

Abstract
O-GlcNAcylation of serine/threonine residues on target proteins occurs dynamically in postmitotic neurons of the hippocampus and may serve to control both the stability and activity of target proteins. Remarkably, the addition and removal of the O-GlcNAc posttranslational modifications are catalyzed by a pair of enzymes, the O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). More than thousands of proteins are modified by O-GlcNAcylation including epigenetic modifying enzymes. A critical target of OGT is the polycomb repressive complex 2 (PRC2) containing the histone lysine methyltransferase EZH2 that mediates trimethylation of lysine 27 on histone H3 (H3K27me3). However, whether OGT and PRC2 activity in the hippocampus couple to regulate gene transcription mechanisms during memory consolidation remains unknown. Here, we found increases in OGT expression and global O-GlcNAcylation levels in dorsal area CA1 of the hippocampus during memory consolidation. Additionally, we observed that OGT exerts control over epigenetic regulation via EZH2-H3K27me3 during memory consolidation. Blocking O-GlcNAc signaling via RNAi within dorsal area CA1 led to the global and site-specific loss of activity-dependent epigenetic plasticity at genes regulated by H3K27me3 and impairment of hippocampus-dependent memory. Together, these findings illustrate a unique epigenetic role of OGT via regulation of histone methylation mediated by EZH2 during memory consolidation of fear conditioned memories.
The posttranslational attachment of β-N-glucosamine (GlcNAc) to serine/threonine residues of proteins (O-GlcNAcylation) is the product of the hexosamine biosynthetic pathway (HBP), which integrates glucose, amino acid, fatty acid, and nucleotide metabolism to generate the donor substrate uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) (Hart and Akimoto 2009; Yang and Qian 2017). O-GlcNAc signaling is highly sensitive to various forms of cellular stress, including heat/cold shock (Kazemi et al. 2010; Yao et al. 2018), hypoxia (Liu et al. 2014), and neuronal excitotoxicity (Zhu et al. 2015). Although the O-GlcNAc axis is only beginning to be described in neuronal alterations, many upstream kinases important for memory consolidation are known to intersect with signaling to OGT and O-GlcNAcylation, including CamKII (Erickson et al. 2013), PKA (Xie et al. 2016), PKC (including PKC-zeta [Miura et al. 2018]), CREB (Rexach et al. 2012; Xie et al. 2016), and many others (Hart et al. 2011). As a result, O-GlcNAcylation has been observed to function as a signal integrator regulating virtually every aspect of cellular function including transcription, translation, signal transduction, and metabolism. Consistent with these observations, disruption of OGT signaling and O-GlcNAcylation processes has been implicated in numerous neurodegenerative diseases, including epilepsy (Sánchez et al. 2019), Alzheimer's disease (Xie et al. 2016), and Parkinson's disease (Wani et al. 2017).
In contrast to other forms of posttranslational glycosylation, which occur largely in the endoplasmic reticulum, O-GlcNAcylation of proteins occurs in the cytoplasm and nucleus (Comer and Hart 2000; Hart and Akimoto 2009). In the nucleus, O-GlcNAcylation occurs on numerous target proteins, consistent with a cellular role as an integrator of signals. In particular, direct O-GlcNAcylation of histones has been reported in both yeast and human cells (Sakabe et al. 2010) and observed to facilitate changes in other epigenetic marks, such as histone monoubiquitination on H2B (Fujiki et al. 2011). Based on these findings, the significance of direct histone O-GlcNAcylation in many mammalian cell types remains in question (Gagnon et al. 2015).
In addition to directly modifying histones and signaling proteins, OGT interacts with and regulates numerous chromatin modifying enzymes (CMEs) including the TET family of DNA hydroxylases (Chen et al. 2013; Dehennaut et al. 2014) and the H3K27 methyltransferase EZH2 (Chu et al. 2014). Learning-induced epigenetic changes mediated by both O-GlcNAc signaling (Lagerlöf and Hart 2014; Wang et al. 2016; Yang et al. 2017; Lagerlöf 2018), as well as OGT-regulated CMEs are critical for long-term memory formation (Jarome and Lubin 2013, 2014); however, the role of OGT in precipitating learning-induced epigenetic changes remains unexplored. In the present study, we observed that OGT is necessary for learning-induced activation of H3K27me3 epigenetic marks and that OGT activity is critical for memory formation.
Results
Fear learning triggers increases in O-GlcNAcylation and OGT protein levels in the hippocampus
We therefore sought to investigate the role of this critical signaling pathway in hippocampus-dependent long-term memory. To ascertain whether the expression of OGT and protein O-GlcNAcylation are dynamically regulated during normal memory formation, we compared hippocampal nuclear lysates from Sprague Dawley rats after training in contextual fear conditioning, a hippocampus-dependent long-term memory paradigm (Fig. 1A). We observed rapidly increased expression of both OGT and protein O-GlcNAcylation, as soon as 1 h, in trained animals relative to naive animals, while animals which received a latent inhibition shock paradigm did not have elevated OGT or O-GlcNAc (Fig. 1B). Similar increases in both OGT and O-GlcNAcylation were observed at 24 h posttraining, suggesting that memory-induced O-GlcNAc signaling persists through the memory consolidation process (Fig. 1C).
Figure 1.
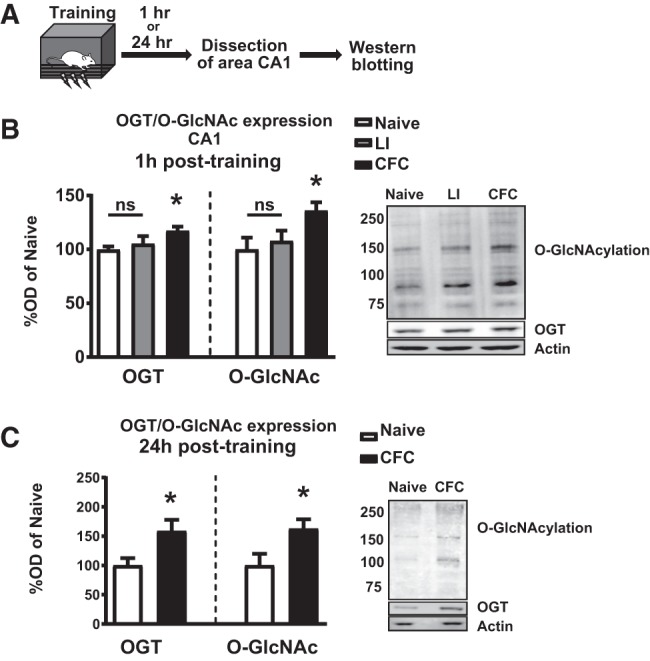
Context fear conditioning leads to persistent up-regulation of OGT and O-GlcNAcylation in the hippocampus. (A) Schematic of training paradigm. (B) Quantification and representative images showing increased OGT (n = 5 for all groups; One-way ANOVA with Fisher's exact test, P = 0.0196) and O-GlcNAc (n = 5 for all groups, One-way ANOVA with Fisher's exact test, P = 0.0274) via western blots 1 h after context fear conditioning but not latent inhibition. (C) Quantification and representative images showing increased OGT (n = 4, 5; Student's t-test, P = 0.0468) and O-GlcNAc (n = 5 for all groups; Student's t-test, P = 0.0396) via western blots 24 h after training in context fear conditioning.
Ogt knockdown prevents memory-induced global increases in O-GlcNAcylation
We next sought to examine the necessity of OGT for long-term memory. To determine whether artificially lowering OGT expression increases global levels of O-GlcNacylation, we directly infused Accell siRNAs (Dharmacon) into the dorsal CA1 and collected tissue around the target infusion site for downstream analysis (Fig. 2A). We confirmed infusion into area CA1 with fluorescently labeled control siRNAs (Fig. 2B). At 5 d postinfusion, we observed significant reductions in both OGT protein expression (Fig. 2C) and OGT mRNA expression in area CA1 but not in the untargeted retrosplenial cortex (Fig. 2D). To verify the sufficiency of siRNA infusions to preclude learning-induced increases in OGT and O-GlcNAc expression, we assayed hippocampal O-GlcNAc and OGT in siRNA-treated animals after training in CFC (Fig. 2E). We observed that animals treated with a nontargeting siRNA had elevated levels of hippocampal O-GlcNAc and OGT after CFC training relative to similarly treated naive controls (Fig. 2F), as observed above in untreated animals (Fig. 1B). Animals treated with OGT siRNAs had significantly reduced expression of hippocampal OGT and O-GlcNAc after training, resembling untrained animals (Fig. 2F). Nup62 expression, an OGT substrate, was not significantly altered during memory consolidation; however, it was significantly down-regulated after treatment with OGT siRNAs (Fig. 2F).
Figure 2.

Knockdown of OGT precludes memory-dependent increases in O-GlcNAcylation. (A) Schematic of intra-CA1 siRNA infusion timeline (B) Fluorescence microscopy images of red-labeled siRNAs. (C) Quantification and representative images of OGT western blots after siRNA knockdown (n = 4 for all groups; Student's t-test, P = 0.0138) (D) Quantification of OGT mRNA after knockdown in CA1 ((n = 4 for all groups; Student's t-test, P = 0.0016) and retrosplenial cortex (negative control; n = 4 for all groups, P = 0.9890) after infusion of siRNA (E) Schematic of intra-CA1 siRNA infusion followed by training in contextual fear conditioning (F) Quantification of OGT, O-GlcNAc, and NUP62 western blots after OGT RNAi and training in context fear conditioning (O-GlcNAc and NUP62: n = 7, 7, 6; One-way ANOVA with Fisher's exact test, * denotes P < 0.05); (OGT: n = 6, 6, 5; One-way ANOVA with Fisher's exact test, * denotes P < 0.05).
Ogt knockdown selectively prevents memory-induced global and site-specific increases in H3K27me3 levels
Several studies have recently reported interactions between O-GlcNAc signaling and other histone posttranslational modifications. Many of these histone marks are known to be important for hippocampal memory formation (Jarome and Lubin 2014). Therefore, we next sought to investigate the potential role of O-GlcNAc signaling on memory-related epigenetic crosstalk. To determine changes in both memory-relevant changes in bulk histone PTMs and site-specific epigenetic alterations, we assayed nuclear histones via both western blots and ChIP-qPCR (Fig. 3A). We observed global changes in expression of numerous memory-related histone PTMs after CFC training, including H2BubiK120, H3K27me3, H3K4me3, and H3K14ac. H3K27me3 was the only mark assayed that was sensitive to pretreatment with OGT siRNA (Fig. 3B). To investigate the necessity of OGT for memory-induced H3K27me3 in vivo in a gene-specific manner, we performed ChIP-qPCR at numerous gene loci. We observed memory-induced decreases in promoter H3K27me3 in several memory-relevant genes, including Bdnf, and Pkc-zeta. These findings are consistent with previously observed increases in Bdnf and Pkc-zeta transcription during memory consolidation, as well as a memory-dependent increase at the promoter of G9a (Fig. 3C). Interestingly, both memory-dependent increases and decreases in promoter H3K27me3 were reversed by pretraining knockdown of OGT (Fig. 3C).
Figure 3.

Context fear conditioning triggers OGT-dependent changes in histone H3 K27 trimethylation. (A) Schematic of intra-CA1 siRNA infusion followed by training in contextual fear conditioning. (B) Quantification of histone western blots 1 h after training in context fear conditioning and OGT knockdown. (H2BubiK120: n = 7, 6, 6; H3K27me3: n = 7, 7, 6; H3K4me3: n = 5, 4, 4; H3K14ac: n = 7, 7, 6); all comparisons made via one-way ANOVA with Fisher's exact test, * denotes P < 0.05). (C) ChIP-qPCR at promoters of memory-related genes 1 h after training in contextual fear conditioning. (G9a: n = 4, 3, 5; Egr1: n = 4, 4, 5; GATA4: n = 4, 5, 5; Bdnf-VI: n = 3, 4, 4; Bdnf-IX: n = 3, 5, 5; Pkm-Zeta: n = 3, 5, 5; Comt: n = 4, 5, 5; Pten: n = 4, 5, 5; all comparisons made via one-way ANOVA with Fisher's exact test, * denotes P < 0.05).
Ogt knockdown regulates EZH2 protein expression levels
Next, we sought to investigate the mechanism by which the infusion of siRNAs significantly repressed the expression of H3K27me3. Western blots confirmed that an obligatory component of the PRC2, the H3K27me3 methyltransferase EZH2, were differentially expressed after manipulation of OGT (Fig. 4A,B). As OGT might act directly either at the level of epigenetic regulation or posttranslationally on the mature EZH2 protein (Sakabe et al. 2010; Chu et al. 2014), we examined the expression of the Ezh2 mRNA via RT-qPCR to determine if Ezh2 was being transcriptionally regulated by OGT. Interestingly, Ezh2 was not differentially expressed at the mRNA level (Fig. 4C) suggesting that transcription of Ezh2 mRNA is intact, and that OGT repressed Ezh2 activity by a posttranscriptional mechanism.
Figure 4.
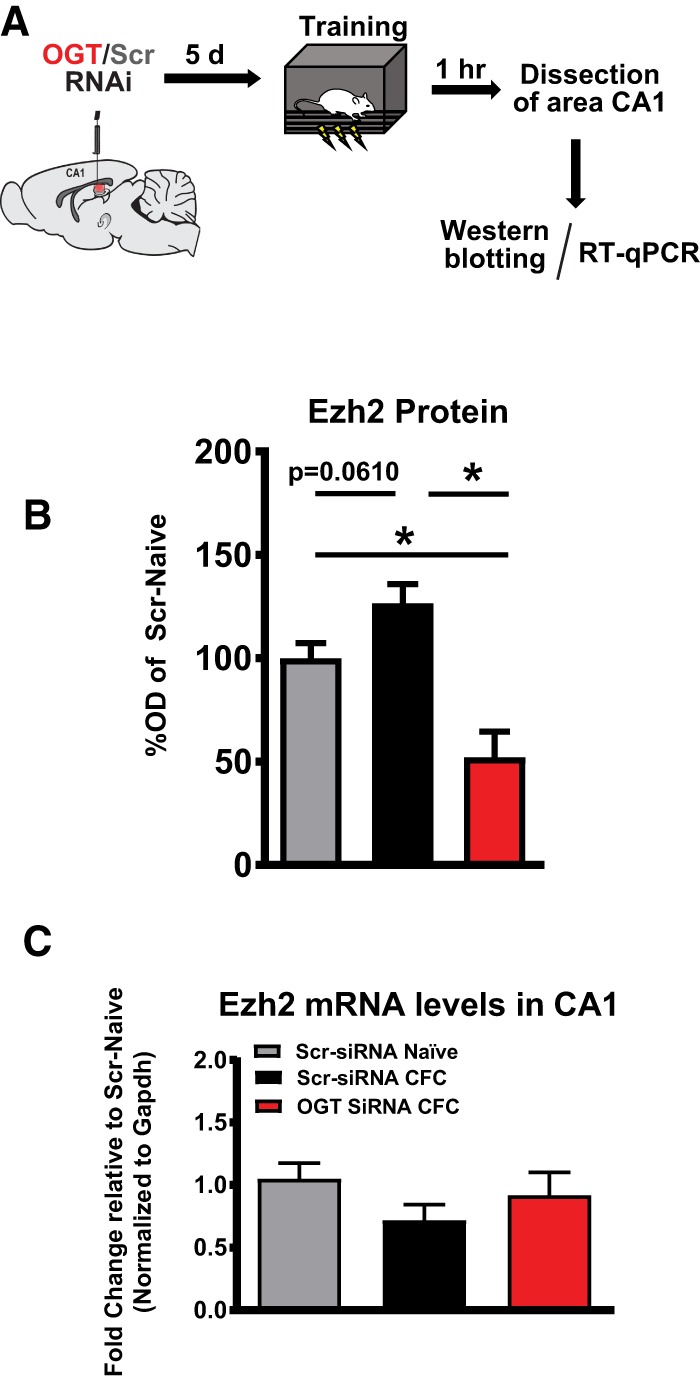
EZH2 protein is dynamically regulated by OGT. (A) Schematic of intra-CA1 siRNA infusion followed by training in contextual fear conditioning. (B) Quantification of EZH2 western blots (n = 6, 6, 5; One-way ANOVA with Fisher's exact test, * denotes P < 0.05) (C) RT-qPCR quantification of (C) Ezh2 mRNA (n = 4, 5, 5; One-way ANOVA with Fisher's exact test).
OGT-dependent induction of Ezh2 is necessary for contextual fear memory
Genetic models have previously demonstrated that forebrain expression of OGT is necessary for memory formation (Wang et al. 2016). These findings led to our investigation of whether intrahippocampal posttranscriptional knockdown of OGT expression is sufficient to regulate long-term memory formation (Fig. 5A). We observed no differences in freezing behavior during CFC training; however, upon returning animals to the training context after 24 h we observed a significant decrease in the freezing response of male rats, indicating impairments in long-term memory (Fig. 5B,C).
Figure 5.
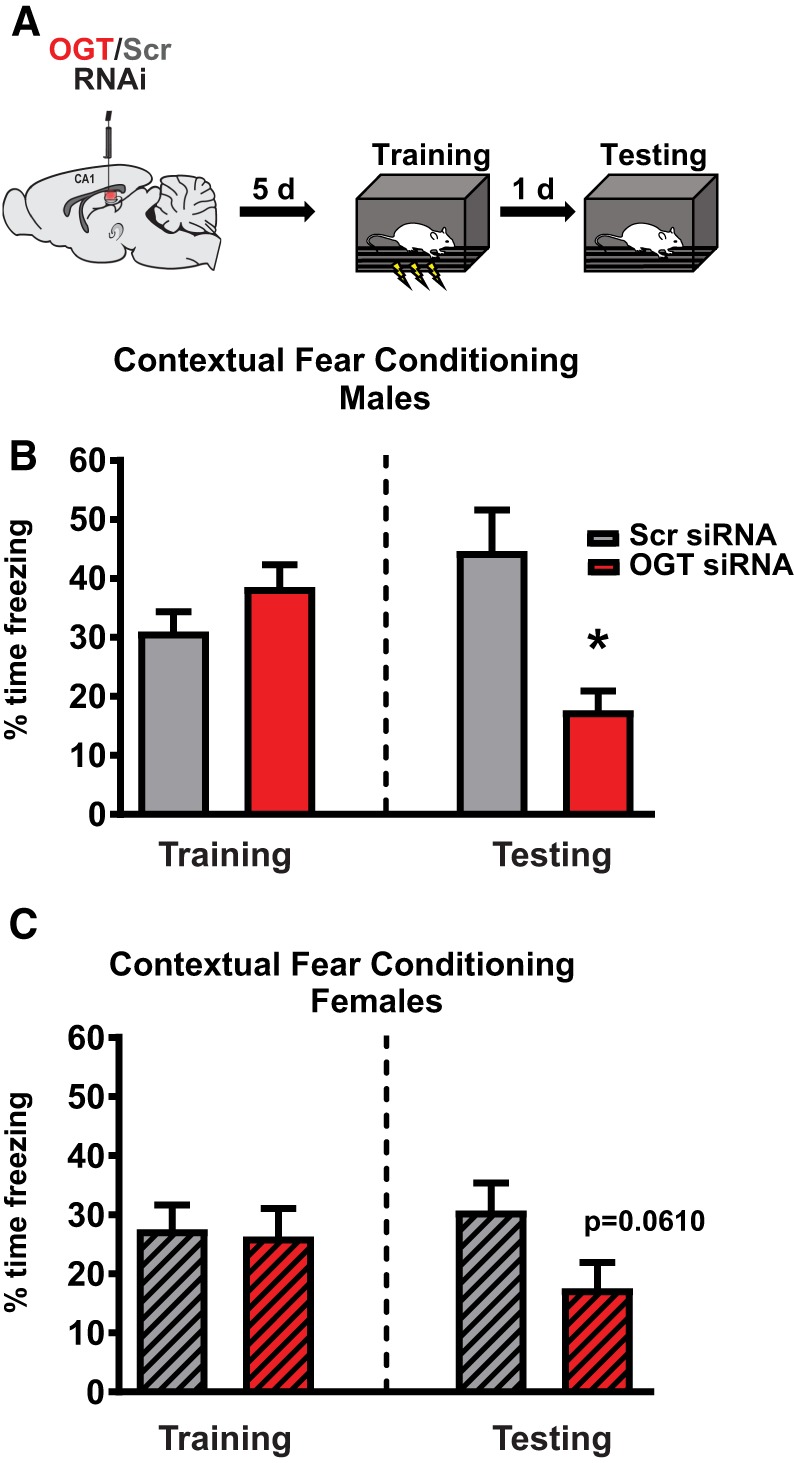
Adult hippocampal expression of Ogt and Ezh2 are necessary for hippocampus-dependent memory formation. (A) Schematic of intra-CA1 Ogt siRNA infusion followed by contextual fear conditioning. (B) Performance of male rats in training phase (n = 6, 7; P = 0.1673) and testing phase (n = 6, P = 0.0057) of contextual fear conditioning after Ogt knockdown. (C) Performance of unsynchronized female rats in training phase (n = 7, 9; P = 0.8516) and testing phase (n = 7; P = 0.0610) of contextual fear conditioning after Ogt knockdown.
Materials and Methods
Subjects
Male and/or female Sprague Dawley rats (Harlan) weighing 250–300 g at the time of arrival were used for all experiments. Rats were singly housed in plastic cages, had free access to water and standard chow, and were maintained on a 12 h light–dark cycle. All procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee and performed in accordance with the National Institutes of Health ethical guidelines.
Chromatin immunoprecipitation
Chromatin immunoprecipitations (ChIP) were performed as described previously (Jarome et al. 2015; Morse et al. 2015). CA1 tissue was fixed in phosphate-buffered saline (PBS) with 1% formaldehyde for 10 min at 37°C and washed extensively in PBS. Samples were homogenized in hypotonic buffer (10 mM KCl, 20 mM HEPES, 1 mM, MgCl, 1 mM DTT) with protease inhibitors and centrifuged at 1350g for 10 min at 4°C to pellet nuclei. Pellets were resuspended in ChIP sonication buffer (1× TE with 1% SDS) with protease inhibitors and chromatin sheared to 300 bp using 25 cycles on a Bioruptor at high power. Lysates were centrifuged at 20,000g for 10 min at 4°C to pellet debris and supernatant collected and DNA content measured using a Nanodrop spectrophotometer. Normalized amounts of DNA were diluted in TE buffer and 2X RIPA buffer (2× PBS, 1% sodium deoxycholate, 2% NP-40, 0.2% SDS) with 2× proteasome inhibitors. Samples were added to Magna ChIP Protein A/G magnetic beads (Millipore). Immunoprecipitations were carried out at 4°C overnight with primary antibody (H3K27me3) or no antibody (control). Immune complexes were collected and washed with low salt buffer (20 mM Tris, pH 8.0, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl), high salt buffer (20 mM Tris, pH 8.1, 0.1% SDS, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA), LiCl immune complex buffer (0.25 M LiCl, 10 mM Tris, pH 8.1, 1% deoxycholic acid, 1% IGEPAL-CA630, 500 mM NaCl, 2 mM EDTA), and twice with TE buffer. Immune complexes were extracted in TE containing 1% SDS and sodium bicarbonate and protein–DNA cross-links were reverted by heating at 65°C overnight. After proteinase K digestion (100 µg; 2 h at 37°C), DNA was extracted by phenol/chloroform/isoamyl alcohol and then ethanol-precipitated. All DNA samples were subjected to quantitative real-time polymerase chain reaction (PCR) using primers specific to the gene of interest. The cycle threshold for each amplicon was taken as a percentage of the input fraction, enrichment over background (no antibody control) was calculated as a fold change of the control group. For western blot analysis of ChIP samples, cross-linking was reverted by heating the samples for 5 min at 95°C and normalized amounts of protein were loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) as described in the western blotting methods below.
Surgery and siRNA infusions
Rats were infused intra-CA1 with Accell siRNAs (Dharmacon) as described previously (Jarome et al. 2015). Fresh aliquots of Accell Ezh2 (catalog #E-094880-00-0005), OGT (catalog #E-080125-00-0005), and nontargeting control (catalog #D-001910-10-05) siRNA stocks (100 µm) (Dharmacon) were resuspended in Accell siRNA resuspension buffer to a concentration of ∼4.5 µm on the day of surgery. A nontargeting red fluorescent Accell siRNA (catalog #D-001960-01-05, Dharmacon) was used to confirm targeting of the CA1 region.
Behavioral procedures
Rats were handled for 3 d followed by 3 d of acclimation to the transport procedure before the start of behavioral training. Rats were then trained to a standard contextual fear-conditioning paradigm in which three shock presentations (0.6 mA, 2 sec, 120 sec Intertrial interval) were delivered over a 7 min period in a novel context. Following the completion of training, rats were returned to their home cages. Testing occurred the following day and consisted of a 5 min exposure to the training context. All behavior was recorded and scored by Med Associates software (Freeze Scan).
Tissue collection
Animals were decapitated, the brain rapidly removed and immediately frozen on dry ice. The CA1 region of the dorsal hippocampus was then dissected using a rat brain matrix (Harvard Apparatus) and subsequently flash-frozen. Tissue was then subjected to histone extraction, nuclear extraction, RNA extraction or chromatin immunoprecipitation protocols as described below. In cases where the tissue was used for both RNA and histone analyses, tissue was first homogenized in the sucrose buffer described in the histone extraction section and then split into two equal parts. One part was used for subsequent histone extraction and the other part was used for RNA extraction. For tissue used for both histone and ChIP analyses, dorsal CA1 hemispheres were dissected and placed into separate tubes. One hemisphere was used for histone extractions and the other for ChIP to account for possible laterality effects. The left or right hemisphere that was used for each procedure (histones or ChIP) was counterbalanced for all groups.
Western blots
Histones were extracted using previously published protocols (Jarome et al. 2015). Briefly, samples were homogenized in sucrose buffer (320 mM) with protease and phosphatase inhibitors. Homogenized samples were centrifuged at 7700g for 1 min and the supernatant collected. This supernatant was used for the analysis of OGT and protein O-GlcNAcylation levels. The resulting pellet was resuspended in 0.4N sulfuric acid and incubated for 30 min on ice, followed by centrifuge at max speed for 10 min. The supernatant was collected and trichloroacetic acid with 4 mg/mL deoxycholic acid was added. Samples were incubated for 30 min on ice, centrifuged, and histones collected by acetone drying. All protein samples were normalized using the Biorad Bradford protein assay.
RT-qPCR
RNA and DNA were extracted from area CA1 using the Qiagen Allprep Kit. RNA (200 ng) was converted to cDNA using the iScript cDNA synthesis kit (Biorad) and reverse transcriptase-polymerase chain reaction (RT-PCR) amplifications for OGT and Ezh2 were performed on the CFX96 real-time PCR system (Biorad). RT-qPCR amplifications were performed at 95°C for 3 min, 50 repeats of 95°C for 10 sec followed by 62.6°C for 30 sec, 95°C for 1 min, 55°C for 1 min and finally held at 4°C. Quantification was performed relative to Gapdh; all data were analyzed using the comparative Ct method.
Antibodies
Antibodies used for western blotting include H2BubiK120 (1:1000, Cell Signaling #5546), H2B (1:1000, Cell Signaling #12364), H3K4me3 (1:500, Millipore #04-745), H3K9me2 (1:500, Millipore #07-441), H3K27me3 (1:1000, Millipore #07-449), H3ac (1:1000, Millipore #06-599), H3 (1:1000, Abcam #ab1791), and Actin (1:1000, Abcam #ab1801). For ChIP studies, H3K27me3 antibodies were used at a concentration of 5 µg per sample.
Statistical analyses
Statistical outliers were determined as those that fell two more standard deviations above/below the group mean and were determined using the Outlier function in Prism software. For molecular data (PCR, westerns), outliers were removed from the final analysis on a case-by-case basis (i.e., based on individual primers, antibodies, etc). For behavioral data, if an animal was an outlier during either training or testing, they were removed from both analyses since the two measures are not independent. All data are presented as the group average with the standard error of the mean and were analyzed using analysis of variance (ANOVA) and Fisher least significant difference (LSD) post-hoc tests or pairwise t-tests as indicated in the figure legends.
Discussion
Protein O-GlcNAcylation functions as an intersection of cell signaling pathways, integrating and controlling diverse aspects of cellular function. While mechanisms of crosstalk between OGT and epigenetic mechanisms have been reported, the relevance of hippocampal OGT expression and OGT-dependent epigenetic regulation for memory formation have yet to be explored. In the present manuscript, we used in vivo posttranscriptional knockdown of OGT via RNAi to investigate the role of hippocampal O-GlcNAc signaling in regulatory control of neuroepigenetic mechanisms. Our group and others have previously reported that expression of the H3K27me3 methyltransferase Ezh2 is critical for performance in hippocampus-dependent memory tasks. In these experiments, we observed that OGT expression is necessary for memory-induced changes in global and site-specific H3K27me3, and that disruption of OGT similarly impairs performance on contextual fear conditioning. The observations that OGT regulates Ezh2 expression (Fig. 4) and H3K27me3 in vivo (Fig. 3) are largely consistent with previous molecular characterizations of an OGT-EZH2 interaction carried out in cultured human cells (Chu et al. 2014); however, we did not observe O-GlcNAcylation effects on expression of H3K9 acetylation/methylation or H3K4 methylation in vivo, as has been previously reported in cultured cells (Zhang et al. 2011).
In the present study, we observed impaired performance in contextual fear conditioning after reducing O-GlcNAcylation via knockdown of OGT. While the present study has achieved some degree of temporal specificity over genetic models, an important consideration given the important developmental roles of OGT (Howerton et al. 2013), future studies should achieve both greater temporal control and cell-type specificity. Furthermore, the reliance of specific histone methylation modifications on a single writer and eraser enzyme, suggests that feedback mechanisms exist that are sensitive to the flux of O-GlcNAcylation. For example, published evidence suggests that feedback mechanisms might involve miR-101 (Jiang et al. 2019), which targets both OGT and EZH2. While we did not observe a change in abundance of the Ezh2 transcript in vivo, evidence from published work suggests that miRNA-mediated posttranscriptional silencing mediates homeostatic control over Ezh2 protein abundance, and moreover, that direct O-GlcNAcylation of EZH2 increases protein stability and mediate increased H3K27me3 (Chu et al. 2014). Nonetheless, our studies confirm a dynamic role for OGT in the regulation of repressive histone lysine methylation during memory formation.
In sum, the present study and other studies in this field are at the beginning stages. As a critical hub in multiple signaling cascades, OGT has been observed to impact numerous memory-relevant signaling mechanisms and likely has a multivalent impact on memory formation. For example, while evidence from the experiments described here suggests that global OGT expression is necessary for long-term memory formation, the activity-induced inhibitory O-GlcNAcylation of CREB at S40 has been reported to act as a molecular brake on both short and long-term memory (Rexach et al. 2012). Thus, in moving forward it will be critical to consider and continue to elucidate the complexities of O-GlcNAcylation in cell signaling.
Importantly, our findings identify another major epigenetic transcriptional mechanism that can be manipulated to rescue or mitigate memory dysfunction associated with numerous neurological disorders. For example, memory deficits are common in patients with epilepsy, especially temporal lobe epilepsy, where memory-related brain structures such as the hippocampus are directly involved in the processes of epileptogenesis (Tramoni-Negre et al. 2017). We have recently reported that O-GlcNAcylation and OGT expression are depleted in the hippocampi of the kainate temporal lobe epilepsy rat model, as well as in human patients with temporal lobe epilepsy (Sánchez et al. 2019). Thus, the idea of OGT signaling as a therapeutic target, merits further discussion, as does the impact of O-GlcNAc modification on epigenetic regulation of gene transcription and hippocampus-dependent long-term memory formation.
Competing interest statement
The authors have no conflicts of interest.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) Grants MH-097909 and NS-090250 to F.D.L., and by the Evelyn F. McKnight Brain Institute at the University of Alabama at Birmingham (UAB). A.A.B. was supported by NIH Institutional training grant NSO61788.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.049023.118.
References
- Chen Q, Chen Y, Bian C, Fujiki R, Yu X. 2013. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493: 561–564. 10.1038/nature11742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C-S, Lo P-W, Yeh Y-H, Hsu P-H, Peng S-H, Teng Y-C, Kang M-L, Wong CL, Juan L-J. 2014. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci 111: 1355–1360. 10.1073/pnas.1323226111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer FI, Hart GW. 2000. O-Glycosylation of nuclear and cytosolic proteins. Dynamic interplay between O-GlcNAc and O-phosphate. J Biol Chem 275: 29179–29182. 10.1074/jbc.R000010200 [DOI] [PubMed] [Google Scholar]
- Dehennaut V, Leprince D, Lefebvre T. 2014. O-GlcNAcylation, an epigenetic mark. Focus on the histone code, TET family proteins, and polycomb group proteins. Front Endocrinol (Lausanne) 5: 155 10.3389/fendo.2014.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, et al. 2013. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502: 372–376. 10.1038/nature12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, et al. 2011. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480: 557–560. 10.1038/nature10656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon J, Daou S, Zamorano N, Iannantuono NVG, Hammond-Martel I, Mashtalir N, Bonneil E, Wurtele H, Thibault P, Affar EB. 2015. Undetectable histone O-GlcNAcylation in mammalian cells. Epigenetics 10: 677–691. 10.1080/15592294.2015.1060387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Akimoto Y. 2009. The O-GlcNAc modification. In Essentials of glycobiology, 2nd ed. (Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, et al. ). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. 2011. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem 80: 825–858. 10.1146/annurev-biochem-060608-102511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howerton CL, Morgan CP, Fischer DB, Bale TL. 2013. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci 110: 5169–5174. 10.1073/pnas.1300065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Lubin FD. 2013. Histone lysine methylation: critical regulator of memory and behavior. Rev Neurosci 24: 375–387. 10.1515/revneuro-2013-0008 [DOI] [PubMed] [Google Scholar]
- Jarome TJ, Lubin FD. 2014. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiol Learn Mem 115: 116–127. 10.1016/j.nlm.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Butler AA, Nichols JN, Pacheco NL, Lubin FD. 2015. NF-κB mediates Gadd45β expression and DNA demethylation in the hippocampus during fear memory formation. Front Mol Neurosci 8: 54 10.3389/fnmol.2015.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Xu B, Li X, Shang Y, Chu Y, Wang W, Chen D, Wu N, Hu S, Zhang S. 2019. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene 38: 301–316. 10.1038/s41388-018-0435-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemi Z, Chang H, Haserodt SA. 2010. O-linked β-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3β-dependent manner. J Biol Chem 285: 39096–39107. 10.1074/jbc.M110.131102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerlöf O. 2018. O-GlcNAc cycling in the developing, adult and geriatric brain. J Bioenerg Biomembr 50: 241–261. 10.1007/s10863-018-9760-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerlöf O, Hart GW. 2014. O-GlcNAcylation of neuronal proteins: roles in neuronal functions and in neurodegeneration. Adv Neurobiol 9: 343–366. 10.1007/978-1-4939-1154-7_16 [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Z, Yu S, Xu J. 2014. Proteasomal degradation of O-GlcNAc transferase elevates hypoxia-induced vascular endothelial inflammatory response. Cardiovasc Res 103: 131–139. 10.1093/cvr/cvu116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Kume M, Kawamura T, Yamamoto K, Hamakubo T, Nishihara S. 2018. O-GlcNAc on PKCζ inhibits the FGF4-PKCζ-MEK-ERK1/2 pathway via inhibition of PKCζ phosphorylation in mouse embryonic stem cells. Stem Cell Rep 10: 272–286. 10.1016/j.stemcr.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse SJ, Butler AA, Davis RL, Soller IJ, Lubin FD. 2015. Environmental enrichment reverses histone methylation changes in the aged hippocampus and restores age-related memory deficits. Biology 4: 298–313. 10.3390/biology4020298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC. 2012. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol 8: 253–261. 10.1038/nchembio.770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K, Wang Z, Hart GW. 2010. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc Natl Acad Sci 107: 19915–19920. 10.1073/pnas.1009023107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez RG, Ryley Parrish R, Rich M, Webb WM, Lockhart RM, Nakao K, Ianov L, Buckingham SC, Broadwater DR, Jenkins A, et al. 2019. Human and rodent temporal lobe epilepsy is characterized by changes in O-GlcNAc homeostasis that can be reversed to dampen epileptiform activity. Neurobiol Dis 124: 531–543. 10.1016/j.nbd.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramoni-Negre E, Lambert I, Bartolomei F, Felician O. 2017. Long-term memory deficits in temporal lobe epilepsy. Rev Neurol 173: 490–497. 10.1016/j.neurol.2017.06.011 [DOI] [PubMed] [Google Scholar]
- Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. 2016. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci 113: 15120–15125. 10.1073/pnas.1606899113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Ouyang X, Benavides GA, Redmann M, Cofield SS, Shacka JJ, Chatham JC, Darley-Usmar V, Zhang J. 2017. O-GlcNAc regulation of autophagy and α-synuclein homeostasis; implications for Parkinson's disease. Mol Brain 10: 32 10.1186/s13041-017-0311-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Jin N, Gu J, Shi J, Sun J, Chu D, Zhang L, Dai CL, Gu JH, Gong CX. 2016. O-GlcNAcylation of protein kinase A catalytic subunits enhances its activity: a mechanism linked to learning and memory deficits in Alzheimer's disease. Aging Cell 15: 455–464. 10.1111/acel.12449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Qian K. 2017. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18: 452–465. 10.1038/nrm.2017.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YR, Song S, Hwang H, Jung JH, Kim S-J, Yoon S, Hur JH, Park JI, Lee C, Nam D, et al. 2017. Memory and synaptic plasticity are impaired by dysregulated hippocampal O-GlcNAcylation. Sci Rep 7: 44921 10.1038/srep44921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Yang Y, Lian S, Shi H, Liu P, Liu Y, Yang H, Li S. 2018. Effects of acute cold stress on liver O-GlcNAcylation and glycometabolism in mice. Int J Mol Sci 19: 9 10.3390/ijms19092815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Roche K, Nasheuer H-P, Lowndes NF. 2011. Modification of histones by sugar β-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. J Biol Chem 286: 37483–37495. 10.1074/jbc.M111.284885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Tao T, Zhang D, Liu X, Ke K, Shen A. 2015. NOS1AP O-GlcNAc modification involved in neuron apoptosis induced by excitotoxicity. Int J Mol Sci 16: 16560–16575. 10.3390/ijms160716560 [DOI] [PMC free article] [PubMed] [Google Scholar]