8 ± 0.2 |
| Li et al.52 | 78.3 |
| Bai and Hancock53 | 77.6 |
| Orsini et al.54 | 76.6 ± 0.6 |
| Kilhamn55 | 75.9 |
| Johansson et al.56 | 75.4 ± 0.6 |
Sequence‐based model. Notice that all other models are based on 3D structural information.
In addition, the accuracy of the predictions of the 3D structure‐based model was assessed in an external set of 52 proteins, which was structurally unrelated to the training data set. The accuracy obtained was 80.8%, while with the method of Dobson and Doig the reported accuracy is 79.0%.47
2.3.4. Pred‐NGlyco
Pred‐NGlyco is a sequence based Random Forest (RF) model for predicting N‐glycosylation sites in peptides and proteins. This model illustrated, for the first time, the applicability of ProtDCal's descriptors to model relevant protein structural data.5 To build the model, 3,508 sequence‐unique windows, with 15 amino acids of length, were extracted from an initial data set of 241 proteins in the OGLYCBASE57 data set. Each window was centered on an asparagine residue and classified in glycosylated (positive) or nonglycosylated (negative). Then, ProtDCal sequence‐based descriptors were computed for each position of these chains.
Feature selection was performed using a Wrapper approach, with a genetic algorithm as implemented in Weka.33 The resulting model was compared via cross‐validation to contemporary N‐glycosylation predictors, such as GPP,58 NetNglyc,58 EnsembleGly,59 and ScanSite.60 The results (Table 3) indicated that, in general, Pred‐NGlyco, EnsembleGly, and GPP outperform the methods NetNGlyc and ScanSite.
Table 3.
Comparison of performance measures for Pred‐NGlyco and other predictors in 10‐fold cross validation test5
| Accuracy (%) | Sensitivity (%) | Specificity (%) | |
|---|---|---|---|
| Pred‐NGlyco | 91.6 | 93.2 | 91.4 |
| GPP | 92.8 | 96.6 | 91.8 |
| NetNGlyc | 76.7 | 43.9 | 95.7 |
| EnsembleGlya | 95.0 | 98.0 | 77.0 |
| ScanSite | 79.8 | 72.7 | 81.9 |
Evaluated in five‐fold cross validation. The specificity value originally reported for EnsembleGly59 actually corresponds to precision.
In addition, the Pred‐NGlyco model was compared using an external test set to the predictor GPP58 (Table 4, the web server associated with EnsembleGly is no longer available). The comparison shows higher performance for the Pred‐NGlyco model with superior values of accuracy, specificity, and precision than those of GPP, while GPP showed slightly better sensitivity.
Table 4.
Performance measures in external test set for Pred‐NGlyco and GPP5
| Accuracy (%) | Sensitivity (%) | Specificity (%) | Precision (%) | |
|---|---|---|---|---|
| GPP | 66.2 | 97.2 | 62.7 | 22.7 |
| Pred‐NGlyco | 87.1 | 93.5 | 86.4 | 43.6 |
Like PPI‐Detect and Enzyme Identifier, Pred‐NGlyco is an example of the value of ProtDCal descriptors to model various biological data.
3. SERVER DETAILS
The server is hosted in an Apache2 webserver and it was implemented in a two‐layer architecture, divided into front‐end and back‐end. The front‐end, written in PHP and JavaScript, is responsible for exchanging information with users. This layer is visualized with HTML5 and Bootstrap framework. All tools were implemented in the Java language using third‐party libraries. The back‐end is formed by a set of Perl scripts that manage job execution on a computer cluster system.
4. CONCLUSIONS
ProtDCal‐Suite is a valuable platform for the machine learning‐based study of protein structure–function relationships. The principal module, ProtDCal, provides scientists with information‐rich features datasets that describe key structural characteristics of proteins. These descriptors are highly suited for the training and evaluation of machine learning models used in the prediction of protein function. The information‐theoretic post‐processing of the generated protein descriptors enables rapid unsupervised feature selection, prior to the creation of the model.
The capability of ProtDCal to generate useful features was assessed in several studies developing novel machine learning‐based tools.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 Here, we present web interfaces for predicting the interaction likelihood of protein–protein and protein–peptide pairs (PPI‐Detect), for identifying enzymes from amino acid sequences or 3D structures (Enzyme Identifier), and for predicting N‐glycosylation sites in peptides and proteins (Pred‐NGlyco).
In future, we will continue incorporating new applications based on ProtDCal features into ProtDCal‐Suite to bring more functionalities to users. A next development will include a tool for the design of antibacterial peptides.
Supporting information
Appendix S1 : Supporting Information
ACKNOWLEDGMENTS
E.S.‐G. acknowledges the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC‐2033—Project ID: 390677874 as well as the support of the Boehringer Ingelheim Foundation (Plus‐3 grant). S. R.‐M. and E.S.‐G. acknowledge funding by the DFG—Project number 316249678—SFB 1279.
Romero‐Molina S, Ruiz‐Blanco YB, Green JR, Sanchez‐Garcia E. ProtDCal‐Suite: A web server for the numerical codification and functional analysis of proteins. Protein Science. 2019;28:1734–1743. 10.1002/pro.3673
Contributor Information
Yasser B. Ruiz‐Blanco, Email: yasser.ruizblanco@uni-due.de.
Elsa Sanchez‐Garcia, Email: elsa.sanchez-garcia@uni-due.de.
REFERENCES
- 1. Alberts B, Bray D, Hopkin K, et al. Protein structure and function Essential cell biology. New York and London: Garland Science, 1997. [Google Scholar]
- 2. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rives AW, Galitski T. Modular organization of cellular networks. Proc Natl Acad Sci U S A. 2003;100:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson D, Cox M. Principles of biochemistry. 4th ed. New York, NY: WH Freeman and Company, 2005. [Google Scholar]
- 5. Ruiz‐Blanco YB, Paz W, Green J, Marrero‐Ponce Y. ProtDCal: A program to compute general‐purpose‐numerical descriptors for sequences and 3D‐structures of proteins. BMC Bioinform. 2015;16:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li ZR, Lin HH, Han LY, Jiang L, Chen X, Chen YZ. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006;34:W32–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen H‐B, Chou K‐C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Analyt Biochem. 2008;373:386–388. [DOI] [PubMed] [Google Scholar]
- 8. Liu B, Liu F, Wang X, Chen J, Fang L, Chou K‐C. Pse‐in‐one: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43:W65–W71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kleandrova VV, Ruso JM, Speck‐Planche A, Dias Soeiro Cordeiro MN. Enabling the discovery and virtual screening of potent and safe antimicrobial peptides. Simultaneous prediction of antibacterial activity and cytotoxicity. ACS Combinat Sci. 2016;18:490–498. [DOI] [PubMed] [Google Scholar]
- 10. Scheraga HA, Rackovsky S. Global informatics and physical property selection in protein sequences. Proc Natl Acad Sci U S A. 2016;113:1808–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simeon S, Li H, Win TS, et al. PepBio: Predicting the bioactivity of host defense peptides. RSC Adv. 2017;7:35119–35134. [Google Scholar]
- 12. García‐Jacas CR, Cabrera‐Leyva L, Marrero‐Ponce Y, Suárez‐Lezcano J, Cortés‐Guzmán F, García‐González LA. GOWAWA aggregation operator‐based global molecular characterizations: Weighting atom/bond contributions (LOVIs/LOEIs) according to their influence in the molecular encoding. Mol Inform. 2018;37:1800039. [DOI] [PubMed] [Google Scholar]
- 13. Johnson D. Biotherapeutics: Challenges and opportunities for predictive toxicology of monoclonal antibodies. Intl J Mol Sci. 2018;19:3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruiz‐Blanco YB, Marrero‐Ponce Y, García‐Hernández E, Green J. Novel “extended sequons” of human N‐glycosylation sites improve the precision of qualitative predictions: An alignment‐free study of pattern recognition using ProtDCal protein features. Amino Acids. 2017;49:317–325. [DOI] [PubMed] [Google Scholar]
- 15. Ruiz‐Blanco YB, Agüero‐Chapin G, García‐Hernández E, Álvarez O, Antunes A, Green J. Exploring general‐purpose protein features for distinguishing enzymes and non‐enzymes within the twilight zone. BMC Bioinform. 2017;18:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Speck‐Planche A, Kleandrova VV, Ruso JM, Cordeiro DS. MNFirst multitarget chemo‐bioinformatic model to enable the discovery of antibacterial peptides against multiple gram‐positive pathogens. J Chem Inform Model. 2016;56:588–598. [DOI] [PubMed] [Google Scholar]
- 17. Corral‐Corral R, Beltrán JA, Brizuela CA, Del Rio G. Systematic identification of machine‐learning models aimed to classify critical residues for protein function from protein structure. Molecules. 2017;22:1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Y, Urolagin S, Niroula A, Ding X, Shen B, Vihinen M. PON‐tstab: Protein variant stability predictor. Importance of training data 1uality. Intl J Mol Sci. 2018;19:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Romero‐Molina S, Ruiz‐Blanco YB, Harms M, Münch J, Sanchez‐Garcia E. PPI‐detect: A support vector machine model for sequence‐based prediction of protein–protein interactions. J Comput Chem. 2019;40:1233–1242. [DOI] [PubMed] [Google Scholar]
- 20. Biggar KK, Ruiz‐Blanco YB, Charih F, et al. MethylSight: Taking a wider view of lysine methylation through computer‐aided discovery to provide insight into the human methyl‐lysine proteome. bioRxiv. 2018;274688. [Google Scholar]
- 21. Hassoun MH. Fundamentals of artificial neural networks. Cambridge: MIT Press, 1995. [Google Scholar]
- 22. Platt JC. Fast training of support vector machines using sequential minimal optimization Advances in kernel methods. Cambridge: MIT Press, 1998; p. 185–208. [Google Scholar]
- 23. Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 24. Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Mol Biol. 2003;10:980–980. [DOI] [PubMed] [Google Scholar]
- 25. Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2006;35:D301–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. wwPDB consortium . Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2018;47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zahn R. The octapeptide repeats in mammalian prion protein constitute a pH‐dependent folding and aggregation site. J Mol Biol. 2003;334:477–488. [DOI] [PubMed] [Google Scholar]
- 28. Kawashima S, Kanehisa M. AAindex: Amino acid index database. Nucleic Acids Res. 2000;28:374–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruiz‐Blanco YB, Marrero‐Ponce Y, Prieto PJ, Salgado J, García Y, Sotomayor‐Torres CM. A Hooke's law‐based approach to protein folding rate. J Theoret Biol. 2015;364:407–417. [DOI] [PubMed] [Google Scholar]
- 30.Dunford, N. and Schwartz, J.T. (1958) Linear Operators, Part I: General Theory. Wiley‐Interscience, New York. [Google Scholar]
- 31. Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. [Google Scholar]
- 32. Sukumar N, Breneman CM. QTAIM in drug discovery and protein modeling The quantum theory of atoms in molecules. Weinheim, Germany: Wiley VCH Verlag, 2007. [Google Scholar]
- 33. Frank E, Hall MA, Witten IH. The WEKA Workbench. Online Appendix for "Data Mining: Practical Machine Learning Tools and Techniques". USA: Morgan Kaufmann Publishers Inc., 2016. [Google Scholar]
- 34. Urias RWP, Barigye SJ, Marrero‐Ponce Y, García‐Jacas CR, Valdes‐Martiní JR, Perez‐Gimenez F. IMMAN: Free software for information theory‐based chemometric analysis. Mol Divers. 2015;19:305–319. [DOI] [PubMed] [Google Scholar]
- 35. Quinlan JR. Induction of decision trees. Machine Learning. 1986;1:81–106. [Google Scholar]
- 36. Lee C, Lee GG. Information gain and divergence‐based feature selection for machine learning‐based text categorization. Inf Process Manage. 2006;42:155–165. [Google Scholar]
- 37. Mosca R, Céol A, Stein A, Olivella R, Aloy P. 3did: A catalog of domain‐based interactions of known three‐dimensional structure. Nucleic Acids Res. 2014;42:D374–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Finn RD, Miller BL, Clements J, Bateman A. iPfam: A database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014;42:D364–D373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blohm P, Frishman G, Smialowski P, et al. Negatome 2.0: A database of non‐interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res. 2014;42:D396–D400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keerthi SS, Shevade SK, Bhattacharyya C, Murthy KRK. Improvements to Platt's SMO algorithm for SVM classifier design. Neural Comput. 2001;13:637–649. [Google Scholar]
- 42. Pitre S, Dehne F, Chan A, et al. PIPE: A protein‐protein interaction prediction engine based on the re‐occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinform. 2006;7:365–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo Y, Yu L, Wen Z, Li M. Using support vector machine combined with auto covariance to predict protein‐protein interactions from protein sequences. Nucleic Acids Res. 2008;36:3025–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu X, Liu B, Huang Z, Shi T, Chen Y, Zhang J. SPPS: A sequence‐based method for predicting probability of protein‐protein interaction partners. PLoS One. 2012;7:e30938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buske C, Kirchhoff F, Munch J. EPI‐X4, a novel endogenous antagonist of CXCR4. Oncotarget. 2015;6:35137–35138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zirafi O, Kim KA, Standker L, et al. Discovery and characterization of an endogenous CXCR4 antagonist. Cell Rep. 2015;11:737–747. [DOI] [PubMed] [Google Scholar]
- 47. Dobson PD, Doig AJ. Distinguishing enzyme structures from non‐enzymes without alignments. J Mol Biol. 2003;330:771–783. [DOI] [PubMed] [Google Scholar]
- 48. Shervashidze N. Scalable graph kernels. Tübingen, Germany: Universität Tübingen, 2012. [Google Scholar]
- 49. Senelle M. Measures on graphs: From similarity to density. Louvain‐la‐Neuve, Belgium: Université catholique de Louvain, 2014. [Google Scholar]
- 50. Shervashidze N, Schweitzer P, van Leeuwen EJ, Mehlhorn K, Borgwardt KM. Weisfeiler‐Lehman Graph Kernels. J Mach Learn Res. 2011;12:2539–2561. [Google Scholar]
- 51. Neumann M, Garnett R, Bauckhage C, Kersting K. Propagation kernels: Efficient graph kernels from propagated information. Mach Learn. 2016;102:209–245. [Google Scholar]
- 52. Li G, Semerci M, Yener B, Zaki MJ. Effective graph classification based on topological and label attributes. Statist Analys Data Mining. 2012;5:265–283. [Google Scholar]
- 53. Bai L, Hancock ER. Depth‐based complexity traces of graphs. Pattern Recogn. 2014;47:1172–1186. [Google Scholar]
- 54. Orsini F, Frasconi P, Raedt LD. Graph invariant kernels IJCAI Proceedings‐International Joint Conference on Artificial Intelligence. USA: AAAI Press / IJCAI, 2015. [Google Scholar]
- 55. Kilhamn J. Fast shortest‐path kernel computations using approximate methods [master of science thesis]. University of Gothenburg; 2015.
- 56. Johansson FD, Frost O, Retzner C, Dubhashi D. Classifying large graphs with differential privacy In: Torra V, Narukawa T, editors. Modeling decisions for artificial intelligence. Switzerland: Springer International Publishing, 2015, 2015; p. 3–17. [Google Scholar]
- 57. Gupta R, Birch H, Rapacki K, Brunak S, Hansen JE. O‐GLYCBASE version 4.0: A revised database of O‐glycosylated proteins. Nucleic Acids Res. 1999;27:370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hamby SE, Hirst JD. Prediction of glycosylation sites using random forests. BMC Bioinform. 2008;9:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caragea C, Sinapov J, Silvescu A, Dobbs D, Honavar V. Glycosylation site prediction using ensembles of support vector machine classifiers. BMC Bioinform. 2007;8:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome‐wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 : Supporting Information