

ABSTRACT
Free‐standing single‐layer β‐sheets are extremely rare in naturally occurring proteins, even though β‐sheet motifs are ubiquitous. Here we report the crystal structures of three homologous, single‐layer, anti‐parallel β‐sheet proteins, comprised of three or four twisted β‐hairpin repeats. The structures reveal that, in addition to the hydrogen bond network characteristic of β‐sheets, additional hydrophobic interactions mediated by small clusters of residues adjacent to the turns likely play a significant role in the structural stability and compensate for the lack of a compact hydrophobic core. These structures enabled identification of a family of secreted proteins that are broadly distributed in bacteria from the human gut microbiome and are putatively involved in the metabolism of complex carbohydrates. A conserved surface patch, rich in solvent‐exposed tyrosine residues, was identified on the concave surface of the β‐sheet. These new modular single‐layer β‐sheet proteins may serve as a new model system for studying folding and design of β‐rich proteins.
Keywords: human gut microbiome, protein folding, secreted proteins, single‐layer β‐sheet proteins, structural genomics, β‐hairpin repeats
1. INTRODUCTION
Many proteins consist of tandem repeats, in which the individual modules adopt a compact globular form known as a domain.1, 2 In structurally simpler cases, smaller repeats of simple topology are linked together to form a larger solenoid‐like protein––examples include the tetratricopeptide (αα), ankyrin (αα), leucine‐rich (αβ), WD40 (ββββ), and hexapeptide (β) motifs.3 These so‐called “repeat proteins” are largely held together by local contacts (resulting in a low overall contact order), rather than long‐range interactions that are common for globular proteins or domains.4, 5 In addition to their involvement in many biological processes, repeat proteins also represent attractive systems for studying protein folding and design due to their unique and relatively simple architecture.6, 7
One of the simplest, yet most uncommon, forms of β‐repeat proteins consists of multiple β‐hairpins assembled into a single‐layer antiparallel β‐sheet, linked together by inter‐strand hydrogen bonds.8 Unlike other folds, no conventional hydrophobic core can exist in single layer β‐sheet proteins. Isolated single‐layer β‐sheets can be found in very small proteins, such as the three‐stranded β‐sheet in the WW domain of Formin binding protein 28 (FBP28)9 and engineered β‐hairpins, such as tryptophan zippers (TrpZip)10 and chignolin.11 These small motifs have been valuable in studying the folding and design of β‐hairpins and larger β‐sheets.12, 13 Single‐layer β‐sheets have also been found as a part of larger proteins, such as the Borrelia burgdorferi outer surface protein A (OspA),14, 15 C‐terminal G‐box domain of the centriolar protein CPAP,16, 17 and ABC toxins.18 Although dominantly composed of antiparallel β‐strands, ABC toxins are also interspersed with occasional α‐helical motifs. In contrast, OspA consists of 21 consecutive antiparallel β‐strands and a terminal α‐helix, which together form a central single‐layer β‐sheet flanked on either end by N‐ and C‐terminal globular domains. The single‐layer β‐sheet in OspA has a well‐defined rigid structure15 and is remarkably thermodynamically stable,19 indicating that proteins lacking a hydrophobic core can still possess some inherent “globular‐like” characteristics. The G‐box domain, although also part of a larger protein, was found to form a single‐layer straight β‐sheet, which is stable by itself.16, 17, 20
Here, we report on crystal structures of three single‐layer β‐sheet proteins (SLBPs) from Bacteroides that are assembled from β‐hairpin repeats: the 9‐stranded BfSLBP (gene: BF3112, PDB ID 3msw) from Bacteroides fragilis NCTC 9343, the 7‐stranded BvSLBP (gene: BVU1206, PDB ID 4r8o) from Bacteroides vulgatus ATCC 8482, and the 7‐stranded PdSLBP from Parabacteroides distasonis ATCC 8503, (gene: BDI3222, PDB ID 4r03). These new structural representatives have allowed identification of a novel Bacteroides‐specific protein family of SLBPs that are implicated in utilization of complex polysaccharides. Moreover, this protein family indicates the emergence of a new class of bacterial proteins that may have evolved to adapt to mammalian habitats and diets.
2. RESULTS
2.1. Target selection and structural determination
The human body is colonized by ~1,000 species of microorganisms, whose total population outnumbers that of human cells by about 10‐fold.21 These microorganisms, known collectively as the human microbiome, have co‐evolved with us and, in some cases, are found only within the human body. The microbiome plays a key role in various aspects of human health, such as nutrition and immunity and, in some cases, are the causative agents of disease. Many new protein families, often specific to the human microbiome, have been identified.22 We have undertaken structural characterization of the secretome of Bacteroides species that prominently populate the human gut. Bacteroides genomes contain large stretches of genetic material that are upregulated in response to the extracellular level of particular polysaccharide moieties. Many of the ensuing secreted molecules are involved in the recruitment or processing of nutrients that are otherwise inaccessible to the human repertoire of digestive enzymes.
Genomic sequences of organisms populating the human gut microbiome were clustered and used as the basis for identifying new protein families specific to the human gut microbiome.22 For structural studies using the high‐throughput structural genomics pipeline implemented at the Joint Center for Structural Genomics (JCSG, http://www.jcsg.org),23, 24 we focused on predicted soluble and secreted family members from representative Bacteroides species whose sequenced genomes were available at the time of target selection (B. uniformis, B. fragilis, B. ovatus, B. vulgatus, B. eggerthii, B. caccae, B. thetaiotaomicron, P. merdae, and P. distasonis). The predicted secreted proteins were identified based on the presence of N‐terminal signal peptides, which were omitted from cloning constructs to facilitate purification and crystallization. The selenomethionine derivative of each target was expressed in Escherichia coli with an N‐terminal TEV‐protease cleavable His6‐tag and purified by metal affinity chromatography. Analytical size exclusion chromatography was used to ensure the purified protein was mono‐disperse before crystallization. Crystals were harvested and screened for diffraction, and the best diffracting crystals were chosen for data collection and structure determination. For this study, 18 homologs were selected for structure characterization. Overall, 17 targets were purified as soluble proteins, six of which produced harvestable crystals. Four crystal structures were determined, three of which were deposited in the PDB (Table S1); the fourth low‐resolution structure served as an MR model to determine the structure of BvSLBP (see below).
Crystal structures of BfSLBP (residues 26–169; PDB ID 3msw) and PdSLBP (residues 29–136; PDB ID 4r03) were determined by the multiple‐wavelength anomalous diffraction (MAD) phasing method. Data processing and refinement statistics are summarized in Tables S2‐S3. In both cases, the asymmetric unit (asu) contains one protein molecule. The structure of BfSLBP was refined to an Rcryst of 19.7% and an Rfree of 22.7% using diffraction data to 1.9 å resolution (Table S2). The model of BfSLBP displays good geometry with an all‐atom clash score of 3.1, and the Ramachandran plot (MolProbity v3)25 has all but one residue in allowed regions. The Ramachandran outlier, Glu102, is located in a loop region involved in a crystallographic contact, and its structure is supported by well‐defined main‐chain electron density (Figure S1). The final model of BfSLBP contains residues 26–163 and Gly0 (the residue that remains after cleavage of the purification tag) and solvent molecules (3 chloride ions, 3 1,2‐propanediol, and 125 waters). The structure of PdSLBP was refined to an Rcryst of 16.6% and an Rfree of 18.8% using diffraction data to 1.5 å resolution, with excellent model geometry (Table S2). The final model of PdSLBP contains all 29–136 residues, as well as Gly0 and solvent molecules.
Structure determination of BvSLBP (residues 26–128; PDB ID 4r8o) by single‐wavelength anomalous diffraction (SAD) or molecular replacement (MR) was not initially successful. Subsequently, a model for the B. uniformis homolog (57% sequence identity) was obtained from a lower resolution 3.3 å MAD dataset and used to determine the BvSLBP structure by MR in space group P3121. The BvSLBP crystal was found to be perfectly twinned, giving rise to an appearance of higher symmetry (P6222 or P6422) for the merged data. The structure of BvSLBP was refined to an Rcryst of 17.7% and an Rfree of 24.4% to 2.5 å resolution (Table S4). The asu contains two protein molecules, consisting of two very similar monomers (Cα RMSD of 0.5 å for residues 30–127), and solvent molecules.
2.2. Overall structures and structural comparison
BfSLBP is composed of nine β‐strands arranged in a single‐layer, antiparallel β‐sheet (β1–β9, Figure 1a), while BvSLBP (Figure 1b) and PdSLBP (Figure 1c) contain seven strands (β1–β7) arranged in a similar manner. The C‐terminus of PdSLBP contains a short helix (two turns) that extends to interact with its inner surface. The β‐strands, each typically ~10 aa in length, form a series of β‐hairpins that are connected by short loops of ~2–7 aa. These hairpin repeats (linked to form a β‐meander motif) are frequently observed in proteins with β‐barrel or β‐propeller folds. Local disruption of hydrogen bonding in β‐sheets to accommodate inserted residues and turns, known as β‐bulges, are found throughout the structure and not only in the edge strands26 (see details for BfSLBP in Figure S2). All three SLBPs have a right‐handed twist, thereby adopting a concave palm shape that forms a potential binding site along the inner concave surface. The presence of classic β‐bulges can attenuate the twist in the β‐sheet,27, 28 and the presence of so many classic β‐bulges even in internal strands may add curvature to the β‐sheet and thus fine‐tune the shape of this inner surface. Since both sides of the β‐sheet are exposed to solvent, these SLBPs do not possess a hydrophobic core, as commonly observed in globular proteins. SLBPs can be categorized as a member of the “curved single‐layer subfold” according to the classification defined by Roche et al.8
Figure 1.
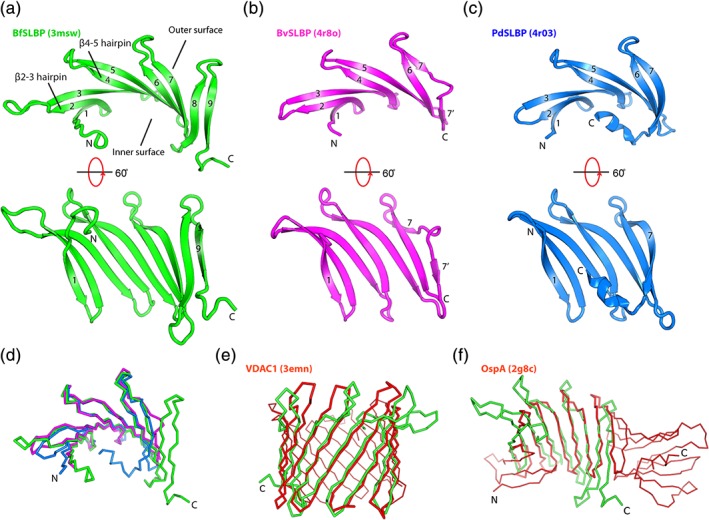
Crystal structures of SLBPs. (a) BfSLBP (green), (b) BvSLBP (magenta), and (c) PdSLBP (blue). The strands are labeled sequentially from 1 to 9. β‐hairpins (βx‐y, where x and y are two adjacent β‐strands forming a hairpin, e.g., β2‐3 hairpin) are referred to as up hairpins (i.e., the loop is on the top, as depicted), or down hairpins (i.e., the loop is at the bottom, as depicted). (d) Superimposition of BfSLBP (green), BvSLBP (magenta), and PdSLBP (blue). (e) Structural comparison between BfSLBP and VDAC1 porin (PDB ID 3emn), and (f) between BfSLBP and OspA (PDB ID 2g8c). BfSLBP is colored green, and the other molecule is red.
The three SLBP structures are very similar (Figure 1d), despite less than 25% pairwise sequence identities. The first seven strands (present in all structures) can be superimposed with an average RMSD of 2.2 å for 95 equivalent Cα atoms. Structural similarity searches using DALI29 indicate that SLBPs display the most significant structural similarity to parts of membrane‐embedded porins. For example, BfSLBP aligns to part (residues 52–175) of the mouse β‐barrel voltage‐dependent anion channel (VDAC; PDB ID 3emn)30 with an RMSD of 3.9 å for 125 Cα atoms (sequence identity of ~6%). Overall, both the bend and twist observed in the BfSLBP β‐sheet are comparable to that of VDAC (Figure 1e). BfSLBP can also be superimposed onto the central β‐sheet of OspA (PDB ID 2g8c, β4–β12, residues 60–164)31 with an RMSD of 3.2 å for 91 Cα atoms (sequence identity ~12%, Figure 1f). SLBPs can only be matched to other β‐sheet proteins (e.g., OspA or lectins) using distant homology recognition algorithms, suggesting that evolutionary relationships, if any, are very distant.
2.3. Conserved β‐hairpin repeats
Repeats within the SLBP amino acid sequences can only be identified with very sensitive sequence comparison algorithms. We therefore analyzed internal repeats in the SLBPs by structural comparisons of the “overlapping” hairpins that comprise the β‐sheet (Figure 1). These hairpins can be divided into two groups, “up” hairpins (β2‐3, β4‐5, β6‐7, and β8‐9) and “down” hairpins (β1‐2, β3‐4, β5‐6, and β7‐8). The up hairpins are structurally highly similar to each other (average pairwise RMSD of only 1.5 å for 22 Cα atoms, Figure 2a,b) and contain conserved sequence motifs (Figure 2c). Most notably, a sequence motif [WM]Nx5‐6[WLF] anchors all up hairpin turns, such that one hydrophobic residue (e.g., Met51 of β2 of BfSLBP) at the end of the first strand makes cross‐strand contacts with the second hydrophobic residue at the beginning of the second strand (e.g., Leu59 of β3 of BfSLBP). The conserved asparagine is involved in hydrogen bonding with the main‐chain amide of the i + 2 residue (Figure S2d). The apparent necessity of aligning these cross‐strand hydrophobic residues imposes a restriction on the geometry of the second β‐strand of each hairpin––as such, the insertion of an additional amino acid in this strand is accommodated through a conserved β‐bulge (the inserted residue is a lysine in all hairpins, except for a serine in β4‐5 for BfSLBP, Figure 2b). In contrast, the down hairpins typically feature shorter connecting loops that are not conserved in sequence and are more varied in structure. The interfaces between β‐strands of the SLBPs are also appreciably larger than the interfaces between the down hairpins (Figure 3a); the up hairpins also contain more hydrogen bonds (Figure 3a). Together, these results suggest that up hairpins comprise the underlying architecture that, by duplication and mutation, gave rise to the overall SLBP architecture.
Figure 2.
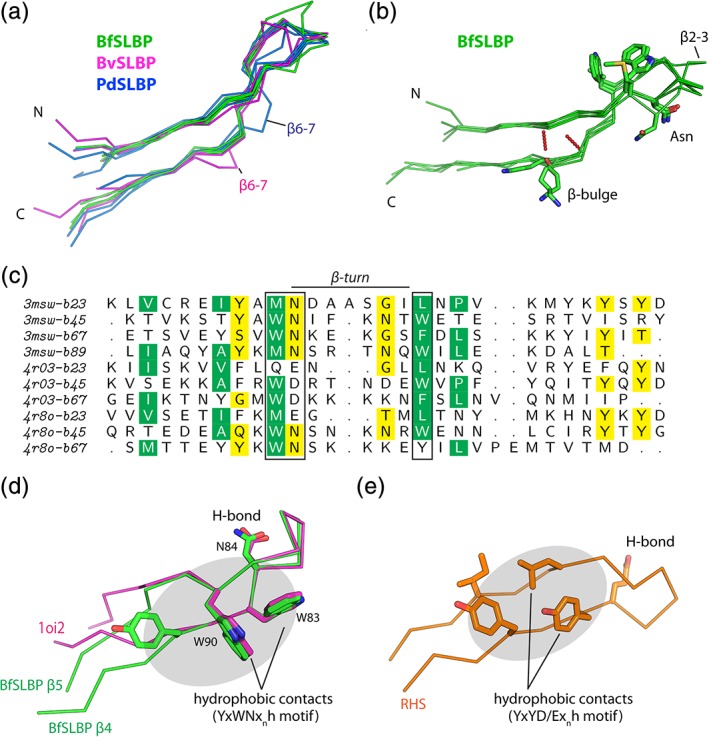
β‐hairpin repeats of SLBPs. (a) Structural comparisons of up hairpins of BfSLBP (green), BvSLBP (magenta), and PdSLBP (blue). (b) Structural superposition of up hairpins in BfSLBP with the highly conserved residues near the turn highlighted as sticks. (c) Structure‐based sequence alignment of up hairpins. Residues shown as sticks around the β‐turn region in (b) are highlighted in boxes (green: hydrophobic residues, yellow: polar residues). (d) Structural comparisons of the β4‐5 hairpin of BfSLBP (green, Figure S2) with a hairpin of E. coli dihydroxyacetone kinase (PDB ID 1oi2, magenta). (e) Representative RHS repeat (residues 297–319) from ABC toxin (PDB ID 4igl, orange), demonstrating the structural similarity of the underlying hairpin‐repeat unit
Figure 3.
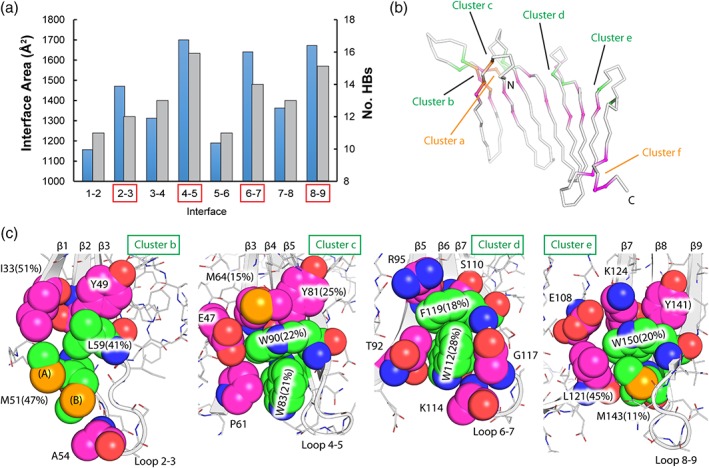
Interactions between β‐hairpins of BfSLBP. (a) Total buried surface area (blue bars, å2) and number of hydrogen bonds (gray bars) within each hairpin interface. Interfaces within up hairpins are denoted by red boxes. (b) Small hydrophobic clusters (cluster b–e in Panel c) present between the up hairpins. The conserved hydrophobic pairs within up hairpins are highlighted in green. Note that these clusters are located on the opposite face of the β‐sheet, as shown in (b). The two conformers of Met51 are labeled as (A) and (B), respectively
Within the up hairpins, the edge‐to‐face (“T‐shaped”) packing of the hydrophobic side‐chain pairs, such as Trp83‐Trp90 or Trp112‐Phe119 hydrophobic pairs in BfSLBP (Figure 2d), is highly similar to that of the TrpZip β‐hairpins (PDB 1le0)10 in which Trp‐Trp interactions are essential for stabilization.32 We identified more than 50 similar T‐shaped Trp‐Trp interacting pairs in other β‐hairpins among ~10,700 nonredundant PDB chains (i.e., chains with a sequence identity <30%). For example, E. coli dihydroxyacetone kinase (PDB 1oi2)33 contains a nearly identical WDxnW hairpin loop to that observed in BfSLBP (Figure 2d). In most cases, bending or termination of these β‐hairpins (near the W + 4 position) is observed, likely due to Trp‐Trp interactions that have a tendency to produce an appreciable twist that may disrupt regular hydrogen bonding.10 Interestingly, the RHS repeats of ABC toxin18 also contain hairpins featuring a YxYD/Exnh motif (where h is a hydrophobic residue), where the second tyrosine and downstream hydrophobic residue make cross‐strand contacts (Figure 2e). Similar cross‐strand hydrophobic interactions (Phe‐Leu ladder) are also observed in the stabilization of OspA.34
2.4. Inter‐β‐hairpin interactions and small hydrophobic clusters
β‐sheets contain a central, tightly packed layer of main‐chain atoms, which are flanked on either face by side chains. In SLBPs, the hydrophobic interactions between stretches of these side chains, or between the side chains and the underlying main‐chain atoms, may have a function analogous to the hydrophobic core of globular proteins.34 The relative solvent exposure of each residue (Figure S3), calculated by GETAREA35 as a ratio of the side‐chain surface area between folded and unfolded (“random coil”) states, indicates that many of the side chains of these hydrophobic residues are buried and well‐protected from solvent. Most of these residues are located toward the ends of β‐strands, forming small inter‐strand hydrophobic clusters as illustrated in Figure 3c, using the structure of BfSLBP as an example. These clusters are often protected at the perimeter by residues like Lys, Arg, or Glu, which bury their aliphatic portions while extending their polar terminal regions into the solvent. Clusters b–e (Figure 3b) are each centered on a cross‐strand hydrophobic pair as discussed above (e.g., Trp83‐Trp90, Trp112‐Phe119, etc.), allowing each up hairpin to interact with the C‐terminal strand of a preceding up hairpin (Figure 3c). Two additional hydrophobic clusters are located near the N‐terminus (Lys30, Phe32, Ile48, and Val62––Cluster a in Figure 3(b,c) as well as the C‐terminus (Leu157, Tyr161, and Ile136––Cluster f in Figure 3b,c), that cement these strands to the central region of the sheet. Overall, these small hydrophobic clusters are strategically located at structurally weak spots (connected to highly mobile regions, see below) and hold these substructures (“micro‐domains”) together. Thus, hydrophobic interactions appear to play a significant role in the stabilization of SLBPs, despite the lack of a unified hydrophobic core.
2.5. Dynamics of SLBPs
We performed computational analysis of BfSLBP to evaluate the thermodynamic properties of the broader SLBPs family. The B‐value distribution across BfSLBP indicates that central β‐strands are relatively rigid and the thermal motions are mostly restricted to the N‐ and C‐termini, as well as the loops connecting conserved β‐hairpins, such as L2‐3, L6‐7, and L8‐9 (Figure 4a left, Figure S3). The regions with highest experimental thermal fluctuations are consistent with predictions from normal mode analysis and molecular dynamics (MD) simulations (Figure 4a right, Figure S3). The MD simulations suggest the fluctuations are slightly greater in solution for the loops and the terminal regions. Overall, the X‐ray and MD results indicate that BfSLBP is a relatively rigid molecule.
Figure 4.
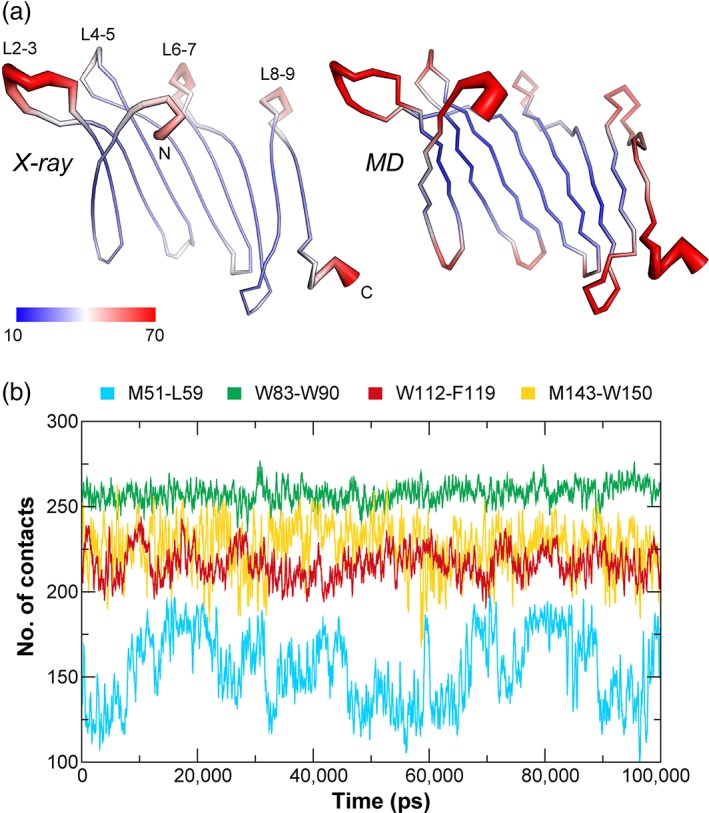
Dynamics of BfSLBP. (a) Mapping of B‐values (left) and computed B‐values derived from MD simulations (right) onto the BfSLBP structure. The molecule is colored in a gradient from blue (low B‐value, thin line) to red (highest B‐values, thick line). (b) Number of contacts between the hydrophobic pair of each up hairpin during the course of a 100 ns MD simulation (with a running average over 200 ps). A “contact” is counted if the distance between two atoms, one from each group, is less than 6 å
BfSLBP is stable during MD simulations, with the overall conformation and total number of hydrogen bonds remaining consistent over time. However, variations in the number of contacts within each hairpin were observed. In particular, the aromatic hydrophobic side‐chain pairs within the up hairpins (Trp83‐Trp90 or Trp112‐Phe119) show greater stability in their orientation and hydrophobic contacts during MD simulations, as compared to the hydrophobic pairs lacking mutual aromatic contributions (Met143‐Trp150 or Met51‐Leu59) (Figure 4b). As expected, methionines in the latter interacting pairs have significant side‐chain mobility and are the primary source of the fluctuations in the contacts. This observation is supported by crystallographic data indicating Met51 has a dual side‐chain conformation (labeled as (A) and (B) in Figure 3c). Overall, the Trp‐Trp interactions maintain the highest numbers of persistent contacts, consistent with the suggestion that Trp‐Trp interaction pairs confer the greatest degree of stabilization to β‐hairpins.32, 36
2.6. A Bacteroides‐specific SLBP family
The analysis above indicates SLBPs are novel proteins in both structure and sequence. The structures reported here were thus used to define a new PFAM protein family PF12930 (DUF3836), which currently contains more than 600 members. These proteins originate from bacteria in the Bacteroidales order, mostly from the Bacteroides genus, except for a few species from the Prevotellaceae or Porphyromonadaceae families. Typically, one to three paralogs are identified in each species. However, as many as seven to nine paralogs can be found in some species, such as in B. cellulosilyticus DSM 14838, a main gut bacterium that degrades cellulose,37 which contains eight paralogs. In addition, over 7,000 homologs have been found in human gut metagenomic datasets, highlighting the enrichment of this family in the human gut microbiome.
Gene expression studies of B. thetaiotaomicron introduced into germ‐free mice that were fed either polysaccharide‐rich or simple‐sugar diets38 demonstrated that the two SLBPs (BT2500 and BT3259) are strongly upregulated in the presence of complex polysaccharides. In another study that compared gene expression levels between young suckling mice (with simple‐sugar diets) and older mice in the weaning period (with complex‐carbohydrate diets), both SLBPs in B. thetaiotaomicron were significantly upregulated (BT2500, 18.4 times; BT3259, 3.3 times).39 During the weaning period, B. thetaiotaomicron adapts to the changing nutrient environment by expressing gene clusters that encode environmental sensors, outer membrane proteins involved in binding and import of glycans, and glycoside hydrolases, that exploit the abundant, plant‐derived polysaccharides in the diet.39 Thus, these microarray studies on B. thetaiotaomicron suggest that SLBPs may play a role in polysaccharide catabolism or sensing.
A multiple sequence alignment of representative SLBP homologs is shown in Figure 5. Structure‐based sequence analysis reveals common characteristics of the SLBP family. (a) all contain a predicted signal peptide within the first 30 residues, suggesting that these proteins function either in the periplasm or outside the cell. (b) The [WM]Nx5‐6[WLF] motif defining the turn regions of the up hairpins is highly conserved among homologs, offering further evidence for the importance of this region. (c) Some homologs are truncated by one or two β‐strands at the C‐terminus and would be expected to form seven‐ or eight‐stranded β‐sheets, as confirmed by the structures of PdSLBP and BvSLBP. (d) Several (Yx)n sequence motifs are present within the β‐strands, including BfSLBP β3 (65YkYsYd70), β7 (125YiYi128), and β8 (139YaYk142) on the inner surface. These tyrosine motifs form small stretches of aromatic side chains that appear to protect and stabilize the free‐standing β‐sheet surface (Figure 6). Importantly, these aromatic‐rich motifs may represent a putative interaction surface on SLBPs (see below).
Figure 5.

Alignment of eight representative SLBPs and molecular phylogenetic analysis using the ML method based on the JTT matrix‐based model (all positions containing gaps or missing data in the alignment were eliminated from the analysis), implemented in MEGA6. The secondary structure and sequence numbering of BfSLBP is shown on top, and sequence motifs are marked below (shown in red). Highly conserved residues are colored blue, while signal peptides and conserved cross‐strand aromatic motifs are highlighted in boxes. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa cluster together is shown next to the branches.
Figure 6.
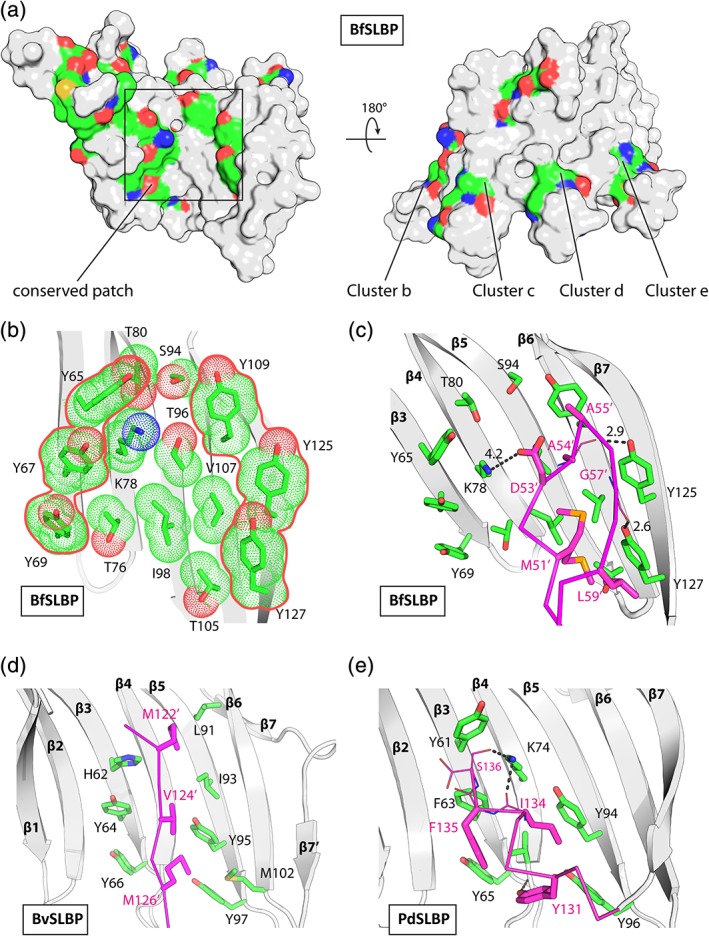
Sequence conservation and conserved surface patches in SLBPs. (a) Mapping of conserved residues onto the BfSLBP surface. The conserved residues are colored by atom types (C: yellow, N: blue, and O: red). (b) A hydrophobic/aromatic site located on the inner surface of BfSLBP. (c) Crystal packing interactions involving the these sites in BfSLBP, (d) BvSLBP, and (e) PdSLBP.
2.7. A conserved surface patch
The highly conserved residues of the SLBP family map mostly near the ends of the β‐strands or inner β‐sheet surface on the BfSLBP structure (Figures 5 and 6a). While the turn residues likely play critical roles in structural stability, those along the solvent exposed β‐sheet surface appears to define a possible binding site delineated at the perimeter by several highly conserved tyrosines (e.g., Tyr65, Tyr67 and Tyr69, Tyr109, and Tyr127 in BfSLBP, Figure 6b). These tyrosines share similar side‐chain conformations and all generally point in the same direction (toward the turns of the up hairpins). The center of the surface contains β‐branched amino acids (valine/threonine, isoleucine) that are common in β‐strands, except for Lys78.
The above identified surface patches are involved the inter‐molecular packing interactions in the crystal structures of all three SLBPs (Figure S4 and Figure 6c–e). In BfSLBP, this tyrosine‐rich region interacts with the L2‐3 loop from a symmetry‐related molecule, burying three hydrophobic residues (Met51′, Ala54′, Gly57′, and Leu59′), while the side chains of Lys78, Tyr125, and Tyr127 hydrogen bond with the carbonyl oxygens of Asp53′, Ala54′, and Gly57′ of the symmetry‐related molecule (Figure 6c). The equivalent BvSLBP region interacts with a β‐strand from the adjacent monomer, burying Met122′, Val124′, and Met126′ (Figure 6d). Moreover, the PdSLBP site interacts with its own C‐terminal helix (burying Ile134 and Phe135), with additional hydrogen bonds involving Lys74 (Figure 6e). These interactions, although likely not physiologically relevant, suggest that these regions could potentially recognize a ligand or a partner protein through a combination of aromatic/hydrophobic interactions, with additional stabilization from hydrogen bonds.
3. DISCUSSION
We have determined crystal structures of a family of unique single layer β‐sheet proteins (Figure 1). These SLBPs contain three or four similar β‐hairpin repeats, suggesting that they evolved from gene duplication of an ancestral β‐hairpin (Figure 2). Further gene duplication and horizontal gene transfer may have facilitated the spread of orthologs and homologs throughout the Bacteroides genus. Protein evolution through sequence divergence could have allowed these organisms to take advantage of their niche environment in the gut, and potentially facilitate the uptake of carbohydrates, as implicated in microarray studies.38, 39
The exact function(s) of SLBPs is currently unknown. Nonetheless, we tentatively identified a conserved surface patch, defined by the rigid, concave surface of the central strands (β3–β7, Figure 6). This surface can be generated with either three or four hairpin repeats, corresponding to the sequence length differences found in SLBP homologs. Overall, the inner surface residues delineated by tyrosine ladders contribute to the formation of a relatively flat hydrophobic binding site. We speculate that this surface may be involved in binding potential substrates (e.g., protein or carbohydrates). Profile‐based sequence comparison using HHpred40 suggests that SLBPs share some sequence similarity (probability ~91%) with that of a hypothetical protein BACUNI_01323 from Bacteroides uniformis ATCC 8492, whose structure we also determined (PDB 4ghb, Figure S5). BACUNI_01323, a member of Pfam DUF4595 (may also be related to UPF0257), has a novel “gated” porin‐like β‐barrel fold that consists of 16 β‐stands and a “plug” domain between strands β12 and β13. It is interesting that BACUNI_01323 also carries a tyrosine‐ladder motif on its concave surface, which is involved in interaction with the helical “plug” domain (Figure S5), reminiscent of the crystal packing interactions observed in SLBPs (Figure 6). Additionally, despite lack of overall structural similarity, the putative binding sites in SLBPs bear some resemblance to the substrate‐binding sites of carbohydrate‐binding modules (CBMs), where aromatic residues (Trp, Tyr, and less often Phe) facilitate hydrophobic stacking and/or hydrogen‐bonding interactions between protein and carbohydrate (Figure S6).41, 42
Analytical size exclusion analysis demonstrated that BfSLBP is a monomer in solution (data not shown), which agrees with the prediction based on crystal packing using the PISA server.43 β‐sheets have a tendency to self‐aggregate or to interact with other proteins using exposed edge strands. It has been suggested that natural β‐sheet proteins prevent unwanted edge‐to‐edge aggregation via negative design, such that the edge strands are structurally altered (e.g., covered by loops, or are irregular by insertions such as β‐bulges) to prevent further edge extension.44 Indeed, the C‐terminal strands of all three SLBPs contained structural irregularities (β‐bulges) that disrupt regular placement of main‐chain hydrogen donor/acceptors (Figure S7). The C‐terminal edge strands of PdSLBP and BvSLBP contain additional inserts (Pro‐Glu or Val) (Figure 2c) and, as a result, are less conserved across the SLBP family (Figure 2a). Further prepending of additional β‐hairpin repeats in SLBPs is prevented by the leading strand. Interestingly, this N‐terminal strand assumes a typical β‐strand geometry (i.e., with regularized, exposed carbonyls) that can be utilized for intermolecular interaction via edge strand extension. For example, the two monomers in the PdSLBP asu interact via their respective β1‐strands to form one extended anti‐parallel β‐sheet (Figure S4).
β‐sheets are a common secondary structural motif in proteins. Thus, elucidating the folding pathways of β‐sheets is critical for understanding the complexities of protein folding.45 β‐hairpins are believed to universally function as critical nucleation sites in the early stages of β‐sheet folding. Moreover, studies of short β‐hairpin‐forming peptides highlight the critical importance of the turn region in the folding process.12 Our analysis suggests that up hairpins can be considered as “micro‐domains” (where each hairpin is held together by strong inter‐strand interactions), which are then linked together by weaker “inter‐domain” interactions (Figure 3a). By analogy to globular proteins, we speculate that the up hairpins fold first, followed by coalescence of the rest of the β‐sheet. Sequence variability among repeating units may affect their folding rates 6 and additional experiments are needed to address this question. Therefore, the SLBP family of proteins, with their modular design, could serve as a new model system for investigating stability, folding, and engineering of β‐hairpins and β‐sheets.
4. MATERIALS AND METHODS
4.1. Protein production
Clones were generated using the Polymerase Incomplete Primer Extension (PIPE) cloning method.46 The gene segments encoding predicted signal peptides using SignalP47 were omitted. The truncated BfSLBP, PdSLBP, and BvSLBP clones were amplified by polymerase chain reaction (PCR) from the corresponding genomic DNA using PfuTurbo DNA polymerase (Stratagene) and I‐PIPE (Insert) primers (Table S5). The expression vector, pSpeedET, which encodes an amino‐terminal tobacco etch virus (TEV) protease‐cleavable expression and purification tag (MGSDKIHHHHHHENLYFQ‐G), was PCR amplified with V‐PIPE (Vector) primers (forward primer: 5′‐taacgcgacttaattaactcgtttaaacggtctccagc‐3′, reverse primer: 5′‐gccctggaagtacaggttttcgtgatgatgatgatgatg‐3′). V‐PIPE and I‐PIPE PCR products were mixed, and the amplified DNA fragments were annealed. E. coli GeneHogs (Invitrogen) competent cells were transformed with the I‐PIPE/V‐PIPE mixture and dispensed on selective LB‐agar plates. The cloning junctions were confirmed by DNA sequencing. Expression was performed in E. coli strain PB1, which was grown in a selenomethionine‐containing medium at 37°C. Selenomethionine was incorporated via inhibition of methionine biosynthesis.48 At the end of fermentation, lysozyme was added to the culture to a final concentration of 250 μg/mL, and the cells were harvested and frozen. After one freeze/thaw cycle, the cells were homogenized and sonicated in lysis buffer [40 mM Tris, 300 mM NaCl, 10 mM imidazole, 1 mM Tris(2‐carboxyethyl)phosphine‐HCl (TCEP), pH 8.0]. Remaining nucleic acids were digested with the addition of 0.4 mM magnesium sulfate and 1 μL of 250 U/μL benzonase nuclease (Sigma) in the lysate. The lysate was clarified by centrifugation at 32,500g for 25 min. The soluble fraction was passed over nickel‐chelating resin (GE Healthcare) pre‐equilibrated with lysis buffer. The resin was then washed with wash buffer [40 mM Tris, 300 mM NaCl, 40 mM imidazole, 10% (v/v) glycerol, 1 mM TCEP, pH 8.0], and the protein was eluted with elution buffer [20 mM Tris, 300 mM imidazole, 10% (v/v) glycerol, 150 mM NaCl, 1 mM TCEP, pH 8.0]. The eluate was buffer‐exchanged with TEV buffer [20 mM Tris, 150 mM NaCl, 30 mM imidazole, 1 mM TCEP, pH 8.0] using a PD‐10 column (GE Healthcare), and incubated with 1 mg of TEV protease per 15 mg of eluted protein for 2 hr at ~20–25°C, followed by digestion overnight at 4°C. The protease‐treated eluate was passed over a nickel‐chelating resin (GE Healthcare) pre‐equilibrated with crystallization buffer (20 mM Tris, 150 mM NaCl, 30 mM imidazole, 1 mM TCEP, pH 8.0) and the resin was washed with the same buffer. The flow‐through and wash fractions were combined and concentrated by centrifugal ultrafiltration (Millipore) for crystallization trials.
The protein oligomeric state in solution was determined using a 1 × 30 cm2 Superdex 200 size exclusion column (GE Healthcare)46 coupled with miniDAWN (Wyatt Technology) static light scattering (SEC/SLS) and Optilab differential refractive index detectors (Wyatt Technology). The mobile phase consisted of 20 mM Tris pH 8.0, 150 mM NaCl, and 0.02% (w/v) sodium azide.
4.2. Crystallization
SLBPs were crystallized using the nanodroplet vapor diffusion method49 with standard JCSG crystallization protocols24 using the robotic CrystalMation platform (Rigaku Automation). Drops composed of 200 nL protein solution mixed with 200 nL crystallization solution in a sitting drop format were equilibrated against a 50 μL reservoir. The detailed crystallization conditions are shown in Tables S2–S4. Initial screening for diffraction was carried out using the Stanford Automated Mounting system50 at the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA).
4.3. Data collection and structure determination
MAD or SAD data were collected at wavelengths corresponding to a selenium MAD experiment at SSRL beamlines (11‐1, 14‐1, and 12‐2). Data processing and structure determination were carried out using automated structure solution protocols developed at the JCSG.51, 52 In brief, the diffraction data were integrated and reduced using XDS and then scaled with the program XSCALE.53 The locations of selenium sites, initial phasing, and identification of the space group were carried out using SHELXD.54 Phase refinement and model building were performed using autoSHARP55 and BUCCANEER.56 The model building and refinement was completed using COOT57 and BUSTER58 or REFMAC5.59 The refinement included experimental phase restraints in the form of Hendrickson–Lattman coefficients (when applicable) and TLS refinement with one TLS group consisting of the whole protein chain in the absence of solvent molecules. Data and refinement statistics are summarized in Table S2–S4.
4.4. MD simulation
MD simulations were performed using GROMACS 4.6.3 with the CHARM22 force field.60 The protein was placed in a rhombic dodecahedron box with the edges at least 10 å away from the protein surface. The system was solvated with water molecules (TIP3P) supplemented with 0.1M NaCl. After energy minimization, position‐restrained MD simulations were carried out in the NVT and NPT ensembles (100 and 500 ps) to equilibrate the system at 298 K and 1 atm. The system was then subjected to unconstrained production MD simulation for 100 ns. Normal mode analysis was carried out using NMwiz.61
4.5. Sequence and structure analysis
Secondary structures and hydrogen bonds were assigned using PROMOTIF.62 Analysis of the stereochemical quality of the model was accomplished using MolProbity.25 Structural superposition was carried out using Matt63 or TMalign.64 Molecular graphics were prepared with PyMOL (http://www.pymol.org/) or VMD.65 Protein sequences were aligned using MAFFT66 and rendered using TeXshade.67 Identification of structural motifs was carried out using SPASM,68 searching against a nonredundant PDB dataset based on PISCES69 (sequence identity <30%, resolution >3.0 å).
4.6. Accession codes
Atomic coordinates and experimental structure factors for BfSLBP, BvSLBP, and PdSLBP have been deposited in the PDB with accession codes 3msw, 4r8o, and 4r03, respectively. The plasmid for producing recombinant BfSLBP is deposited in the PSI:Biology‐Materials Repository (http://dnasu.asu.edu) with clone ID BfCD00327505.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1 SLBP homologs selected for structure determination
Table S2. Crystallization, data collection and refinement statistics for BfSLBP (PDB 3msw)
Table S3. Crystallization, data collection and refinement statistics for PdSLBP (PDB ID 4r03)
Table S4. Crystallization, data collection and refinement statistics for BvSLBP (PDB ID 4r8o)
Table S5. Primers used for cloning
ACKNOWLEDGMENTS
The authors thank the members of the JCSG high‐throughput structural biology pipeline for their contribution to this work. MD simulations were carried out using the GPU cluster of the Institute for Computational & Mathematical Engineering (ICME) at Stanford. This work was supported by the NIH, National Institute of General Medical Sciences (NIGMS), Protein Structure Initiative (U54 GM094586). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE‐AC02‐76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). Genomic DNA from B. fragilis NCTC 9343 (ATCC Number 25285D) were obtained from the American Type Culture Collection (ATCC). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS, NCRR, or NIH.
Xu Q, Biancalana M, Grant JC, et al. Structures of single‐layer β‐sheet proteins evolved from β‐hairpin repeats. Protein Science. 2019;28:1676–1689. 10.1002/pro.3683
Qingping Xu and Matthew Biancalana contributed equally to this work.
Funding information National Institutes of Health, Grant/Award Number: U54 GM094586
REFERENCES
- 1. Hartl FU, Hayer‐Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. [DOI] [PubMed] [Google Scholar]
- 2. Dill KA, MacCallum JL. The protein‐folding problem, 50 years on. Science. 2012;338:1042–1046. [DOI] [PubMed] [Google Scholar]
- 3. Di Domenico T, Potenza E, Walsh I, et al. RepeatsDB: A database of tandem repeat protein structures. Nucleic Acids Res. 2014;42:D352–D357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrade MA, Perez‐Iratxeta C, Ponting CP. Protein repeats: Structures, functions, and evolution. J Struct Biol. 2001;134:117–131. [DOI] [PubMed] [Google Scholar]
- 5. Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. [DOI] [PubMed] [Google Scholar]
- 6. Main ER, Lowe AR, Mochrie SG, Jackson SE, Regan L. A recurring theme in protein engineering: The design, stability and folding of repeat proteins. Curr Opin Struct Biol. 2005;15:464–471. [DOI] [PubMed] [Google Scholar]
- 7. Kloss E, Courtemanche N, Barrick D. Repeat‐protein folding: New insights into origins of cooperativity, stability, and topology. Arch Biochem Biophys. 2008;469:83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roche DB, Viet PD, Bakulina A, Hirsh L, Tosatto SCE, Kajava AV. Classification of β‐hairpin repeat proteins. J Struct Biol. 2018;201:130–138. [DOI] [PubMed] [Google Scholar]
- 9. Macias MJ, Gervais V, Civera C, Oschkinat H. Structural analysis of WW domains and design of a WW prototype. Nat Struct Biol. 2000;7:375–379. [DOI] [PubMed] [Google Scholar]
- 10. Cochran AG, Skelton NJ, Starovasnik MA. Tryptophan zippers: Stable, monomeric beta‐hairpins. Proc Natl Acad Sci U S A. 2001;98:5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Honda S, Yamasaki K, Sawada Y, Morii H. 10 residue folded peptide designed by segment statistics. Structure. 2004;12:1507–1518. [DOI] [PubMed] [Google Scholar]
- 12. Lewandowska A, Oldziej S, Liwo A, Scheraga HA. β‐hairpin‐forming peptides; models of early stages of protein folding. Biophys Chem. 2010;151:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pantoja‐Uceda D, Santiveri CM, Jimenez MA. De novo design of monomeric β‐hairpin and β‐sheet peptides. Methods Mol Biol. 2006;340:27–51. [DOI] [PubMed] [Google Scholar]
- 14. Pham TN, Koide A, Koide S. A stable single‐layer β‐sheet without a hydrophobic core. Nat Struct Biol. 1998;5:115–119. [DOI] [PubMed] [Google Scholar]
- 15. Li H, Dunn JJ, Luft BJ, Lawson CL. Crystal structure of Lyme disease antigen outer surface protein A complexed with an Fab. Proc Natl Acad Sci U S A. 1997;94:3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cottee MA, Muschalik N, Wong YL, et al. Crystal structures of the CPAP/STIL complex reveal its role in centriole assembly and human microcephaly. Elife. 2013;2:e01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatzopoulos GN, Erat MC, Cutts E, et al. Structural analysis of the G‐box domain of the microcephaly protein CPAP suggests a role in centriole architecture. Structure. 2013;21:2069–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Busby JN, Panjikar S, Landsberg MJ, Hurst MR, Lott JS. The BC component of ABC toxins is an RHS‐repeat‐containing protein encapsulation device. Nature. 2013;501:547–550. [DOI] [PubMed] [Google Scholar]
- 19. Koide S, Bu Z, Risal D, et al. Multistep denaturation of Borrelia burgdorferi OspA, a protein containing a single‐layer β‐sheet. Biochemistry. 1999;38:4757–4767. [DOI] [PubMed] [Google Scholar]
- 20. Cutts EE, Inglis A, Stansfeld PJ, Vakonakis I, Hatzopoulos GN. The centriolar protein CPAP G‐box: An amyloid fibril in a single domain. Biochem Soc Trans. 2015;43:838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ellrott K, Jaroszewski L, Li W, Wooley JC, Godzik A. Expansion of the protein repertoire in newly explored environments: Human gut microbiome specific protein families. PLoS Comput Biol. 2010;6:e1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lesley SA, Kuhn P, Godzik A, et al. Structural genomics of the Thermotoga maritima proteome implemented in a high‐throughput structure determination pipeline. Proc Natl Acad Sci U S A. 2002;99:11664–11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elsliger MA, Deacon AM, Godzik A, et al. The JCSG high‐throughput structural biology pipeline. Acta Cryst. 2010;F66:1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen VB, Arendall WB 3rd, Headd JJ, et al. MolProbity: All‐atom structure validation for macromolecular crystallography. Acta Cryst. 2010;D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chan AW, Hutchinson EG, Harris D, Thornton JM. Identification, classification, and analysis of β‐bulges in proteins. Protein Sci. 1993;2:1574–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richardson JS. The anatomy and taxonomy of protein structure. Adv Protein Chem. 1981;34:167–339. [DOI] [PubMed] [Google Scholar]
- 28. Chan AW, Hutchinson EG, Harris D, Thornton JM. Identification, classification, and analysis of beta‐bulges in proteins. Protein Sci. 1993;2:1574–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ujwal R, Cascio D, Colletier JP, et al. The crystal structure of mouse VDAC1 at 2.3 å resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci U S A. 2008;105:17742–17747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Makabe K, Tereshko V, Gawlak G, Yan S, Koide S. Atomic‐resolution crystal structure of Borrelia burgdorferi outer surface protein A via surface engineering. Protein Sci. 2006;15:1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu L, McElheny D, Huang R, Keiderling TA. Role of tryptophan‐tryptophan interactions in Trpzip β‐hairpin formation, structure, and stability. Biochemistry. 2009;48:10362–10371. [DOI] [PubMed] [Google Scholar]
- 33. Siebold C, Garcia‐Alles LF, Erni B, Baumann U. A mechanism of covalent substrate binding in the X‐ray structure of subunit K of the Escherichia coli dihydroxyacetone kinase. Proc Natl Acad Sci U S A. 2003;100:8188–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yan S, Gawlak G, Makabe K, Tereshko V, Koide A, Koide S. Hydrophobic surface burial is the major stability determinant of a flat, single‐layer β‐sheet. J Mol Biol. 2007;368:230–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- 36. Santiveri CM, Jimenez MA. Tryptophan residues: Scarce in proteins but strong stabilizers of β‐hairpin peptides. Biopolymers. 2010;94:779–790. [DOI] [PubMed] [Google Scholar]
- 37. Robert C, Chassard C, Lawson PA, Bernalier‐Donadille A. Bacteroides cellulosilyticus sp. nov., a cellulolytic bacterium from the human gut microbial community. Int J Syst Evol Microbiol. 2007;57:1516–1520. [DOI] [PubMed] [Google Scholar]
- 38. Sonnenburg JL, Xu J, Leip DD, et al. Glycan foraging in vivo by an intestine‐adapted bacterial symbiont. Science. 2005;307:1955–1959. [DOI] [PubMed] [Google Scholar]
- 39. Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–36279. [DOI] [PubMed] [Google Scholar]
- 40. Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate‐binding modules: Fine‐tuning polysaccharide recognition. Biochem J. 2004;382:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hashimoto H. Recent structural studies of carbohydrate‐binding modules. Cell Mol Life Sci. 2006;63:2954–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 44. Richardson JS, Richardson DC. Natural β‐sheet proteins use negative design to avoid edge‐to‐edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hughes RM, Waters ML. Model systems for β‐hairpins and β‐sheets. Curr Opin Struct Biol. 2006;16:514–524. [DOI] [PubMed] [Google Scholar]
- 46. Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994. [DOI] [PubMed] [Google Scholar]
- 47. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. [DOI] [PubMed] [Google Scholar]
- 48. Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP‐12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. [DOI] [PubMed] [Google Scholar]
- 49. Santarsiero BD, Yegian DT, Lee CC, et al. An approach to rapid protein crystallization using nanodroplets. J Appl Cryst. 2002;35:278–281. [Google Scholar]
- 50. Cohen AE, Ellis PJ, Miller MD, Deacon AM, Phizackerley RP. An automated system to mount cryo‐cooled protein crystals on a synchrotron beamline, using compact samples cassettes and a small‐scale robot. J Appl Cryst. 2002;35:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu Q, Abdubek P, Astakhova T, et al. Structure of the γ‐D‐glutamyl‐L‐diamino acid endopeptidase YkfC from Bacillus cereus in complex with L‐Ala‐γ‐D‐Glu: insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Cryst. 2010;F66:1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van den Bedem H, Wolf G, Xu Q, Deacon AM. Distributed structure determination at the JCSG. Acta Cryst. 2011;D67:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sheldrick GM. A short history of SHELX. Acta Cryst. 2008;A64:112–122. [DOI] [PubMed] [Google Scholar]
- 55. Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Cryst. 2003;D59:2023–2030. [DOI] [PubMed] [Google Scholar]
- 56. Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Cryst. 2006;D62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 57. Emsley P, Cowtan K. COOT: model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 58. Blanc E, Roversi P, Vonrhein C, Flensburg C, Lea SM, Bricogne G. Refinement of severely incomplete structures with maximum likelihood in BUSTER‐TNT. Acta Crystallogr. 2004;D60:2210–2221. [DOI] [PubMed] [Google Scholar]
- 59. Murshudov GN, Skubak P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. 2011;D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pronk S, Pall S, Schulz R, et al. GROMACS 4.5: A high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bakan A, Meireles LM, Bahar I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hutchinson EG, Thornton JM. PROMOTIF––A program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Menke M, Berger B, Cowen L. Matt: Local flexibility aids protein multiple structure alignment. PLoS Comput Biol. 2008;4:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Y, Skolnick J. TM‐align: A protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res. 2005;33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(33–38):27–38. [DOI] [PubMed] [Google Scholar]
- 66. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Beitz E. TEXshade: Shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics. 2000;16:135–139. [DOI] [PubMed] [Google Scholar]
- 68. Kleywegt GJ. Recognition of spatial motifs in protein structures. J Mol Biol. 1999;285:1887–1897. [DOI] [PubMed] [Google Scholar]
- 69. Wang G, Dunbrack RL Jr. PISCES: A protein sequence culling server. Bioinformatics. 2003;19:1589–1591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1 SLBP homologs selected for structure determination
Table S2. Crystallization, data collection and refinement statistics for BfSLBP (PDB 3msw)
Table S3. Crystallization, data collection and refinement statistics for PdSLBP (PDB ID 4r03)
Table S4. Crystallization, data collection and refinement statistics for BvSLBP (PDB ID 4r8o)
Table S5. Primers used for cloning