

Abstract
Disordered domains are long regions of intrinsic disorder that ideally have conserved sequences, conserved disorder, and conserved functions. These domains were first noticed in protein–protein interactions that are distinct from the interactions between two structured domains and the interactions between structured domains and linear motifs or molecular recognition features (MoRFs). So far, disordered domains have not been systematically characterized. Here, we present a bioinformatics investigation of the sequence–disorder–function relationships for a set of probable disordered domains (PDDs) identified from the Pfam database. All the Pfam seed proteins from those domains with at least one PDD sequence were collected. Most often, if a set contains one PDD sequence, then all members of the set are PDDs or nearly so. However, many seed sets have sequence collections that exhibit diverse proportions of predicted disorder and structure, thus giving the completely unexpected result that conserved sequences can vary substantially in predicted disorder and structure. In addition to the induction of structure by binding to protein partners, disordered domains are also induced to form structure by disulfide bond formation, by ion binding, and by complex formation with RNA or DNA. The two new findings, (a) that conserved sequences can vary substantially in their predicted disorder content and (b) that homologues from a single domain can evolve from structure to disorder (or vice versa), enrich our understanding of the sequence ➔ disorder ensemble ➔ function paradigm.
Keywords: disorder–function relationships, disorder‐to‐structure transitions, intrinsically disordered domains
Short abstract
Abbreviations
- CATH
class, architecture, topology, and homology
- CDF
cumulative distribution function
- CH
charge–hydropathy
- IDPs or IDRs
intrinsically disordered proteins or regions
- kip
kinase inhibitor protein
- NMA–NIA
normalized monomeric area and normalized interface area
- NMR
nuclear magnetic resonance
- PDB
Protein Data Bank
- PDD
probable disordered domain
- SCOP
Structural Classification of Proteins
- VSL2b
disorder predictor trained on V = variously characterized proteins with S = short and/or L = long IDRs, version 2b
1. INTRODUCTION
Molecular biologists and geneticists use domain for a contiguous set of amino acids having a particular function, for example, the autoinhibitory domain1, 2 and the transactivation domain.3, 4 Such domains are often located within intrinsically disordered protein regions and undergo disorder‐to‐structure transitions upon binding to their partners.4, 5 In contrast, structural biologists initially used domain to describe a protein unit that folds autonomously6 and has functional autonomy and/or evolutionary conservation.7, 8
Domain databases based on evolutionary conservation include Pfam,9, 10 SMART,11 and the Conserved Domain Database (CDD).12 Domain databases based on structural autonomy include the Structural Classification of Proteins (SCOP)13 and Class, Architecture, Topology, and Homology (CATH).14 Finally, a database based on both evolutionary conservation and structural autonomy is SUPERFAMILY.15
In contrast to structured proteins, intrinsically disorder proteins and regions (IDPs and IDRs) lack stable structures in solution, existing instead as highly dynamic ensembles with thousands of conformations.16, 17, 18, 19, 20 Many IDPs and IDRs undergo disorder‐to‐structure transitions upon binding partners.16, 17, 18, 19, 20, 21 These disorder‐to‐structure changes upon binding are often incomplete with flanking or looping IDRs22 that sometimes contribute positively or negatively to the binding constant. Such complexes are called fuzzy.23, 24 Some fuzzy complexes remain entirely disordered yet bind with high affinity.25 Structure‐to‐disorder transitions upon binding have also been observed for some proteins,26, 27, 28 with at least one protein showing simultaneous structural changes in both directions for different regions.26
IDPs and IDRs are abundant in all domains of life.29, 30, 31 IDPs, IDRs, and their various interactions with partners of all types32 are critically involved in many biological processes, such as molecular recognition, signaling, regulation, and cell cycle control20, 33, 34, 35, 36, 37 among many others.38, 39
In one study, the large majority of IDRs exhibited significantly less sequence conservation than the structured regions of the same proteins.40 Follow‐up studies have shown that conserved‐sequence IDRs are also common.41, 42 Conserved‐sequence IDRs were more recently called constrained disorder 42 and are correlated with tissue‐specific alternative splicing and cell regulation.43 One IDR is conserved in length and dynamic behavior but has negligible sequence conservation;44 such segments are called conserved (or flexible 42) disorder. Some IDR sequences are not conserved and change from being predicted‐to‐be disordered to being predicted‐to‐be structured; these regions are called non‐conserved.42 Finally, insertions and deletions (IDELs) are more often disordered than structured,45, 46, 47 and some IDR deletions are observed to occur in paralogues.48 In summary, from an evolutionary point of view, there are four types of IDRs: (a) conserved‐sequence (or constrained) disorder; (b) variable‐sequence (or flexible) disorder; (c) non‐conserved disorder, and (d) INDELs.
IDPs and IDRs carry out molecular recognition,49 and disorder predictors have been used to identify specific IDP and IDR loci that bind to globular protein partners.50 IDR‐located binding sites have been identified by disorder prediction,34, 51, 52 and, alternatively, by linear sequence motifs.53, 54, 55 Globular protein partner binding by IDPs and IDRs is common across the three domains of life.56 These prediction‐based studies have focused on short segments (5–15 residues in length) located within longer IDRs or even within IDPs.
Much longer IDRs (≥20–30 residues in length) bound to globular protein partners, and the previous work on conserved‐sequence disorder41 shows that such regions have conserved functions, conserved sequences, and conserved disorder, and therefore, by analogy to structured domains, such IDPs or IDRs were called disordered domains.57 An especially interesting disordered domain example is found in p27kip1, p21Waf1/Cip1/Sdi, and p57kip2.58, 59, 60 All three of these proteins have domains that are entirely disordered by both prediction and experiment and play similar key regulatory roles in controlling the cell cycle.
Like p27kip1 and its homologues, many Pfam domains contain sequences with 100% predicted intrinsic disorder.57, 61, 62 It is noteworthy that the Pfam database recently added “disordered” as a specific entry type,10 suggesting that the definition of protein domains should now also include those that are intrinsically disordered.
In the current work, we analyzed the sequences, the induced structures, and the functions for a set of Pfam disordered domains to explore their sequence–disorder–function relationships. The results presented herein highlight interesting distinctions between disordered and structured domains and improve our understanding of protein sequence–disorder–function relationships.
2. RESULTS
2.1. Analysis of probable disordered domains
The workflow for analysis of probable disordered domains (PDDs) is shown in Figure 1. As indicated in Section 4, 206 PDDs were obtained based on the set of PDD seed sequences from a previous study.61 First, as indicated in Figure 1 on the left, the homologues to the PDD sequences were collected, giving a total of 19,577 sequences. These homologues were collected from the seed proteins for each Pfam domain having at least one PDD sequence. These homologues in the seed alignment were used to correlate sequence conservation with predicted disorder and to investigate the sequence–disorder relationships. Not used here are the many additional homologous sequences matching the full alignment that could have been found by application of the hidden Markov model predictors developed for each Pfam domain; if such domains were included, the number of PDDs could be greatly increased. Next, as indicated in Figure 1 on the right, 2,548 different structures from 110 domains were collected for members having available structures in PDB. Their likely mechanisms of disorder‐to‐structure transition were examined to further investigate disorder–function relationships.
Figure 1.
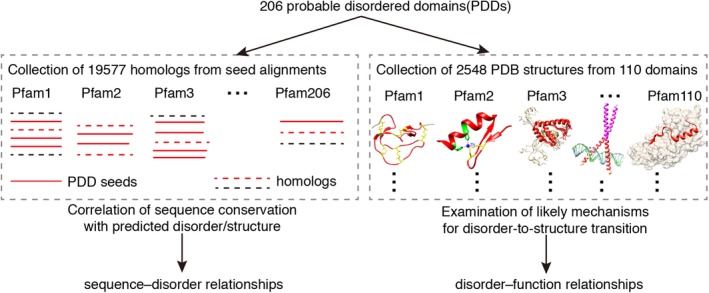
Workflow for analysis of probable disordered domains in Pfam database
2.2. Sequence–disorder relationships: Unexpected wide range of predicted disorder/structure among homologous sequences
Disorder prediction was applied to all the collected seed members for the PDDs. It was expected that the additional homologous sequences of the PDD seeds would also show highly predicted disorder. Such a result was indeed commonly observed (Figure 2a), with 78 (38%) of the Pfam domains having predicted disorder (defined by an average vsl2b score >0.6 for all of the associated seed proteins, and with an additional 69 (33%) of the Pfam, having predicted disorder PDD for more than half of the seed proteins and with substantial predicted disorder in most of the remaining seed proteins (see Section 4 and Figure S1 for more details). Thus, a total of 147 (71%) Pfam domains were found to be all or mostly predicted‐to‐be‐disordered as expected. Totally unexpected was that finding that four Pfam domains (2%) were found to contain equal numbers of predicted to be disordered and predicted to be structured members and that 55 Pfam domains (27%) were found to contain mostly predicted‐to‐be‐structured members with only a few predicted disorder members (Figure 2a).
Figure 2.
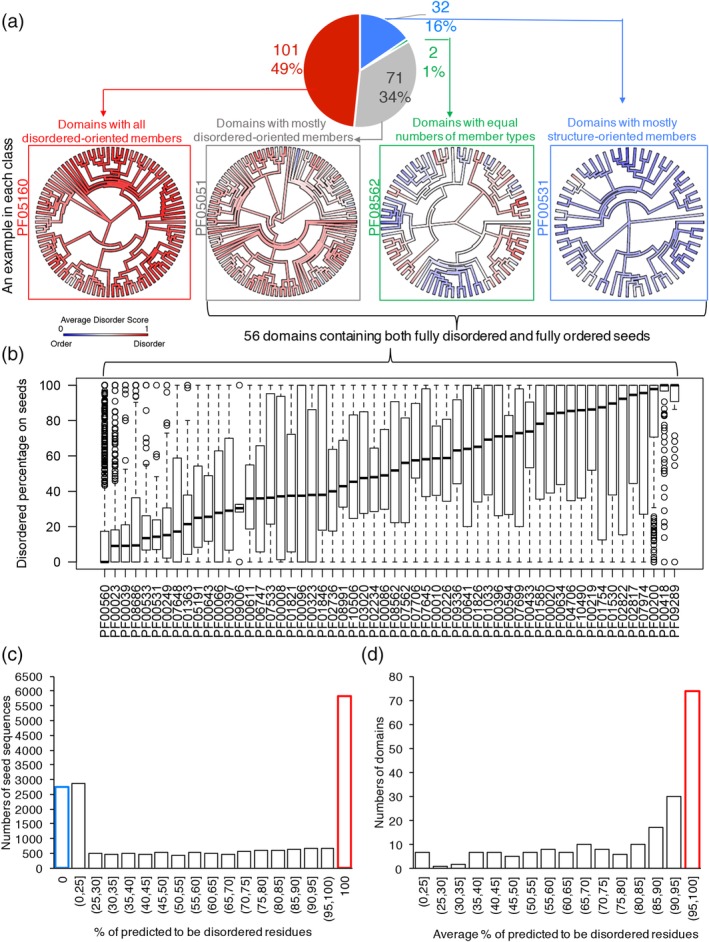
Significant diversity of predicted disorder among the homolog sequences within PDDs. (a) The pie chart showing the numbers and percentage of domains having different compositions of seed members. In each group, examples were presented as the Pfam phylogenetic trees mapped with average VSL2b disordered score into each sequence. (b) A set of 56 PDDs having both 100% predicted‐to‐be‐disordered and 100% predicted‐to‐be‐structured members. The distribution of predicted disorder percentage for each domain is shown as boxplots. (c) Distribution of predicted disorder percentage of all the 19,577 seeds. (d) Distribution of average predicted disorder percentage for 206 domains. PDD, probable disordered domain; VSL2B, disorder predictor trained on V = variously characterized proteins with S = short and/or L = long IDRs, version 2b
Thus, four groups having distinct seed member compositions were observed as shown in the pie chart of Figure 2a. In each group, one domain example is presented by mapping the average VSL2b score into Pfam phylogenetic trees, where red and blue indicate predicted‐to‐be‐disordered and predicted‐to‐be‐structured seeds, respectively. In the latter three groups, there were 56 domains that contained both fully predicted‐to‐be‐disordered (100% residues with VSL2b score >0.5) and fully predicted‐to‐be‐structured seeds (100% residues with VSL2b scores <0.5, Figure 2b).
Overall, the disorder prediction showed significant diversity, as shown in Figure 2c for all the seeds (predicted disorder percentage ranged from 0 to 100%) and in Figure 2d across the domains (average predicted disorder percentage ranged from 13 to 100%), respectively. Among the 19,577 seeds, only 30% of them (5,833) were PDD (highlighted in the red bar in Figure 2c), and 14% (2,755) of them were predicted to be fully structured (0% residues predicted to be disordered, highlighted in the blue bar in Figure 2c). Also, only 74 of the 206 domains (36%) were predicted to be >95% disordered (highlighted in red bar in Figure 2d). The wide range of predicted disorder or structure for the homologous sequences within a domain is completely unexpected. This observation was validated on a larger set of representative proteomes reduced to a sequence identity of 55%. The disorder contents in both sets showed close agreement across all domains studied (Figure S2), which suggests the seed sequence set does not contain a biased order–disorder content. These striking differences of predicted disorder or structure suggested that, for these domains, their sequences are likely more conserved than their structures. Thus, the widely held view that structure is more strongly conserved than sequence63, 64, 65, 66, 67 is clearly not followed by all IDP or IDR domains, which instead often show the unexpected contrary behavior that sequence is more strongly conserved than structure.
Further investigation of sequence conservation indeed shows that conserved positions have higher disorder prediction scores on average than non‐conserved positions in PDDs (Figure S3a). The structured domain set showed similar pattern (Figure S3b). Therefore, many PDDs contain conserved sequences and conserved disorder, which is similar to structured domains having conserved sequences and conserved structure.
The evolutionary origins of these structural differences were investigated by calculating the disorder content of the last common ancestor of each domain (Figure 3). In many cases, the estimated disorder content of the domain ancestor is consistent with overall disorder content of domain examples, which indicates that different structural differences may be an ancient feature of these domains. However, in a few cases, disorder is significantly different in the domain ancestor than in the domain examples, either higher in three cases (Figure 3: PF00560, PF01821, PF00096) or lower in one case (Figure 3: PF00641). This seems to indicate a structural shift from disorder to order, or order to disorder, in these domains across evolutionary history.
Figure 3.
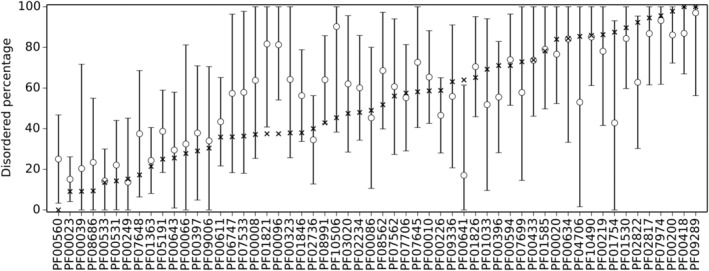
Estimated disorder content of the last common ancestor of each of the 56 PDDs having both 100% predicted‐to‐be‐disordered and 100% predicted‐to‐be‐structured members. The estimated percentage disordered residues of the ancestor (circles) and the 95% confidence interval of the estimate (error bars) are plotted along with the median percentage disordered residues (X) for comparison. PDD, probable disordered domain
2.3. Unusual disorder–function relationships: Disorder‐to‐structure transition depends upon various mechanisms
Given the wide range of disorder and structure within a domain, we first examined the distribution of disorder prediction for the identified PDB structures. This was done to exclude the possibility that domains could form structures simply because they are predicted‐to‐be structured members. Histograms in Figure 4a confirmed that majority of identified structures were predicted to be disordered on their sequences, with only a small set (109, 11.7%) predicted to be fully structured. An obvious question is why do predicted‐to‐be‐disordered proteins have structures in PDB?
Figure 4.
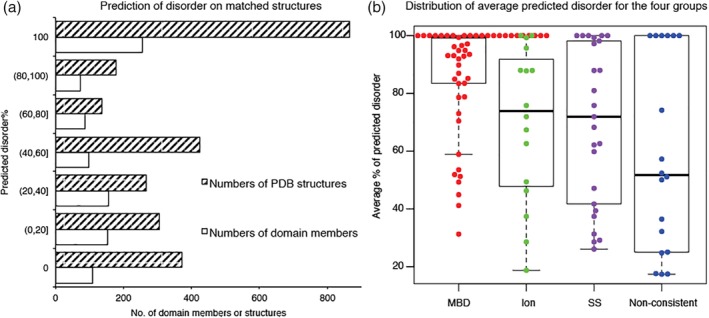
Identified structures were mostly predicted to be disordered on their sequences. (a) Prediction of disorder on matched structures. (b) Distribution of average predicted disorder for the four groups. MBD: macromolecular‐binding domains. Ion: ion‐binding. SS: disulfide bonds. Non‐consistent: domains without consistent mechanism of structural formation
Regarding the existence of structures for the predicted‐to‐be‐disordered proteins, three mechanisms were found that could potentially bring about disorder‐to‐structure transitions: (a) disulfide bond formation; (b) ion binding; and (c) macromolecular binding. In addition, there is a fourth group for which the stabilization mechanisms are uncertain or inconsistent. For each group, the numbers of domains and structures and the range of the average predicted disorder are summarized in Table 1. Percentage of disordered residues for each structure was averaged over all structures for each domain type. The distribution of average percentage of predicted disorder in the four different groups is shown in Figure 4b. The first three groups had significantly higher predicted disorder than the fourth, and many of domains in the first three groups have experimental evidence supporting disorder‐to‐structure transitions that depend on the indicated stabilizing factors. Particularly, the intrinsic disorder for the macromolecular‐binding domains was verified by searching for experimental evidence in literature. The stabilization factors for the fourth group are discussed in more detail in the Supporting Information (Table S2 and the text below the table). Here, we discuss the first three groups.
Table 1.
Likely mechanism for predicted‐to‐be‐disordered domains to form PDB structures
| Groups of stabilization factors | Numbers of domains | Numbers of PDB chains | Range of average predicted disorder% (median) |
|---|---|---|---|
| Disulfide bonds | 25 | 267 | 26–100% (72%) |
| Ion‐binding | 16 | 419 | 19–100% (74%) |
| Macromolecular‐binding | 60 | 1,084 | 12–100% (100%) |
| Uncertain stabilization mechanisms | 7 | 16 | 52–100% (100%) |
| 11 | 357 | 17–74% (34%) |
Abbreviation: PDB, Protein Data Bank.
2.3.1. Disorder‐to‐structure transitions upon disulfide bond formation or ion binding
Disulfide bonds are formed by oxidation of thiol groups between cysteines in a relatively oxidizing environment. Because the interior of cells is a reducing environment, disulfide bonds are usually not found in intracellular proteins but instead are found in extracellular, secreted, and periplasmic proteins,68 although disulfide bonds can also be formed in cytoplasmic proteins under certain conditions,69 such as oxidative stress.70 Among the 25 disulfide‐bond‐containing domains, most of them (22 domains) are secreted proteins (annotation from UniProt), two (PF06747, PF05051) are found in the intermembrane space of mitochondria, and the remaining one (PF09256) is an extracellular domain found in membrane receptors; therefore, the environment of this example is similar to that of a secreted protein. Some secreted proteins in gram negative bacteria have been shown to be unfolded inside the cell because structure‐inducing disulfide bonds remain unconnected in the reducing conditions and then adopt structures outside the periplasm as a result of disulfide bond formation in the oxidative environment.71 In eukaryotes, disulfide bond formation in secreted proteins involves the endoplasmic reticulum and is more complicated but still depends on movement from a reducing environment to a more oxidative environment.72 Furthermore, recent work in bacteria shows that their secreted proteins need to be at least partially unfolded inside the cell to interact with the secretion apparatus and become folded outside the cell, where the extra‐cellular folding is brought about not just by disulfide bond formation but by a variety of mechanisms.73 Thus, secreted proteins represent a new cohort that are transiently disordered and have been called “delayed folding proteins.”74 The observation that the delayed folding of many secreted proteins is regulated by disulfide bond formation confirms our previous conjecture62 that disordered domains that become folded by disulfide bond formation are likely secreted proteins that remain unstructured inside the cell and then gain structure outside the cell following secretion.69, 72, 73, 74
Proteins with ion binding functions are significantly enriched in IDPs.75 Various types of ions, such as Ca2+, Zn2+, Cd2+, Co2+, Mg2+, Cu2+, and Na+, have been identified in the current study. Among these, Zn2+ binding is the most commonly found type. This is consistent with our previous findings that zinc fingers are the second among the top 20 domains that are strongly correlated with predicted disorder,35 and zinc is the third among the top 20 ligand keywords that are strongly correlated with predicted disorder.38 The folding of a typical zinc finger depends on the binding of Zn2+—the conformation changes from an unfolded state into a highly folded structure in the presence of zinc.35, 76 In addition, Gla domains (PF00594) are mostly disordered in the absence of calcium ions and undergo a disorder‐to‐structure transition upon calcium binding, suggesting that calcium ions are required for proper Gla‐domain folding.77
2.3.2. Disorder‐to‐structure transition upon DNA/RNA/protein binding
A set of domains that function via induced folding upon DNA or RNA binding was collected. Evidence of disorder‐to‐structure transition upon DNA/RNA binding is known for all the examples in this study (summarized in Table S3). For instance, DNA‐induced folding of the basic regions of HLH domains (Figure 5) is well documented. HLH domains are developmental regulators of transcription and generally function as dimers, with each monomer containing a basic region (~18 residues) necessary for DNA binding, a helix–loop–helix motif (~34 residues), and a leucine‐zipper region (~30 residues) (Figure 5). The basic region was predicted to be disordered (Figure 5a), which is supported by NMR data demonstrating that this region of the Max protein is poorly folded in the absence of DNA78 (Figure 5b), but acquires an α‐helical conformation upon binding to DNA (Figure 5c). This activity has also been reported in MyoD79 and USF proteins,80 supporting our predictions of HLH disorder (Figure 5a) and suggesting that the disordered basic region is required for DNA recognition.
Figure 5.
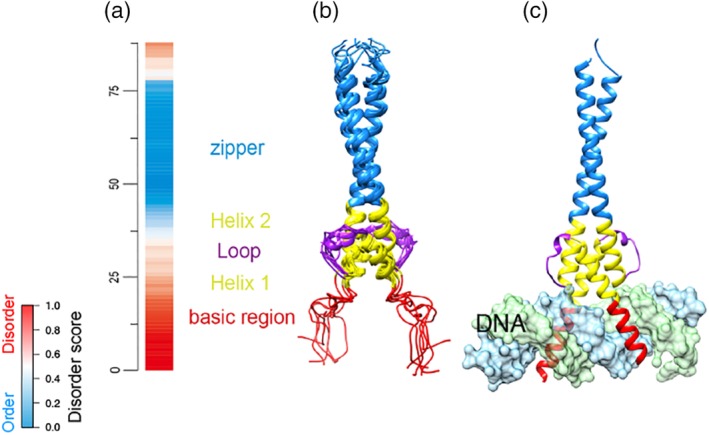
DNA‐induced folding of the disordered basic region from HLH domain of human Max protein. (a) VSL2b disorder prediction for the sequences. Red: disorder; blue: order; white: ambiguous. (b) NMR structure of the HLH domain in the absence of DNA (PDB id: 1R05). Basic region: 1–18 residues. Helix–loop–helix: 19–53 residues. Leucine zipper: 54–87 residues. (c) X‐ray structure of the HLH domain bound with DNA (PDB id: 1NKP). The two structures represent the same regions (23–102 residues) from human Max protein (UniProt ID: P61244). DNA chains are colored in light blue and green. NMR, nuclear magnetic resonance; PDB, PDB, Protein Data Bank; VSL2B, disorder predictor trained on V = variously characterized proteins with S = short and/or L = long IDRs, version 2b
Predicted disorder or structure was examined in more detail by sequence alignment and structural superposition for Methly‐CpG binding domain (MBD, PF01429). MBD includes members predicted to be structured and fully disordered (from 1 to 100% disorder). The multiple sequence alignment suggests 15 highly conserved residues, several of which are directly involved in methyl‐CgG binding based on available PDB structures (Figure 6). Obvious conformational changes were observed when comparing the least and most disordered members (Figure 6a,d).
Figure 6.
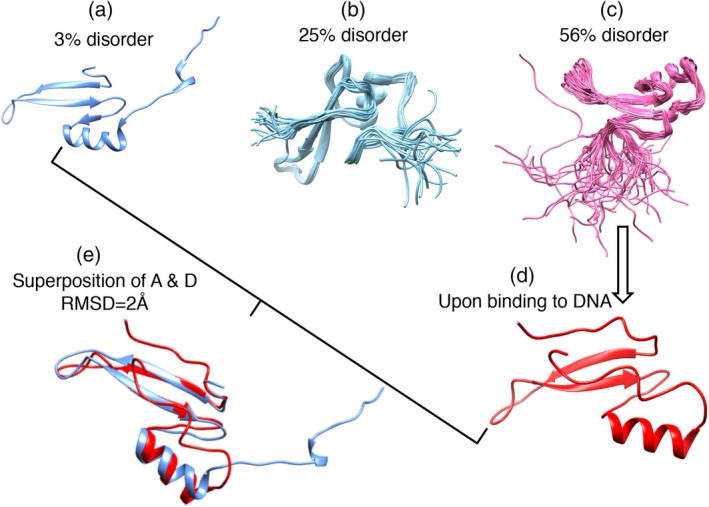
Diversity of structure/disorder in the MBD domain (PF01429). Top: multiple sequence alignment was carried out by T‐coffee and displayed by ESPript 3.0. Predicted disorder for the sequences increases from top (structure) to bottom (full disorder). The asterisk (*) at the bottom of the alignment indicates highly conserved DNA‐binding sites four available structures were shown below. (a) A structural member (PDB id: 3VXV, chain A). (b) A mediate disordered member (PDB id: 2MOE, chain A). (c) A higher disordered member (PDB id: 1QK9, chain A). (d) A fully disordered member in its DNA bound state (PDB id: 6CCG, chain A). (e) Superposition of the least and fully disordered member (sequence identity 42%) in their bound state. The binding partners are not shown here. MBD, methly‐CpG binding domain; PDB, PDB, Protein Data Bank
Protein‐binding PDDs contained two groups: disordered protein binding and structured protein binding. All of the PDDs found here were predicted to be disordered by structure‐based normalized monomeric area and normalized interface area (NMA–NIA) analysis (see Supporting Information for more detail), and most of them were also predicted to be highly disordered (>75% of the residues) by the sequence‐based VSL2b predictor, suggesting a strong consistency between these two methods (Figure S4a). Most PDDs were bound to structured partners. Selected examples were labeled by their PDB ids, and their structures in the binding complexes were shown in Figure S4b–g. All the disordered domain structures lack globularity, and experimental evidence of intrinsic disorder in the absence of their binding interactions was found for 56 examples (summarized in Table S3).
Overall, the apparent contradiction between the prediction of disorder and existence of PDB structures can be reconciled for the majority of the examples because of the various stabilization factors described above that bring about disorder‐to‐structure transitions. These transitions make disordered domains distinct from well‐structured domains with regard to the mechanisms underlying of protein–protein and protein–nucleic acid interactions.
3. DISCUSSION
3.1. A surprisingly wide range of predicted disorder was observed among the homologues within many individual domains
Prediction of disorder or structure is strikingly different for homologous sequences within many of the different Pfam domains. Sets of seed proteins varied from 13% (PF00023, Ank) to 100% disorder on average among the domains. Of these, only ~36% (75 of 206 domains) were predicted to be >95% disordered on average. When examining the disorder prediction of individual members for a given domain, the percentage range was even wider, ranging from fully ordered to fully disordered. We did not expect that proteins suggested to be homologues by hidden Markov models in Pfam would show such a wide range of predicted disorder. These results are certainly contrary to the general view that structure is more highly conserved than sequence.63, 64, 65, 66, 67
3.2. Disordered domains form structures as a result of various mechanisms
Various mechanisms cause disordered domains to form structures, including disulfide bonds, ion coordination, macromolecular partner binding, and many others. These major three factors are known to assist protein folding and were used to divide disordered domains in previous work.62 The examination of structural characteristics provides reasonable explanations of prediction of disorder and formation of structure for PDDs. This investigation suggests that future disorder predictors should consider these specific types of intrinsic disorder, which are likely to be distinct from IDPs or IDRs that do not undergo disorder‐to‐structure transitions.
3.3. Different disorder amount is likely related to crucial functional differences
Given the significantly different amount of predicted disorder among members in the same domain, it raises the question whether this correlates with their distinct functions. Experiment studies that correlate the disorder amount with functional differences remain largely un‐investigated, although distinct functions between different domain members are known for many examples. For instance, the Gγ domain (PF00631) has two members (Gγ1 and Gγ2) with significantly different amounts of predicted disorder (100 and 33%, respectively). Evidence shows that both Gγ1 and Gγ2 bind to the same partner (Gβ1), forming β1γ1 and β1γ2 dimers having significant differences in their functions.81, 82 An experimental investigation of whether there is truly a correlation between functional differences and intrinsic disorder in Gγ would be of great interest.
This work raises important questions about homologous domains that contain both predicted‐to‐be‐disordered and predicted‐to‐be‐structured members. For example, what are the underlying selective advantages that influence a predicted‐to‐be‐disordered domain to evolve into a predicted‐to‐be‐structured domain or vice versa? These conserved‐sequence‐associated changes in structure from disorder‐to‐structure or vice versa might be associated factors such as gain‐or‐loss‐of‐function arising from the altered structural tendency or such as increased resistance or increased sensitivity to regulatory protease digestion. Also, what are the detailed changes in sequence that enable a sequence to change from predicted‐to‐be‐structured into predicted‐to‐be‐disordered (or vice versa) while still being indicated to be conserved by hidden Markov models? Work on these questions is in progress.
In summary, here, we provide further evidence for the existence of intrinsically disordered domains. We show that many disordered Pfam domains have conserved sequences, conserved disorder, and conserved functions, which is analogous to the definition of structured domains, which have conserved sequences, conserved structures, and conserved functions. Our findings strongly support disordered domains as a new type of protein functional element that is distinct from classic structured domains. Our findings also suggest that disordered domains have the unexpected capacity to evolve from disorder to structure (or vice versa) over time.
4. MATERIALS AND METHODS
4.1. Datasets
4.1.1. Structured protein dataset
Of note, 664 structured proteins were derived from non‐redundant (sequence identity <25%) X‐ray crystallography structures from PDB. These are single‐chain monomers having no missing density, no disulfide bonds/ions/small molecules, and not including secreted proteins and coiled coils (because these proteins often predicted to be disordered).
4.1.2. IDP and IDR dataset
One hundred and thirteen IDPs and 692 IDRs were obtained from DisProt.39 Only regions longer than 20 consecutive residues were included for further analysis.
4.1.3. PDD dataset
The initial disordered dataset contained a set of domain seed members for which all of their residues were predicted by VSL2b to have scores >0.50.61 The term “domain” here refers to a Pfam‐A entry. Each Pfam entry includes a set of representative members of the family, hidden Markov models built from the seed alignment, and the automatically generated full alignment containing the proteins belonging to the entry. The term “seed” here refers to an individual protein sequence (in part or in whole) from the seed alignment. The term “member” here refers to an individual protein from the seed alignment, or full alignment, and the difference is spelled out.
The previously used requirement that all residues have VSL2b scores >0.5061 is too restrictive and misses many experimentally confirmed IDPs. Two methods have been used to estimate whole protein disorder, the charge–hydropathy plot,83, 84 and the cumulative distribution function.34, 85 However, neither of these methods has been extended for the problem of distinguishing structured protein regions from IDRs, and many disordered Pfam domains are IDRs rather than IDPs. Thus, we developed a new approach using the average value of per‐residue VSL2b predictions of disorder that we tested for both IDPs and IDRs. In plots of the histograms of the VSL2b average values for the 664 structured proteins, the 113 IDPs and the 692 IDRs in the dataset described above, values in the range of 0.4–0.6 gave good separations of the structured proteins from both the IDPs and IDRs as shown in Figure S1. Not one of the structured protein exhibited an average VSL2b score >0.60; therefore, we used this value here.
For the set of PDD sequences using VSL2b scores >0.60, 206 domain entries were identified and considered as PDDs. Each chosen domain had at least one seed that was identified as a PDD by this criterion.
4.2. Disorder prediction
Disorder prediction was used to examine whether or not additional homologues of the PDD sequences were also highly predicted to be disordered. All the seed sequences of the 206 domains were collected and predicted for disorder on the whole parent Uniprot sequences using the PONDR VSL2b predictor. Seed sequences were chosen because these are manually selected by Pfam for significant sequence identity with each other and more likely to contain additional disordered homolog within a domain having PDD seeds. VSL2b was used because it showed the best overall performance for long IDRs (≥30 consecutive residues) in a comprehensive comparison of 19 predictors.86 Because the length of disordered domains was proposed to be over 20–30 residues,57 it is expected that VSL2b would show good accuracy. We also chose this predictor for the reason of consistency (it was used in our previous work61 for the statistical analysis).
4.3. Classify PDDs by distinct seed compositions
To examine the seed member composition for each PDD, average disorder scores by VSL2b on seed sequences were used to partition the domains into groups. The sequences with average disorder score >0.6 were defined as predicted‐to‐be‐disordered seed members, and those with average disorder score ≤0.6 were defined as predicted to be ordered seed members. In each domain, the numbers of the two types of seeds were calculated, and four groups were classified: (a) domains containing all predicted‐to‐be‐disordered seeds; (b) domains containing mostly predicted‐to‐be‐disordered seeds; (c) domains containing equal numbers of seed types (number difference <2); (d) domains containing mostly predicted‐to‐be‐structured seeds.
Evolutionary relationships between ordered and disordered seeds were examined with domain sequence‐based phylogenies. Phylogenies were built from Pfam model domain alignments using FastTree87 and rooted by the midpoint method.88 The maximum likelihood disorder content and 95% confidence intervals of the last common ancestor of each domain family, according to phylogeny midpoint, was determined.89 Plots of domain families were branch length‐scaled by node depth for display, but ancestors were colored according to their estimated disorder content using the original branch lengths. Phylogenetic analysis and plotting was performed with the R package phytools.90
4.4. Examination of structure/disorder–function relationship
To examine the structure/disorder–function relationship for the PDDs, X‐ray and NMR structures in PDB were identified and possible mechanisms of disorder‐to‐structure transition were provided. Matched PDB structures for the domains were from members in Pfam full alignment. As the focus was on the PDDs, structures with multiple domains on the target chains were removed for further analysis. Short matched peptides (<20 residues) were also not included. Sequence‐based disordered predictions were applied to the whole parent UniProt sequences of the structural members, and the average percentage of predicted disorder was calculated for each matched domain region.
Next, likely mechanisms of structural formation for these predicted‐to‐be‐disordered domains were used to partition them into four groups. These are: (a) domains form structures because of disulfide bonds; (b) domains that form structures because of ion binding; (c) domains that form structures because of macromolecular binding; and (d) domains form structures with no consistent mechanism for structure formation. The method for partitioning into these groups is the same as that used in our previous work.62
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the contents of this article.
Supporting information
Appendix S1 Supplemental Methods and Materials
Figure S1 Using average VSL2b score to determine the threshold for IDPs, IDRs and structured proteins.
Figure S2 Consistence of disorder prediction between RP55 and Seed sets for the 206 domains.
Figure S3 Sequence Conservation in PDDs and structured domains
Figure S4 NMA–NIA boundary distances divided protein‐binding interactions into disordered or structured protein binding
Table S1 Percentage of IDPs, IDRs and structured proteins included within three different thresholds of average VSL2b score
Table S2 Likely stabilization factors for highly predicted‐to‐be‐disordered monomer structures
Table S3 Evidence of intrinsic disorder for macromolecular‐binding disordered domains
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31670851, 31470821, 91530320, and 31600671), as well as the National Key Research and Development Program of China (No. 2016YFC1306605). We thank the Celeste Brown (University of Idaho) for her critical review and insightful discussion regarding the aspects of this manuscript dealing with molecular evolution and phylogenetic analysis.
Zhou J, Oldfield CJ, Yan W, Shen B, Dunker AK. Intrinsically disordered domains: Sequence ➔ disorder ➔ function relationships. Protein Science. 2019;28:1652–1663. 10.1002/pro.3680
Funding information National Natural Science Foundation of China, Grant/Award Numbers: 31670851, 31470821, 91530320, and 31600671, 31600671, 91530320, 31470821, 31670851; National Key Research and Development Program of China, Grant/Award Number: 2016YFC1306605
Contributor Information
Bairong Shen, Email: bairong.shen@scu.edu.cn.
A. Keith Dunker, Email: kedunker@iu.edu.
REFERENCES
- 1. Smith MK, Colbran RJ, Soderling TR. Specificities of autoinhibitory domain peptides for four protein kinases. Implications for intact cell studies of protein kinase function. J Biol Chem. 1990;265:1837–1840. [PubMed] [Google Scholar]
- 2. Pufall MA, Graves BJ. Autoinhibitory domains: Modular effectors of cellular regulation. Annu Rev Cell Dev Biol. 2002;18:421–462. [DOI] [PubMed] [Google Scholar]
- 3. Lees JA, Fawell SE, Parker MG. Identification of constitutive and steroid‐dependent transactivation domains in the mouse oestrogen receptor. J Steroid Biochem. 1989;34:33–39. [DOI] [PubMed] [Google Scholar]
- 4. Kumar R, Litwack G. Structural and functional relationships of the steroid hormone receptors' N‐terminal transactivation domain. Steroids. 2009;74:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yeon JH, Heinkel F, Sung M, Na D, Gsponer J. Systems‐wide identification of cis‐regulatory elements in proteins. Cell Syst. 2016;2:89–100. [DOI] [PubMed] [Google Scholar]
- 6. Wetlaufer DB. Nucleation, rapid folding, and globular intrachain regions in proteins. Proc Natl Acad Sci U S A. 1973;70:697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Copley RR, Goodstadt L, Ponting C. Eukaryotic domain evolution inferred from genome comparisons. Curr Opin Genet Dev. 2003;13:623–628. [DOI] [PubMed] [Google Scholar]
- 8. Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA. Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol. 2004;14:208–216. [DOI] [PubMed] [Google Scholar]
- 9. Sonnhammer EL, Eddy SR, Durbin R. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. [DOI] [PubMed] [Google Scholar]
- 10. Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marchler‐Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: A database of conserved domain alignments with links to domain three‐dimensional structure. Nucleic Acids Res. 2002;30:281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. [DOI] [PubMed] [Google Scholar]
- 14. Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH—A hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. [DOI] [PubMed] [Google Scholar]
- 15. Gough J, Chothia C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002;30:268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re‐assessing the protein structure‐function paradigm. J Mol Biol. 1999;293:321–331. [DOI] [PubMed] [Google Scholar]
- 17. Dunker AK, Lawson JD, Brown CJ, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. [DOI] [PubMed] [Google Scholar]
- 18. Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. [DOI] [PubMed] [Google Scholar]
- 19. Uversky VN, Dunker AK. Understanding protein non‐folding. Biochim Biophys Acta. 2010;1804:1231–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oldfield CJ, Dunker AK. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem. 2014;83:553–584. [DOI] [PubMed] [Google Scholar]
- 21. Spolar RS, Record MT Jr. Coupling of local folding to site‐specific binding of proteins to DNA. Science. 1994;263:777–784. [DOI] [PubMed] [Google Scholar]
- 22. Mohan A, Oldfield CJ, Radivojac P, et al. Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006;362:1043–1059. [DOI] [PubMed] [Google Scholar]
- 23. Tompa P, Fuxreiter M. Fuzzy complexes: Polymorphism and structural disorder in protein‐protein interactions. Trends Biochem Sci. 2008;33:2–8. [DOI] [PubMed] [Google Scholar]
- 24. Fuxreiter M, Simon I, Bondos S. Dynamic protein‐DNA recognition: Beyond what can be seen. Trends Biochem Sci. 2011;36:415–423. [DOI] [PubMed] [Google Scholar]
- 25. Borgia A, Borgia MB, Bugge K, et al. Extreme disorder in an ultrahigh‐affinity protein complex. Nature. 2018;555:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muller CW, Schlauderer GJ, Reinstein J, Schulz GE. Adenylate kinase motions during catalysis: An energetic counterweight balancing substrate binding. Structure. 1996;4:147–156. [DOI] [PubMed] [Google Scholar]
- 27. Fong JH, Shoemaker BA, Garbuzynskiy SO, Lobanov MY, Galzitskaya OV, Panchenko AR. Intrinsic disorder in protein interactions: Insights from a comprehensive structural analysis. PLoS Comput Biol. 2009;5:e1000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitrea DM, Kriwacki RW. Cryptic disorder: An order‐disorder transformation regulates the function of nucleophosmin. Pac Symp Biocomput. 2012;152–163. [PubMed] [Google Scholar]
- 29. Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 30. Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. [DOI] [PubMed] [Google Scholar]
- 31. Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–149. [DOI] [PubMed] [Google Scholar]
- 32. Peng Z, Wang C, Uversky VN, Kurgan L. Prediction of disordered RNA, DNA, and protein binding regions using DisoRDPbind. Methods Mol Biol. 2017;1484:187–203. [DOI] [PubMed] [Google Scholar]
- 33. Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. [DOI] [PubMed] [Google Scholar]
- 34. Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. [DOI] [PubMed] [Google Scholar]
- 35. Vucetic S, Xie H, Iakoucheva LM, et al. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van der Lee R, Buljan M, Lang B, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xie H, Vucetic S, Iakoucheva LM, et al. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Piovesan D, Tabaro F, Micetic I, et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017;45:D1123–D1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown CJ, Takayama S, Campen AM, et al. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. [DOI] [PubMed] [Google Scholar]
- 41. Chen JW, Romero P, Uversky VN, Dunker AK. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J Proteome Res. 2006;5:879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bellay J, Han S, Michaut M, et al. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011;12:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Colak R, Kim T, Michaut M, et al. Distinct types of disorder in the human proteome: Functional implications for alternative splicing. PLoS Comput Biol. 2013;9:e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Daughdrill GW, Narayanaswami P, Gilmore SH, Belczyk A, Brown CJ. Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J Mol Evol. 2007;65:277–288. [DOI] [PubMed] [Google Scholar]
- 45. Fukuchi S, Homma K, Minezaki Y, Nishikawa K. Intrinsically disordered loops inserted into the structural domains of human proteins. J Mol Biol. 2006;355:845–857. [DOI] [PubMed] [Google Scholar]
- 46. Brown CJ, Johnson AK, Dunker AK, Daughdrill GW. Evolution and disorder. Curr Opin Struct Biol. 2011;21:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Light S, Sagit R, Ekman D, Elofsson A. Long indels are disordered: a study of disorder and indels in homologous eukaryotic proteins. Biochim Biophys Acta. 2013;1834:890–897. [DOI] [PubMed] [Google Scholar]
- 48. Yruela I, Contreras‐Moreira B, Dunker AK, Niklas KJ. Evolution of protein ductility in duplicated genes of plants. Front Plant Sci. 2018;9:1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–384. [DOI] [PubMed] [Google Scholar]
- 50. Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- 51. Dosztanyi Z, Meszaros B, Simon I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Disfani FM, Hsu WL, Mizianty MJ, et al. MoRFpred, a computational tool for sequence‐based prediction and characterization of short disorder‐to‐order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–i83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome‐wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Puntervoll P, Linding R, Gemund C, et al. ELM server: A new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31:3625–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Davey NE, Shields DC, Edwards RJ. SLiMDisc: Short, linear motif discovery, correcting for common evolutionary descent. Nucleic Acids Res. 2006;34:3546–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yan J, Dunker AK, Uversky VN, Kurgan L. Molecular recognition features (MoRFs) in three domains of life. Mol Biosyst. 2016;12:697–710. [DOI] [PubMed] [Google Scholar]
- 57. Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN. Close encounters of the third kind: disordered domains and the interactions of proteins. Bioessays. 2009;31:328–335. [DOI] [PubMed] [Google Scholar]
- 58. Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin‐dependent‐kinase inhibitor bound to the cyclin A‐Cdk2 complex. Nature. 1996;382:325–331. [DOI] [PubMed] [Google Scholar]
- 59. Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1‐phase progression. Genes Dev. 1999;13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 60. Galea CA, Nourse A, Wang Y, Sivakolundu SG, Heller WT, Kriwacki RW. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1. J Mol Biol. 2008;376:827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Williams RW, Xue B, Uversky VN, Dunker AK. Distribution and cluster analysis of predicted intrinsically disordered protein Pfam domains. Intrinsic Disord Prot. 2013;1:e25724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou J, Oldfield CJ, Huang F, Yan W, Shen B, Dunker AK. Intrinsic disorder in conserved Pfam domains In: Hamid R, Arabnia FGT, Tran Q‐N, Yang M, editors. International Conference on Bioinformatics & Computational Biology. Las Vegas: CSREA, 2018; p. 3–9. [Google Scholar]
- 63. Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. [DOI] [PubMed] [Google Scholar]
- 65. Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. [DOI] [PubMed] [Google Scholar]
- 66. Wood TC, Pearson WR. Evolution of protein sequences and structures. J Mol Biol. 1999;291:977–995. [DOI] [PubMed] [Google Scholar]
- 67. Illergard K, Ardell DH, Elofsson A. Structure is three to ten times more conserved than sequence—A study of structural response in protein cores. Proteins. 2009;77:499–508. [DOI] [PubMed] [Google Scholar]
- 68. Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847. [DOI] [PubMed] [Google Scholar]
- 69. Saaranen MJ, Ruddock LW. Disulfide bond formation in the cytoplasm. Antioxid Redox Signal. 2013;19:46–53. [DOI] [PubMed] [Google Scholar]
- 70. Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–21758. [DOI] [PubMed] [Google Scholar]
- 71. Denoncin K, Collet JF. Disulfide bond formation in the bacterial periplasm: Major achievements and challenges ahead. Antioxid Redox Signal. 2013;19:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hudson DA, Gannon SA, Thorpe C. Oxidative protein folding: From thiol‐disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic Biol Med. 2015;80:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tsirigotaki A, Chatzi KE, Koukaki M, et al. Long‐lived folding intermediates predominate the targeting‐competent secretome. Structure. 2018;26:695–707. [DOI] [PubMed] [Google Scholar]
- 74. Zhou J, Dunker AK. Regulating protein function by delayed folding. Structure. 2018;26:679–681. [DOI] [PubMed] [Google Scholar]
- 75. Lobley A, Swindells MB, Orengo CA, Jones DT. Inferring function using patterns of native disorder in proteins. PLoS Comput Biol. 2007;3:e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bombarda E, Grell E, Roques BP, Mely Y. Molecular mechanism of the Zn2+‐induced folding of the distal CCHC finger motif of the HIV‐1 nucleocapsid protein. Biophys J. 2007;93:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Freedman SJ, Blostein MD, Baleja JD, Jacobs M, Furie BC, Furie B. Identification of the phospholipid binding site in the vitamin K‐dependent blood coagulation protein factor IX. J Biol Chem. 1996;271:16227–16236. [DOI] [PubMed] [Google Scholar]
- 78. Sauve S, Tremblay L, Lavigne P. The NMR solution structure of a mutant of the Max b/HLH/LZ free of DNA: Insights into the specific and reversible DNA binding mechanism of dimeric transcription factors. J Mol Biol. 2004;342:813–832. [DOI] [PubMed] [Google Scholar]
- 79. Wendt H, Thomas RM, Ellenberger T. DNA‐mediated folding and assembly of MyoD‐E47 heterodimers. J Biol Chem. 1998;273:5735–5743. [DOI] [PubMed] [Google Scholar]
- 80. Ferre‐D'Amare AR, Pognonec P, Roeder RG, Burley SK. Structure and function of the b/HLH/Z domain of USF. EMBO J. 1994;13:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fawzi AB, Fay DS, Murphy EA, Tamir H, Erdos JJ, Northup JK. Rhodopsin and the retinal G‐protein distinguish among G‐protein beta gamma subunit forms. J Biol Chem. 1991;266:12194–12200. [PubMed] [Google Scholar]
- 82. Wildman DE, Tamir H, Leberer E, Northup JK, Dennis M. Prenyl modification of guanine nucleotide regulatory protein gamma 2 subunits is not required for interaction with the transducin alpha subunit or rhodopsin. Proc Natl Acad Sci U S A. 1993;90:794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Uversky VN, Gillespie JR, Fink AL. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. [DOI] [PubMed] [Google Scholar]
- 84. Huang F, Oldfield CJ, Xue B, et al. Improving protein order‐disorder classification using charge‐hydropathy plots. BMC Bioinform. 2014;15(Suppl 17):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xue B, Oldfield CJ, Dunker AK, Uversky VN. CDF it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009;583:1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z. Length‐dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006;7:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Price MN, Dehal PS, Arkin AP. FastTree 2—Approximately maximum‐likelihood trees for large alignments. PLoS One. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Farris JS. Estimating phylogenetic trees from distance matrices. Am Natural. 1972;106:645–668. [Google Scholar]
- 89. Felsenstein J. Phylogenies and the comparative method. Am Natural. 1985;125:1–15. [Google Scholar]
- 90. Revell LJ. Phytools: An R package for phylogenetic comparative biology. Methods Ecol Evol. 2012;3:217–223. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplemental Methods and Materials
Figure S1 Using average VSL2b score to determine the threshold for IDPs, IDRs and structured proteins.
Figure S2 Consistence of disorder prediction between RP55 and Seed sets for the 206 domains.
Figure S3 Sequence Conservation in PDDs and structured domains
Figure S4 NMA–NIA boundary distances divided protein‐binding interactions into disordered or structured protein binding
Table S1 Percentage of IDPs, IDRs and structured proteins included within three different thresholds of average VSL2b score
Table S2 Likely stabilization factors for highly predicted‐to‐be‐disordered monomer structures
Table S3 Evidence of intrinsic disorder for macromolecular‐binding disordered domains