

Abstract
Objective
The spectrum of UDP-glucuronyl transferase A1 (UGT1A1) variants in hereditary unconjugated hyperbilirubinemia varies markedly between different ethnic populations. This study evaluated the UGT1A1 genotypes in hyperbilirubinemia patients from southeastern China.
Methods
We enrolled 60 patients from southeastern China (44 men and 16 women; age range: 3–76 years) with unconjugated hyperbilirubinemia and performed genetic analysis of the UGT1A1 gene by direct sequencing.
Results
For patients with Gilbert syndrome, 85% (47/55) harbored pathogenic variants of UGT1A1⁎60. Both UGT1A1⁎28 and UGT1A1⁎81 were detected in the promoter region of UGT1A1. Additionally, 83% (20/24) of patients with Gilbert syndrome heterozygous for UGT1A1⁎60 had an association with heterozygous variation of UGT1A1⁎28 or UGT1A1⁎81, while 91% (21/23) of Gilbert syndrome patients homozygous for UGT1A1⁎60 had biallelic variations of UGT1A1⁎28 and UGT1A1⁎81. We detected 213 UGT1A1 allelic variants, including six novel variations, with the most frequent allele being the UGT1A1⁎60, followed by UGT1A1⁎28 and UGT1A1⁎6. All of the patients showed multiple sites of variants in UGT1A1; however, variation number was not associated with bilirubin levels (P>0.05).
Conclusions
The spectrum of UGT1A1 variants in southeastern Chinese patients was distinct from other ethnic populations. Our findings broaden the knowledge concerning traits associated with UGT1A1 variants and help profile genotype–phenotype correlations in hyperbilirubinemia patients.
1. Introduction
Hereditary unconjugated hyperbilirubinemia is autosomal recessive disorder and can be categorized as Crigler–Najjar syndrome type I (CN-I; OMIM#218800), Crigler–Najjar syndrome type II (CN-II; OMIM#606785), or Gilbert syndrome (GS; OMIM#143500) based on serum bilirubin levels. The concentration of serum total bilirubin (TBIL) in CN-I, CN-II, and GS ranges from 513 μM to 855 μM, 102.6μM to 342 μM, and 17 μM to 85 μM, respectively [1]. These hyperbilirubinemias result from increased water-insoluble unconjugated bilirubin in the liver in the absence of liver dysfunction or hemolysis [2]. The common clinical presentation in hyperbilirubinemia patients is jaundice, and in CN-I patients, jaundice is apparent from birth and progressively accumulates to present a risk of kernicterus [3]. Under normal conditions, unconjugated bilirubin is conjugated to water-soluble bilirubin-glucuronide conjugates and secreted into bile [4].
UDP-glucuronyl transferase (UGT), encoded by UGT1A1, is the only enzyme in liver that glucuronidates bilirubin. Hereditary unconjugated hyperbilirubinemia, including CN-I, CN-II, and GS, is, respectively, caused by mutations in UGT1A1 (OMIM∗191740), which is a member of the UGT1 superfamily and located on chromosome (2q37). The UGT1A1 promoter contains a TATA-box sequence, with an open reading frame of 1062 bp length [5, 6]. UGT1A1 enzyme activity can be increased by phenobarbital administration, which induces UGT1A1 expression by binding to the phenobarbital-responsive module (PBREM) in the distal enhancer element [7]. To date, >130 variants in both the regulatory and coding regions of UGT1A1 have been identified in hereditary hyperbilirubinemia patients [8], with variations identified in CN-I, CN-II, and GS reducing UGT1A1 enzyme activity to 0%, 10%, and 30%, respectively [9–11].
The spectrum of UGT1A1 variants varies markedly in different populations. In Caucasian populations, the most common genotype is a TA insertion in the TATA-box sequence of the UGT1A1 gene (UGT1A1∗28), resulting in A(TA)7TAA instead of the normal A(TA)6TAA sequence [12, 13]. In Western countries, the allelic frequency of the TA insertion can be as high as 0.4 [14, 15], and in Asian countries, such as Japan, the most common variation is the UGT1A1∗6 variant in exon 1, resulting in a p.Gly71Arg substitution [16]; however, few studies have reported UGT1A1 variants in hyperbilirubinemia patients from China [17, 18]. Allelic differences in UGT1A1 in a Chinese population with hyperbilirubinemia are expected; therefore, the present study investigated the allelic frequency and distribution of UGT1A1 variants in southeastern Chinese patients with hyperbilirubinemia.
2. Methods
2.1. Patients
Sixty patients with unconjugated hyperbilirubinemia from southeast China were enrolled at The Affiliated Hospital of Hangzhou Normal University between 2016 and 2018. All patients showed TBIL levels ≥17.1 μM, with normal liver enzymes and no evidence of hemolysis. The patients included 44 men and 16 women (age range: 3–76 years), with most originally suspected as having hyperbilirubinemia because of apparent jaundice, whereas others were admitted during conventional health checks. The patients enrolled were all checked negative for viral hepatitis, including serology tests for hepatitis A virus (HAV), hepatitis B virus (HBV), hepatitis C virus (HCV), hepatitis D virus (HDV), and hepatitis E virus (HEV). Other hepatic diseases which may cause hyperbilirubinemia were excluded, including hemolysis, alcoholic liver disease, and autoimmune liver disease. All subjects included in this study had normal levels of liver enzymes (ALT:1-52 U/L; AST:1-40 U/L). Previous/past drug history of potentially hepatotoxic medications was also excluded. Abdominal ultrasound images for all patients were normal, and no treatment was administered when the biomedical parameters were obtained. Serum TBIL levels in all 60 patients ranged from 28.8 μM (1.68 mg/dL) to 301.2 μM (17.61 mg/dL), with none showing TBIL levels ≥ 30 mg/dL, as seen in CN-I. Based on serum TBIL levels, 55 patients were divided into the GS group (hyperbilirubinemia: 17–85 μM), three into the CN-II group (hyperbilirubinemia: 102.6–342 μM), and two into the Intermediate group (borderline CN-II and GS).
Written informed consent was obtained from participants or their legal guardians. The study was approved by the Ethics Committee of the Affiliated Hospital of Hangzhou Normal University.
2.2. Genomic DNA Extraction and Mutation Analysis
Genomic DNA was extracted from the peripheral blood leukocytes of all patients using a genomic DNA purification kit (Qiagen, Hilden, Germany). All exon, flanking-intron, promoter, and PBREM regions of UGT1A1 were amplified from genomic DNA. Primers were designed using Primer Premier 5 software (http://www.premierbiosoft.com/primer design/) according to the reference cDNA sequence of UGT1A1 (NM_000463). Polymerase chain reaction (PCR) analysis was performed using ~100 ng genomic DNA under the following conditions: initial denaturation for 5 min at 95°C, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 58°C for 1 min, and elongation at 72°C for 1 min, with a final elongation at 72°C for 5 min. PCR products were directly sequenced on an ABI3730XL sequencer (Applied Biosystems, Foster City, CA, USA). Primers sequences used to amplify UGT1A1 DNA fragments were listed as Table S1.
2.3. Statistical Analysis
Statistical tests were performed using SPSS (v.17.0; SPSS Inc., Chicago, IL, USA). Continuous variables [age, alanine aminotransferase (ALT), aspartate aminotransferase (AST), TBIL, direct bilirubin (DBIL), and unconjugated bilirubin (IBIL)] were evaluated using the Kolmogorov–Smirnov test or the Shapiro–Wilk test for normal distribution analysis. Continuous variables that were normally distributed were expressed as the mean ± standard deviation and compared by one-way analysis of variance. Continuous variables not normally distributed were presented as the median and range and compared using the Kruskal–Wallis H test. Categorical variables were analyzed using the Chi-square test. A P<0.05 was considered significant.
3. Results
3.1. Patient Characteristics Based on the c.-3279T>G Genotype
Demographic information and biochemical parameters are presented in Table 1. Among the 55 GS patients, 43% (24/55) patients harbored one c.-3279T>G variation (UGT1A1∗60), 42% (23/55) harbored two c.-3279T>G variations, and 15% (8/55) showed no c.-3279T>G variation. Based on the c.-3279T>G genotype, we subdivided GS patients into three groups: heterozygotes with one c.-3279T>G variation, homozygotes with two c.-3279T>G variations, and wild-type (no c.-3279T>G variation harbored). Forty-one GS patients were male, including 19 heterozygotes, 17 homozygotes, 5 wild-types. There was no significant difference in gender distribution among the three subgroups of GS patients (P=0.54).
Table 1.
Demographic information and biochemical parameters in Gilbert patients.
| Total | -3279T>G | -3279T>G | -3279T>G | P | |
|---|---|---|---|---|---|
| Heterozygote | Homozygote | Wildtype | |||
| N | 55 | 24(43%) | 23(42%) | 8(15%) | |
| Sex(M/F) | 41M/14F | 19M/5F | 17M/6F | 5M/3F | 0.64 |
| Age, y | 34(3~76) | 33.5(3~66) | 34.0(21~61) | 46.0(18~76) | 0.25 |
| ALT(U/L) | 26.73±11.85 | 27.87±12.76 | 26.13±12.34 | 25.00±7.76 | 0.80 |
| AST(U/L) | 24.05±12.02 | 27.83±16.73 | 20.47±4.97 | 23.00±4.95 | 0.10 |
| ALB(g/L) | 46.7(41.3~52.1) | 47.3(41.3~52.1) | 46.4(44.4~51.8) | 45.0(42.8~49.0) | 0.18 |
| GGT(U/L) | 18.93±6.96 | 18.42±7.24 | 18.0±6.26 | 24.12±7.06 | 0.09 |
| TBil(μmol/L) | 43.9(28.8~82.9) | 44.3(30.1~70.2) | 42.4(28.8~82.9) | 38.0(32.1~57.9) | 0.39 |
| DBil(μmol/L) | 11.84±3.33 | 12.17±3.40 | 11.93±3.21 | 10.64±3.62 | 0.53 |
| IBil(μmol/L) | 30.7(21.4~70.4) | 32.0(22.2~53.1) | 30(21.4~70.4) | 29.4(23.9~48.7) | 0.32 |
Wild-type TT; Heterozygote TG; Homozygote GG
Variables were checked by Kolmogorov-Smirnov test or Shapiro-Wilk test for normal distribution analysis. Normally distributed data are expressed as mean±SD and compared by one-way ANOVA. Not normally distributed data were presented as median and range and were compared by Kruskal-Wallis H test. Categorical variables were analyzed using Chi-square test.
The age at onset in our patients with hyperbilirubinemia ranged from 3 to 76 years, and among the three subgroups of GS patients, there was no significant difference in onset age (P=0.25). Additionally, differences in levels of ALT (P=0.80), AST (P=0.10), albumin (P=0.18), and gamma-glutamyltransferase (P=0.09) were not significant; however, TBIL and especially IBIL levels were beyond the normal range in all GS patients, although we found no significant difference in these levels among the three subgroups. Moreover, we also detected one or two c.-3279T>G variations carried by our Intermediate patients but not CN-II patients. These findings indicated that c.-3279T>G variation is essential for the pathogenesis of mild hyperbilirubinemia.
3.2. Variants in the Proximal Promoter Region of UGT1A1
As noted, 85% patients (47/55) of GS patients harbored one or two c.-3279T>G variations in the PBREM region of UGT1A1 (Figure 1(a)). Table 2 shows that, of the GS patients heterozygous for the c.-3279T>G variation (n=24), 50% (12/24) were also heterozygous for A(TA)7TAA (UGT1A1∗28), 33.3% (8/24) were heterozygous for a c.-64G>C variation (UGT1A1∗81), one patient harbored a biallelic TA insertion, and 12.5% (8/24) showed no variations in the promoter region. These results indicated that 83.3% of GS patients heterozygous for the c.-3279T>G variation also harbored heterozygous variation in the UGT1A1 promoter region (Figure 1(a′)), suggesting that c.-3279T>G heterozygosity is mostly accompanied by heterozygous variations in the UGT1A1 promoter in our patient cohort.
Figure 1.

Incidence of the c.-3279T>G genotype in GS patients. (a) Incidence of the c.-3279T>G genotype in GS patients. (a′) Incidence of different genotypes in GS patients heterozygous for c.-3279T>G. (b′) Incidence of different genotypes in GS patients homozygous for c.-3279T>G.
Table 2.
Association of c.-3279T>G in PBREM with TA insertion or c.-64G>C in promoter region of UGT1A1 in Gilbert patients.
| GS (n=55) |
c.-3279 T>G in PBREM | ||
|---|---|---|---|
| Wild-type | Heterozygote | Homozygote | |
| n=8 | n=24 | n=23 | |
| A(TA)7TAA | |||
| Heter | 0 | 12(50%) | 2(8.7%) |
| Homo | 0 | 1(4.2%) | 14(60.9%) |
| c.-64G>C | |||
| Heter | 0 | 8(33.3%) | 0 |
| Homo | 0 | 0 | 1(4.3%) |
| A(TA)7TA&c.-64G>C | |||
| 0 | 0 | 6(26.1%) | |
| Others | |||
| 8 | 3(12.5%) | 0 | |
Wild-type TT; Heterozygote TG; Homozygote GG
In GS patients homozygous for the c.-3279T>G variation (n=23), 61% (14/23) were also homozygous for A(TA)7TAA, 4% (1/23) were homozygous for the c.-64G>C variation, 26% (6/23) harbored a TA insertion and the c.-64G>C variation, and two patients were heterozygous for the TA insertion. These results indicated that 91% of GS patients homozygous for the c.-3279T>G variation also harbored biallelic variations in the UGT1A1 promoter region (Figure 1(b′)), suggesting that c.-3279T>G homozygosity was frequently associated with homozygous variations in the UGT1A1 promoter. Furthermore, in our Intermediate patients harboring the c.-3279T>G variation, we also detected a TA insertion. These findings demonstrated that the c.-3279T>G genotype was closely accompanied by A(TA)7TAA or c.-64G>C genotype in the UGT1A1 promoter, indicating that variants of the c.-3279T>G and A(TA)7TAA or c.-64G>C represented the principal genotype associated with GS in this cohort.
3.3. Novel Variants
A total of 213 allelic variants at six sites in UGT1A1 were detected in our patient cohort, including variants in the PBREM, proximal promoter, and coding regions (exons 1, 3, 4, and 5). The most common variants were c.-3279T>G in the PBREM region, with an allele frequency of 34.3% (UGT1A1∗60, 73/213), followed by A(TA)7TAA in the promoter region (UGT1A1∗28, 52/213) and p.Gly71Arg in exon 1 (UGT1A1∗6, 37/213). Six novel variants were detected (Figure 2 and Table 3), including p.Asp259Glu, p.Ile268Val, c.1084+1G>T, p.Glu463Lys, p.Val491Met, and p.Arg522Stop, with all of these located in or adjacent to the coding region (Figure 3). Allelic number of these novel alleles has not been noted by UGT Nomenclature. Also linkage disequilibrium analysis was performed among all UGT1A1 variants detected in this cohort (Figure 4).
Figure 2.

Novel variants found in 60 patients with hyperbilirubinemias.
Table 3.
UGT1A1 variants found in all 60 patients with hyperbilirubinemias.
| Gene Region | Nucleotide Change | Amino acid Change | rs Number in dbSNP database | No. of alleles | Allele Frequency (%) |
1000g_CHB MAF (%) |
P value |
|---|---|---|---|---|---|---|---|
| Enhancer | |||||||
| PBREM | -3279 T>G | rs4124874 | 73 | 34.3 | 27.20 | 2.27E-06∗ | |
| Promoter | |||||||
| -64 G>C | rs873478 | 17 | 7.98 | 3.40 | 0.02316∗ | ||
| TATA box | A(TA)6TAA> A(TA)7TAA |
rs3064744 | 52 | 24.4 | 12.90 | 1.05E-17∗ | |
| Exon1 | |||||||
| c.211 G>A | p.Gly71Arg | rs4148323 | 37 | 17.4 | 22.80 | 0.102251 | |
| c.625 C>T | p.Arg209Trp | rs72551343 | 2 | 0.94 | 0.00 | 0.052645 | |
| c.686 C>A | p.Pro229Glu | rs35350960 | 8 | 3.75 | 0.50 | 0.000572∗ | |
| c.777 C>G | p.Asp259Glu | Novel | 1 | 0.47 | NA | NA | |
| c.802 A>G | p.Ile268Val | Novel | 1 | 0.47 | NA | NA | |
| Exon3 | |||||||
| c.1084 G>A c.1084+1 G>T |
p.Gly362Ser | rs755218546 Novel |
1 1 |
0.47 0.47 |
0 NA |
0.171234 NA |
|
| Exon4 | |||||||
| c.1091 C>T | p.Pro364Leu | rs34946978 | 9 | 4.22 | 2.40 | 0.018437∗ | |
| Exon5 | |||||||
| c.1387 G>A | p.Glu463Lys | Novel | 1 | 0.47 | NA | NA | |
| c.1456 T>G | p.Tyr486Asp | rs34993780 | 6 | 2.82 | 0 | 0.000735∗ | |
| c.1470 C>T | p.Asp490Asp | rs114123636 | 1 | 0.47 | 0.50 | 0.652817 | |
| c.1471 G>A | p.Val491Met | Novel | 1 | 0.47 | NA | NA | |
| c.1567 C>T | p.Arg522Stop | Novel | 2 | 0.94 | NA | NA |
dbSNP: database of Single Nucleotide Polymorphism(https://www.ncbi.nlm.nih.gov/SNP/);
1000g_CHB MAF: Minor allele frequency of Han population in Beijing, China in 1000 genomes database(http://www.1000genomes.org).
Figure 3.

The distribution of variants in 60 patients with hyperbilirubinemias. Variants in UGT1A1 regulatory regions are shown as nucleotide changes. Variants in the UGT1A1 coding region are shown as amino acid substitutions. Novel variants are indicated in red.
Figure 4.
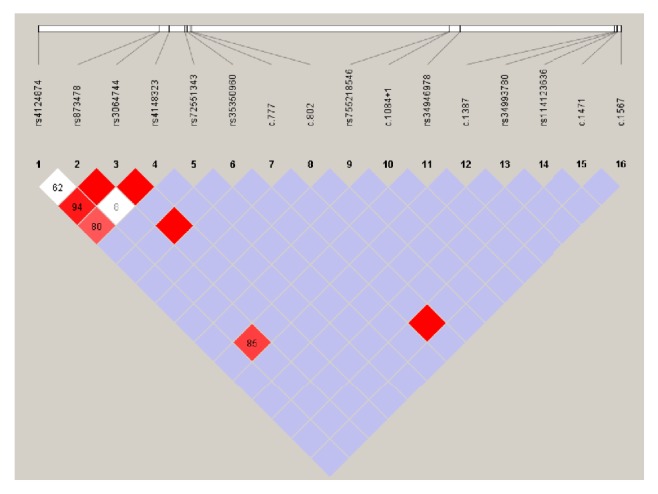
Linkage disequilibrium analysis of the UGT1A1 variants detected in this cohort. Pairwise LD map, a denser color indicates greater linkage.
3.4. Multiple Variants
All of the patients harbored at least two sites of sequence variations associated with UGT1A1. Thirteen patients, including 11 GS and two CN-II patients, harbored variations at two sites (Table S2), 15 patients, including 14 GS and one Intermediate patient, harbored variations at three sites (Table S3), 19 patients, including 18 GS and one CN-II patient, harbored variations at four sites (Table S4), and 12 patients, including 11 GS and one Intermediate patient, harbored variations at five sites (Table S5). Additionally, we detected variations at six sites in one GS patient homozygous for a combination of UGT1A1∗60, UGT1A1∗28, and UGT1A1∗27. However, associations between levels of serum TBIL and the number of variations did not differ significantly between each group (Figure 5).
Figure 5.
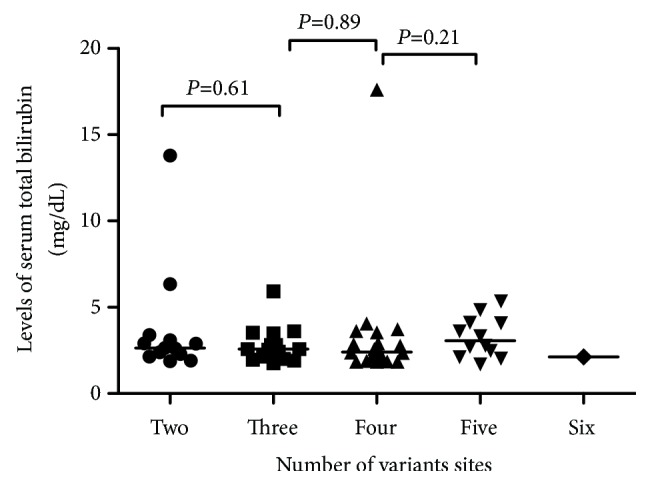
Association between levels of serum total bilirubin and the number of variants in 60 patients with hyperbilirubinemia. Analysis of two groups using the Mann-Whitney U test revealed no significant associations. Lines indicate the median of each group.
4. Discussion
In this study, we identified UGT1A1 variants in 60 patients with unconjugated hyperbilirubinemias, including 55 GS patients, three CN-II patients, and two Intermediate patients, based on their bilirubin levels. None of patients displayed bilirubin levels ≥ 30 mg/dL, suggesting the absence of CN-I. CN-I syndrome is extremely rare and can be fatal due to kernicterus [19, 20], with UGT1A1 enzyme activity in CN-I either absent or greatly attenuated [10].
GS is a mild, prolonged hyperbilirubinemia syndrome, with a prevalence ranging from 3% to 13% [21]. UGT1A1∗28 is the most common pathogenic variant found in GS patients, with an allelic frequency of 0.4 in Western populations [14] and often linked with UGT1A1∗60 variant [22]. In the present study, UGT1A1∗60 was the most common variant found, with an allelic frequency of 0.34, which exceeded that in the Japanese population (allele frequency, 0.17) [23]. Additionally, we found that UGT1A1∗28 was the second most common variant, with an allelic frequency of 0.24. Moreover, we detected the UGT1A1∗81 (c.-64G>C) in the UGT1A1 proximal promoter region, which has not been reported previously in an Asian population. In our GS patients, the UGT1A1∗60 was also mostly accompanied by UGT1A1∗28 or UGT1A1∗81, suggesting that the genotype of UGT1A1∗60 accompanied with UGT1A1∗28 or UGT1A1∗81 was essential for GS pathogenesis in this cohort, whereas in our CN-II patients, we did not detect this accompanying. This may be due to the limited number of patients enrolled in this group.
The missense variant of UGT1A1∗6 (p.Gly71Arg), resulting from a G>A substitution in exon 1 of UGT1A1,was the third most common pathogenic variant found in our cohort, with an allelic frequency of 0.17. This variant was identified in both GS and CN-II patients; however, a genotype heterozygous for UGT1A1∗60/UGT1A1∗28 (or UGT1A1∗81) was detected in most of the patients harboring UGT1A1∗6 (18/19 patients). Five GS patients were identified as homozygous for UGT1A1∗6. These findings indicated that the p.Gly71Arg variant could be cause of hyperbilirubinemia in this cohort not only through its linkage with variants in the UGT1A1 regulatory regions but also in isolation.
We identified six novel UGT1A1-associated variants in our hyperbilirubinemia patients, including four missense variants, one nonsense variant, and one splicing variant. In silico analysis using SIFT, Polyphen-2, and MutationTaster [24–26] predicted the variants of p.Asp259Glu, p.Glu463Lys, and p.Val491Met as being likely pathogenic while p.Ile268Val was predicted as benign (data not shown). Additionally, the p.Arg522Stop variant was predicted as pathogenic, resulting in a truncated UGT1A1 protein potentially causing nonsense-mediated mRNA decay [27]. Moreover, the c.1084+1G>T variation disrupts the splicing-donor site of intron 3 in UGT1A1 and was predicted to cause the expression of abnormal UGT1A1 transcripts. All of these novel variants were found in the GS patients in our cohort, except for p.Arg522X, which was carried by one CN-II patient with a serum TBIL level of 301.2 μM (17.6 mg/dL). These findings broaden the spectrum of UGT1A1 variants associated with hyperbilirubinemia syndrome.
The spectrum of variants identified in this study was distinct from that reported previously. We detected 213 allelic variants at six sites associated with UGT1A1 in our patient cohort, with all of the patients harboring multiple variants sites. However, isolated heterozygous mutations were not detected, strongly supporting recessive inheritance of hyperbilirubinemia [2]. Furthermore, we found that the number of variants was unrelated to TBIL levels. In our CN-II and Intermediate patients, the more variant sites detected in coding regions, the more severity of hyperbilirubinemia presented, and in Gilbert patients, when we compared subgroups that harbored one coding variation site in total two sites harbored group and total five sites harbored group, we found that the more number of variations detected in promoter region, the higher levels of serum bilirubin presented (data not shown). These data suggested that allele frequency and distribution might be essential factors associated with the severity of hyperbilirubinemia. A Japanese study reported that variants located in UGT1A shared exons (exons 2 through 5) are present in 14.1% of GS patients (9/64) [28], whereas a Taiwanese study reported that variants located in UGT1A1 shared exons were absent from GS patients [29]. In the present study, we found that 29.1% of GS patients (16/55) harbored variants located in UGT1A1 shared exons. These results provide novel insight into population genetics associated with hyperbilirubinemia syndrome; however, further studies are required to elucidate the mechanisms associated with these variants.
In total, our study broadens the knowledge concerning traits associated with UGT1A1 variations and helps profile genotype–phenotype correlations in hyperbilirubinemia patients. Based on the finding that most Gilbert patients harbored variants located in promoter or exon 1 and most CN-II patients harbored variants located in exons 2 through 5, our study emphasizes the value of UGT1A1 genotypes in differential diagnosis of Gilbert and CN-II in everyday clinical practice. Also, our project addressed the genetic traits in hyperbilirubinemia patients from southeast China and will contribute to establishing genetic testing as a feasible and cost-effective tool to perform large-scale hyperbilirubinemia screening in the general population.
Acknowledgments
This study was supported by a Natural Science Foundation of Zhejiang Province grant to Ling Gong (LY18H160027), a Chinese Medical Science and Technology Planning Project of Zhejiang Province grant to Ling Gong (2018ZB105), and a Research Fund Project of the Affiliated Hospital of Hangzhou Normal University grant to Xiao-xiao Mi (Grant number: none). The authors would like to thank colleagues from the Affiliated Hospital of Hangzhou Normal University for their support and collaboration.
Abbreviations
- UGT1A1:
UDP-glucuronyl transferase A1
- GS:
Gilbert syndrome
- CN-I:
Crigler–Najjar syndrome type I
- CN-II:
Crigler–Najjar syndrome type II
- PBREM:
Phenobarbital-responsive module
- TBIL:
Total bilirubin levels
- DBIL:
Direct bilirubin levels
- IBIL:
Indirect bilirubin levels
- ALT:
Alanine aminotransferase
- AST:
Aspartate aminotransferase
- ALB:
Albumin
- GGT:
Gamma-Glutamyltransferase.
Contributor Information
Jun-ping Shi, Email: 20131004@hznu.edu.cn.
Ling Gong, Email: gongllyy22@163.com.
Data Availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Affiliated Hospital of Hangzhou Normal University research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent
Informed written consent was obtained from the patients for publication of this article and accompanying images.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
All authors have contributed significantly. Xiao-xiao Mi, Ling Gong, and Jun-ping Shi conceived and designed the project with great input from Jian Yan, Xiao-jie Ma, Ge-li Zhu, Yi-dan Gao, Wen-jun Yang, Xiao-wen Kong, and Gong-ying Chen. Xiao-xiao Mi and Jian Yan conducted mutations analysis. Xiao-jie Ma, Ge-li Zhu, Yi-dan Gao, Wen-jun Yang, Xiao-wen Kong, and Gong-ying Chen provided the clinical data. Xiao-xiao Mi wrote the manuscript with input from Jun-ping Shi and Ling Gong.
Supplementary Materials
Table S1: primer sequences used for amplicons of the UGT1A1 gene. Table S2: patients with variations at two sites. Table S3: patients with variations at three sites. Table S4: patients with variations at four sites. Table S5: patients with variations at five sites.
References
- 1.Radlovic N. Hereditary hyperbilirubinemias. Srpski Arhiv za Celokupno Lekarstvo. 2014;142(3-4):257–260. doi: 10.2298/SARH1404257R. [DOI] [PubMed] [Google Scholar]
- 2.Erlinger S., Arias I. M., Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146(7):1625–1638. doi: 10.1053/j.gastro.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 3.Arias I. M., Gartner L. M., Cohen M., Ezzer J. B., Levi A. Chronic nonhemolytic unconjugated hyperbilirubinemia with glucuronyl transferase deficiency. American Journal of Medicine. 1969;47(3):395–409. doi: 10.1016/0002-9343(69)90224-1. [DOI] [PubMed] [Google Scholar]
- 4.Fujiwara R., Haag M., Schaeffeler E., Nies A. T., Zanger U. M., Schwab M. Systemic regulation of bilirubin homeostasis: potential benefits of hyperbilirubinemia. Hepatology. 2018;67(4):1609–1619. doi: 10.1002/hep.29599. [DOI] [PubMed] [Google Scholar]
- 5.Drenth J. P., Peters W. H., Jansen J. B. From gene to disease; unconjugated hyperbilirubinemia: gilbert's syndrome and Crigler-Najjar types I and II. Nederlands Tijdschrift voor Geneeskunde. 2002;146:1488–1490. [PubMed] [Google Scholar]
- 6.Kraemer D., Scheurlen M. Gilbert disease and type I and II crigler-najjar syndrome due to mutations in the same UGT1A1 gene locus. Medizinische Klinik. 2002;97(9):528–532. doi: 10.1007/s00063-002-1180-6. [DOI] [PubMed] [Google Scholar]
- 7.Sugatani J., Kojima H., Ueda A., et al. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology. 2001;33(5):1232–1238. doi: 10.1053/jhep.2001.24172. [DOI] [PubMed] [Google Scholar]
- 8.Canu G., Minucci A., Zuppi C., Capoluongo E. Gilbert and crigler najjar syndromes: an update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells, Molecules, and Diseases. 2013;50(4):273–280. doi: 10.1016/j.bcmd.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Arias I. M., London I. M. Bilirubin glucuronide formation in vitro; demonstration of a defect in gilbert's disease. Science. 1957;126:563–564. doi: 10.1126/science.126.3273.563. [DOI] [PubMed] [Google Scholar]
- 10.Ritter J. K., Yeatman M. T., Ferreira P., Owens I. S. Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient. The Journal of Clinical Investigation. 1992;90(1):150–155. doi: 10.1172/JCI115829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seppen J., Bosma P. J., Goldhoorn B. G., et al. Discrimination between Crigler-Najjar type I and II by expression of mutant bilirubin uridine diphosphate-glucuronosyltransferase. The Journal of Clinical Investigation. 1994;94(6):2385–2391. doi: 10.1172/JCI117604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosma P. J., Chowdhury J. R., Barker C., et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. The New England Journal of Medicine. 1995;333(18):1171–1175. doi: 10.1056/NEJM199511023331802. [DOI] [PubMed] [Google Scholar]
- 13.Beutler E., Gelbart T., Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proceedings of the National Acadamy of Sciences of the United States of America. 1998;95(14):8170–8174. doi: 10.1073/pnas.95.14.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monaghan G., Ryan M., Seddon R., Hume R., Burchell B. Genetic variation in bilirubin UDP-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet. 1996;347(9001):578–581. doi: 10.1016/S0140-6736(96)91273-8. [DOI] [PubMed] [Google Scholar]
- 15.Bosma P. J. Inherited disorders of bilirubin metabolism. Journal of Hepatology. 2003;38(1):107–117. doi: 10.1016/S0168-8278(02)00359-8. [DOI] [PubMed] [Google Scholar]
- 16.Mikami H., Ogasawara M., Matsubara Y., et al. Molecular analysis of methylmalonyl-CoA mutase deficiency: identification of three missense mutations in mut 0 patients. Journal of Human Genetics. 1999;44(1):35–39. doi: 10.1007/s100380050103. [DOI] [PubMed] [Google Scholar]
- 17.Li L., Deng G., Tang Y., Mao Q. Spectrum of UGT1A1 variations in Chinese patients with crigler-najjar syndrome type II. PLoS ONE. 2015;10, article e0126263(5) doi: 10.1371/journal.pone.0126263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L., Li M., Zhang L., et al. Differences in UGT1A1 gene mutations and pathological liver changes between Chinese patients with gilbert syndrome and crigler-najjar syndrome type II. Medicine. 2017;96, article e8620(45) doi: 10.1097/MD.0000000000008620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crigler J. F., Jr., Najjar V. A. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10(2):169–180. [PubMed] [Google Scholar]
- 20.Crigler J. F., Jr., Najjar V. A. Congenital familial nonhemolytic jaundice with kernicterus; a new clinical entity. A.M.A. American journal of diseases of children. 1952;83(2):259–260. [PubMed] [Google Scholar]
- 21.Owens D., Evans J. Population studies on Gilbert's syndrome. Journal of Medical Genetics. 1975;12(2):152–156. doi: 10.1136/jmg.12.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maruo Y., Addario D. C., Mori A., et al. Two linked polymorphic mutations (A(TA)7TAA and T-3279G) of UGT1A1 as the principal cause of Gilbert syndrome. Human Genetics. 2004;115(6):525–526. doi: 10.1007/s00439-004-1183-x. [DOI] [PubMed] [Google Scholar]
- 23.Sugatani J., Yamakawa K., Yoshinari K., et al. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochemical and Biophysical Research Communications. 2002;292(2):492–497. doi: 10.1006/bbrc.2002.6683. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei I. A., Schmidt S., Peshkin L., et al. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sim N.-L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P. C. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Research. 2012;40(1):W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarz J. M., Cooper D. N., Schuelke M., Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nature Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 27.Neu-Yilik G., Kulozik A. E. NMD: multitasking between mRNA surveillance and modulation of gene expression. Advances in Genetics. 2008;62:185–243. doi: 10.1016/S0065-2660(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi K., Kobayashi Y., Tamaki S., et al. Genetic polymorphisms of bilirubin uridine diphosphate-glucuronosyltransferase gene in Japanese patients with Crigler-Najjar syndrome or Gilbert's syndrome as well as in healthy Japanese subjects. Journal of Gastroenterology and Hepatology. 2004;19(9):1023–1028. doi: 10.1111/j.1440-1746.2004.03370.x. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh S., Wu Y., Lin D., Chu C., Wu M., Liaw Y. Correlation of mutational analysis to clinical features in Taiwanese patients with Gilbert's syndrome. American Journal of Gastroenterology. 2001;96(4):1188–1193. doi: 10.1111/j.1572-0241.2001.03699.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: primer sequences used for amplicons of the UGT1A1 gene. Table S2: patients with variations at two sites. Table S3: patients with variations at three sites. Table S4: patients with variations at four sites. Table S5: patients with variations at five sites.
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its supplementary information files].