

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is associated with robust activity of the coagulation system. To determine mechanisms by which clotting factors influence PDAC tumor progression, we generated and characterized C57B1/6-derived KPC (KRasG12D, TRP53R172H) cell lines. Tissue factor (TF) and protease-activated receptor-1 (PAR-1) were highly expressed in primary KPC pancreatic lesions and KPC cell lines similar to expression profiles observed in biopsies of patients with PDAC. In allograft studies, tumor growth and metastatic potential were significantly diminished by depletion of TF or Par-1 in cancer cells or by genetic or pharmacologic reduction of the coagulation zymogen prothrombin in mice. Notably, PAR-1–deleted KPC cells (KPC-Par-1KO) failed to generate sizable tumors, a phenotype completely rescued by restoration of Par-1 expression. Expression profiling of KPC and KPC-Par-1KO cells indicated that thrombin–PAR-1 signaling significantly altered immune regulation pathways. Accordingly, KPC-Par-1KO cells failed to form tumors in immune-competent mice but displayed robust tumor growth comparable to that observed with control KPC cells in immune-compromised NSG mice. Immune cell depletion studies indicated that CD8 T cells, but not CD4 cells or natural killer cells, mediated elimination of KPC-Par-1KO tumor cells in C57Bl/6 mice. These results demonstrate that PDAC is driven by activation of the coagulation system through tumor cell–derived TF, circulating prothrombin, and tumor cell–derived PAR-1 and further indicate that one key mechanism of thrombin/PAR-1–mediated tumor growth is suppression of antitumor immunity in the tumor microenvironment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly fatal disease with an average 5-year survival rate of 8% (1). The aggressive and invasive nature of the malignancy and poor diagnostic tools contribute to the extreme mortality rate since most patients present with late-stage disease. Although the majority of PDAC tumors harbor activating KRAS and dominant-negative P53 mutations (2), directly targeting KRAS or TP53 has proven to be challenging as a treatment paradigm. The dismal survival profiles highlight the urgency to better understand the molecular mechanisms behind tumor growth and metastasis for developing effective therapeutic strategies.
PDAC has the highest rate of cancer-associated venous thromboembolism (VTE; ref. 3) and VTE is decidedly correlated with disease aggressiveness (4). Unfortunately, the molecular interplay between thrombosis and PDAC is not fully understood. What is known is that PDAC tumor cells express high levels of tissue factor (TF) and patients with high TF exhibit increased rates of VTE (5). Elevated plasma TF activity also has been observed in patients with PDAC, again correlating with increased incidence of thrombosis (6). Notably, high TF expression is driven by the same KRAS and P53 mutations that initiate cellular transformation and tumor progression (7).
TF expression by tumor cells serves as a critical link between cancer and cancer-associated thrombosis (8). The assumption that the pathologic role of the TF–thrombin axis in cancer is limited to thrombosis has been replaced by the belief that TF, thrombin, and downstream targets may also promote cancer progression (9). TF levels correlate with disease histologic grade, and high TF expression in tumor specimens is an important negative predictor of PDAC patient survival (6, 10). Thrombin can also drive multiple aspects of tumor biology. Pharmacologic reduction of thrombin limits colon cancer severity and thrombin inhibitors can block metastasis of various cancer cell lines (e.g., fibrosarcomas, lung carcinomas; refs. 11, 12). Despite data indicating that the TF-thrombin axis may promote cancer progression, the identification of specific mechanisms and thrombin targets [e.g., fibrinogen, protease-activated receptor (PAR)-1, −3, −4] largely remain open questions.
We sought to determine the potential role of thrombin signaling via tumor cell–derived PARs in PDAC progression. Through multiple genetic and pharmacologic approaches, our study demonstrates that PAR-1 expressed by PDAC tumor cells serves as a critical downstream effector of the TF–thrombin cascade to promote tumor growth and metastasis. More importantly, a key mechanism by which PAR-1 promotes pancreatic cancer progression appears to be linked to suppression of the host immune system, underlining a novel connection between the coagulation signaling cascade and antitumor immunity.
Materials and Methods
Cell culture, plasmid constructs, shRNA interference, and CRISPR gene editing
C57Bl/6 mouse pancreatic acinar cells (Ac) were isolated as described previously (13). KPC tumor cell lines were generated from individual primary tumors derived from KPC (KrasG12D/+, p53R172H/+, ElasCreER/+) mice at Purdue University. Cells were maintained in RPMI1640 medium containing 10% FBS and 1% penicillin/streptomycin. Lentivirus encoding shTF and shPAR-1 were obtained from Sigma (TF: TRCN0000375847 and TF B: TRCN0000366885; PAR-1: TRCN0000226149). The dimeric CRISPR RNA-guided Fokl nuclease strategy was used to edit the murine Par-1 gene as described previously (14). Stable doxycycline (Dox) inducible, re-expression clones were generated using the mouse Par-1 open reading frame with a C-terminal Myc epitope tag cloned into the Tet-One plasmid (Clontech, 634301). All cell lines were genetically authenticated by the ATCC and pathogen-tested by IDEXX Laboratories. Results of pathogen testing, including mycoplasma analysis, were negative.
Subcutaneous or orthotopic tumor growth and lung metastasis assays
For subcutaneous tumor studies cells were injected in the intrascapular region at a concentration of 2.5 × 105 in 100 μL sterile PBS. Tumors were measured over time and tumor volume was calculated as: Volume = (Length × Width2)/2. Orthotopic injections were performed at a concentration of 5 × 104 cells in 20 μL sterile PBS. In experiments of pharmacological targeting of prothrombin, C57Bl/6 mice received weekly intraperitoneal injections of 50 mg/kg F2 antisense oligonucleotide (ASO) “gapmer” (5′-attccatagtgtaggtcctt-3′) in 200 μl of sterile PBS or a control ASO (5′-ccttccctgaaggttcctcc-3′) for a total of 3 weeks prior to subcutaneous tumor cell injection and during the course of tumor growth. In the experiments of activating Tet-PAR-1 expression in vivo, mice were provided Dox chow (ENVIGO, TF 08541) with food replaced every 6 days. For experimental metastasis assays, 5 × 104 cells in 200 μL PBS were administered by tail vein injection. Three weeks after injection the lung tissue was harvested, weighed, and analyzed by histology. Immune cell depletion was performed using targeting monoclonal antibodies to CD8a (clone 2.43), CD4 (clone GK1.5), or natural killer (NK) cells (clone PK136) injected intraperitoneally twice weekly starting 1 day prior to the injection of KPC tumor cells. Successful depletion (>95% loss) of immune cell subpopulations was confirmed at the time of tumor harvest by flow cytometry of mouse splenocytes using antibodies APC-CD4 (L3T4; eBioscience), Alexa Flour 488-CD8a (Clone: 53-6.7; Biolegend), FITC-NK1.1 (Clone PK136; eBioescience), and PE-CD49b (DX5; BD Pharmingen). All procedures described above were conducted following the Guide for the Care and Use of Laboratory Animals of the NIH and approved by the Cincinnati Children’s Hospital Medical Center or the Purdue University IACUC Committees.
In vitro analysis of tumor cells and tumors
Statistical analysis
qRT-PCR, tumor mass, and lung mass data were analyzed by unpaired Student t test, Welch correction was applied when unequal variances were presented between groups. Cell growth doubling time and colony numbers were analyzed using one-way ANOVA. Tumor volumes and fluorescence intensity over time data were analyzed by repeated measures ANOVA or stated otherwise. All data are presented as the mean ± SEM. The statistics of mouse survival was calculated by the Kaplan–Meier log rank analysis with Chi square = 26.76. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
Results
TF expression by transformed cells contributes to PDAC tumor growth
PDAC patient biopsies revealed high TF protein levels in epithelial cells of early PanIN lesions and mature PDAC tumors (Fig. 1A; Supplementary Fig. S1), whereas TF protein was not detected in acinar or duct cells of histologically normal pancreas tissue (Fig. 1A). Numerous cancer gene expression datasets document high TF expression in pancreatic cancer and high TF expression correlates with significantly poorer patient survival (Supplementary Fig. S2). To determine if the same pattern occurs in a mouse PDAC model, we examined pancreas tissue of “KPC” mice (KrasG12D/+, p53R172H/+, ElasCreER/+) that develop PDAC disease, which mimics the genetic, biochemical, immunologic, and histologic features of PDAC in patients (15, 16). Similar to PDAC patient tissue, robust TF staining was observed in epithelial cells of mouse PanIN and PDAC lesions but was absent in wild-type (WT) pancreas cells (Fig. 1B). Similar high TF expression was observed in “KPC” pancreata in which the Cre-expression was driven by the Pdx-1 promoter (Supplementary Fig. S3). As predicted, high TF transcript levels were also detected in KPC-derived PanIN and PDAC tissues with little to no detection of TF transcripts in normal WT pancreas or isolated WT Ac (Fig. 1C).
Figure 1.
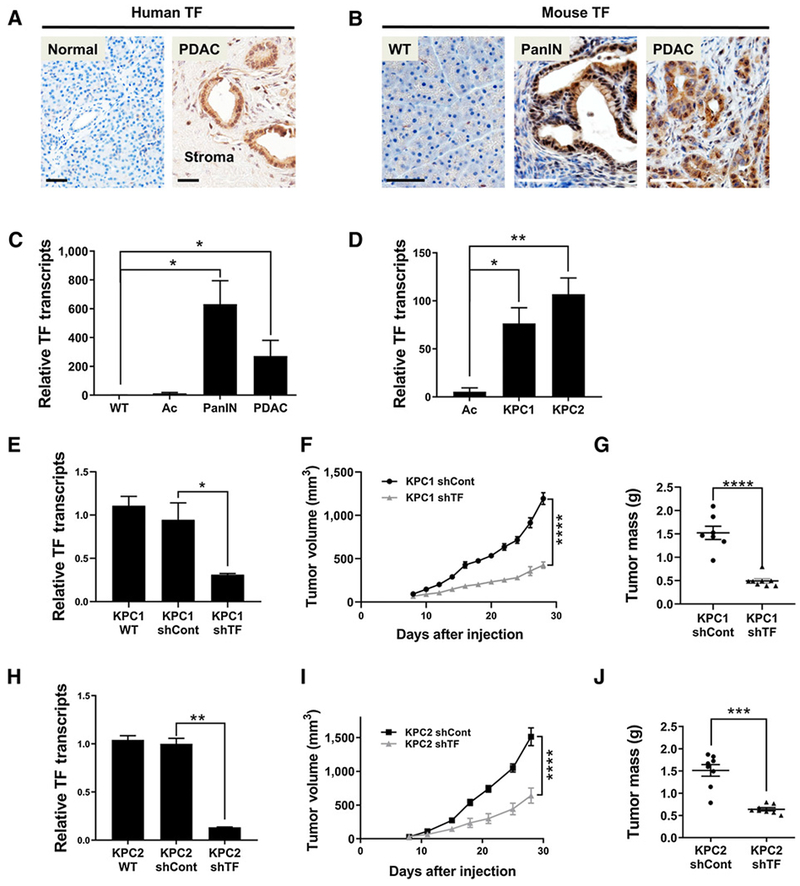
TF expression by transformed pancreatic cells contributes to PDAC tumor growth. A, Human TF IHC (brown) in normal pancreas and PDAC patient samples. B, Mouse TF IHC in normal pancreas (WT) and in PanIN lesions and PDAC tumors in the KPC model. C, qRT-PCR analysis of mouse TF transcripts in WT pancreas, isolated Ac, PanIN, and PDAC tissue (one-tailed t test, N = 3 per group). D, qRT-PCR analysis of TF transcripts in isolated Ac preparations and in the mouse PDAC tumor cell lines KPC1 and KPC2 (N = 3 per group). E, qRT-PCR of TF transcripts in KPC1 WT, KPC1 shControl, and KPC1 shTF “knockdown” cell lines. F and G, Analysis of tumor growth following subcutaneous injection of KPC1 shControl and KPC1 shTF lines in WT mice (N = 8 mice per group). H, qRT-PCR of TF transcripts in KPC2 WT, KPC2 shControl, and KPC2 shTF “knockdown” cell lines. I and J, Analysis of tumor growth following subcutaneous injection of KPC2 shControl and KPC2 shTF lines in WT mice (N = 8 mice per group). Tumor volume was measured over time and tumor mass quantified at 4 weeks of growth. Scale bars, 50 μm (A and B). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Tumor cell lines (termed KPC1, KPC2) were generated from 2 separate KPC tumor bearing mice. High TF gene expression was maintained in these lines relative to normal pancreatic Ac (Fig. 1D). To analyze TF function, stable TF-specific shRNA knockdown KPC lines were generated. shTF lines exhibited an approximate 90% reduction in TF transcript levels, whereas KPC1 or KPC2 cells expressing control shRNAs showed no change in TF expression (Fig. 1E and H; Supplementary Fig. S4A). Notably, KPC1 and KPC2 shControl cells and shTF cells were morphologically indistinguishable and had virtually identical proliferation rates. In contrast, subcutaneous tumor growth in immunocompetent C57Bl/6 mice revealed a dramatic decrease in tumor volume over time for both the KPC1 shTF cells and the KPC2 shTF cells when compared with the KPC shControl cells (Fig. 1F and I; Supplementary Fig. S4B), resulting in a 2.5-fold reduction in final tumor mass (Fig. 1G and J; Supplementary Fig. S4C). Together, these results suggest that TF expression is critical in promoting PDAC tumor progression.
Prothrombin promotes PDAC tumor growth
To test whether TF primarily promotes PDAC tumor growth by driving thrombin activation within the tumor microenvironment (TME), we evaluated tumor growth of KPC2 cells in genetically modified mice (termed flllow) that express 10% of normal circulating prothrombin levels (17). KPC2 tumors were significantly smaller in flllow mice when compared with WT mice (Fig. 2A and B). As a complementary approach, prothrombin expression was pharmacologically reduced (Fig. 2C) using a highly selective murine prothrombin ASO (18). As expected, a control ASO had no effect on KPC2 tumor growth, whereas lowering prothrombin levels with the prothrombin ASO resulted in a significantly reduced KPC2 tumor mass (Fig. 2D). To determine whether reduction of prothrombin could limit tumor growth in the pancreas, orthotopic injections of KPC2 cells were performed. Here, reduction of circulating prothrombin by prothrombin ASO after injection of the tumor cells resulted in significantly smaller KPC2 tumors at 21 days (Fig. 2E and F). Thus, genetically (flllow) or pharmacologically (ASO) reducing prothrombin levels significantly diminishes KPC tumor growth even in contexts in which prothrombin reduction occurs following the seeding of tumor cells within the pancreas.
Figure 2.
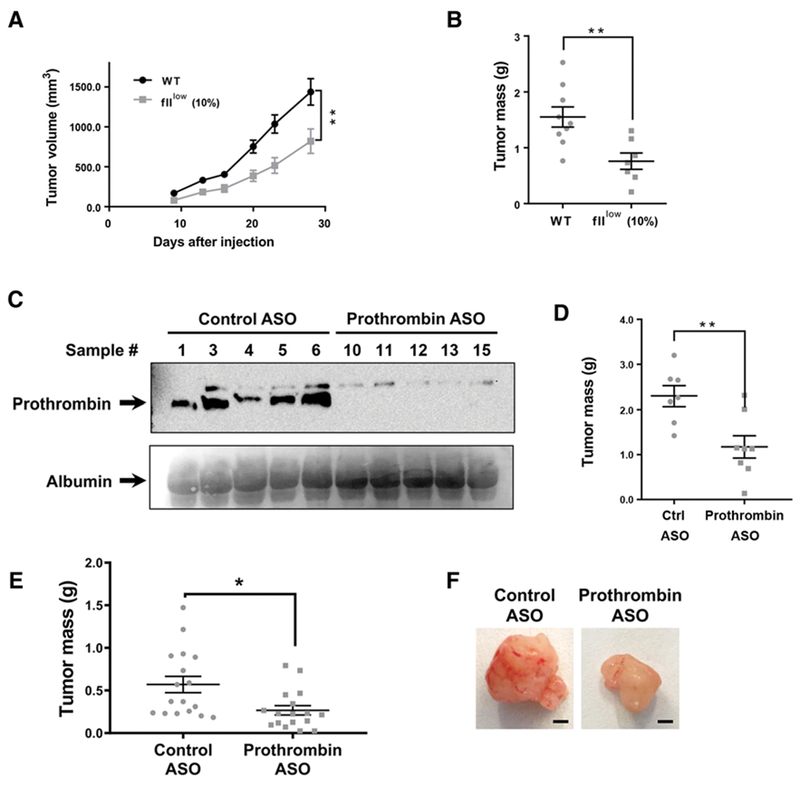
Prothrombin promotes PDAC tumor growth. A and B, Analysis of tumor growth following subcutaneous injection of KPC2 WT cells into C57BI/6 WT or fIIlow mice (N = 7-8 mice per group). Tumor volume was measured over time and tumor mass established for isolated tumors following 4 weeks of growth. C and D, WT C57Bl/6 mice were administered a prothrombin-specific ASO gapmer or a nontargeting control gapmer and analyzed for KPC2 cell tumor growth following subcutaneous injections. C, Immunoblot analysis for prothrombin of plasma from ASO-treated mice. Albumin levels as detected by Ponceau S staining of the blot was used as a loading control. D, Tumor mass was determined for isolated tumors following 4 weeks of growth. E and F, Analysis of the final tumor mass 21 days following orthotopic injection of KPC2 cells into the pancreas of C57Bl/6 mice treated with a control or prothrombin-specific ASO weekly following injection of the tumor cells. *, P < 0.05; **, P < 0.01. Scale bar, 2 mm.
Cancer cell–derived PAR-1 drives PDAC tumor growth
To establish if thrombin may promote PDAC tumor growth through PARs expressed by tumor cells, transcript levels of the Par gene family were first quantified. WT pancreas tissue exhibited nearly undetectable expression of all Par family members (Fig. 3A). In contrast, primary mouse PDAC tumor tissue and the KPC1 and KPC2 tumor cell lines expressed high levels of Par-1. Despite the undetectable expression of Par-3 in KPC tumor cell lines and normal pancreata, gene transcripts for Par-2 and −4 were also elevated in primary PDAC tumors and KPC tumor cell lines compared with normal pancreata expression but at significantly lower levels than observed for Par-1 (Fig. 3A). Notably, transcript levels of the other thrombin sensitive PARs (Par-3, Par-4) were substantially lower than those for Par-1, suggesting that PAR-1 is likely the primary tumor cell target of thrombin. Indeed, IHC confirmed robust PAR-1 protein in the majority of PDAC patient samples and in mouse PanIN and PDAC epithelial lesions (Fig. 3B and C; Supplementary Figs. S1 and S3).
Figure 3.
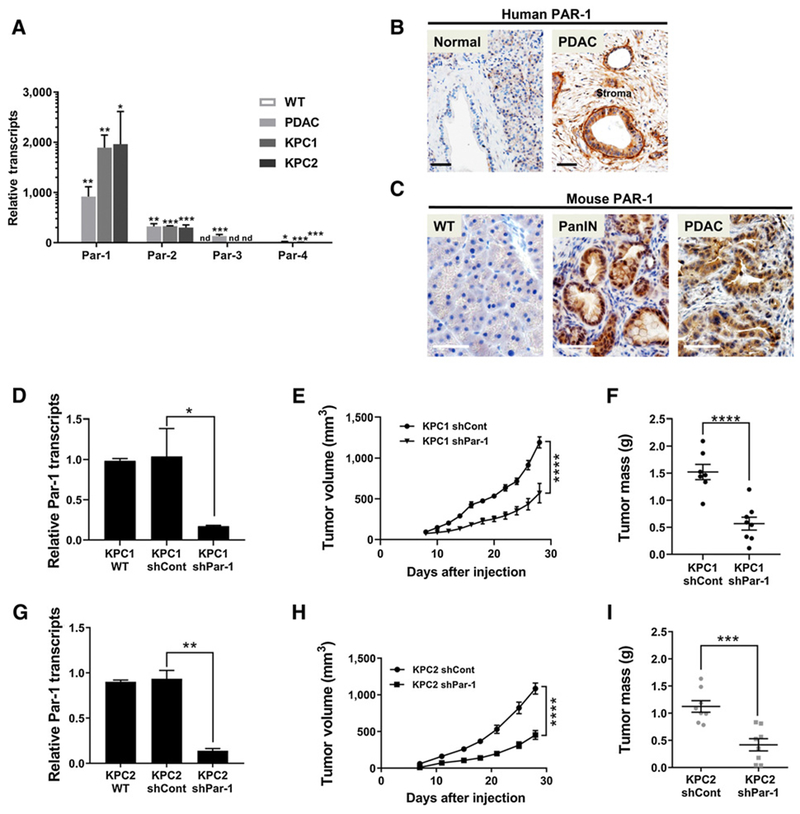
Par-1 is highly expressed in transformed PDAC epithelial cells, and reduction of PAR-1 diminishes PDAC tumor growth. A, qRT-PCR analysis of transcripts for Par-1, −2, −3, and −4 in mouse WT pancreas, PDAC, KPC1, and KPC2 samples (N = 3 per group). Statistical significance was calculated comparing relative transcript levels of each gene in WT pancreata vs. PDAC, KPC1, and KPC2 groups. B and C, PAR-1 IHC in normal human pancreas or PDAC patient samples and in mouse WT pancreas, PanIN lesions, and PDAC. D, qRT-PCR analysis for Par-1 transcripts in KPC1 WT, KPC1 shControl, and KPC1 shPar-1 “knockdown” cell lines (N = 3). E and F, Subcutaneous allograft assays using KPC1 shCont and KPC1 shTF lines in C57BL/6 mice. G, qRT-PCR analysis for Par-1 transcripts in KPC2 WT, KPC2 shControl, and KPC2 shPar-1 “knockdown” cell lines (N = 3). H and I, Subcutaneous allograft assays using KPC2 shCont and KPC2shTF lines in C57BL/6 mice. Tumor sizes were measured over time and tumor mass quantified at the day of sacrifice (N = 8 mice per group). Scale bars, 50 μm (B and C). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; nd, not detected.
To determine if tumor cell PAR-1 is critical for influencing tumor growth, stable shRNA knockdown KPC1 and KPC2 lines were generated (Fig. 3D and G). Following subcutaneous injection into C57Bl/6 mice, the shPar-1 cells displayed reduced tumor growth (Fig. 3E and H), resulting in a significantly lower final tumor mass (Fig. 3F and I). Interestingly, proliferation indices of KPC2 shControl, KPC2 shPar-1, and KPC2 shTF cells grown in vitro were similar (Supplementary Fig. S5A and S5B), suggesting that the TF-thrombin axis drives PDAC tumor growth through PAR-1 activation on PDAC tumor cells, but the mechanism is distinct from simple modulation of tumor cell proliferation.
We next generated KPC2 Par-1 knockout cell lines (Par-1KO1, Par-1KO8) to completely eliminate PAR-1 from the cell surface using a derivative of CRISPR-Cas9. A deletion in the 5′ portion of exon 1 resulted in loss of the initiator methionine codon and signal peptide sequence (Supplementary Fig. S5C and S5D). qRT-PCR analysis suggested that these deletions resulted in substantial transcript instability (Fig. 4A). To confirm the absence of functional PAR-1 protein, KPC2 WT, KPC2 cells transfected with Cas9 cDNA (KPC2 Cas9) and KPC2 PAR-1KO1 and KPC2 PAR-1KO8 lines were treated ± thrombin. Thrombin stimulation led to induction of phospho-ERK activation for KPC2 WT and KPC2 Cas9 control cells, whereas KPC2 Par-1KO1 and Par-1KO8 cells failed to respond to thrombin, confirming the absence of functional PAR-1 signaling in these cells (Fig. 4B). Elimination of Par-1 from KPC cells did not alter the expression of other Par receptors (Supplementary Fig. S5E), although TF expression was elevated in the Par-1KO1 line but not in the Par-1KO8 line (Supplementary Fig. S5F).
Figure 4.
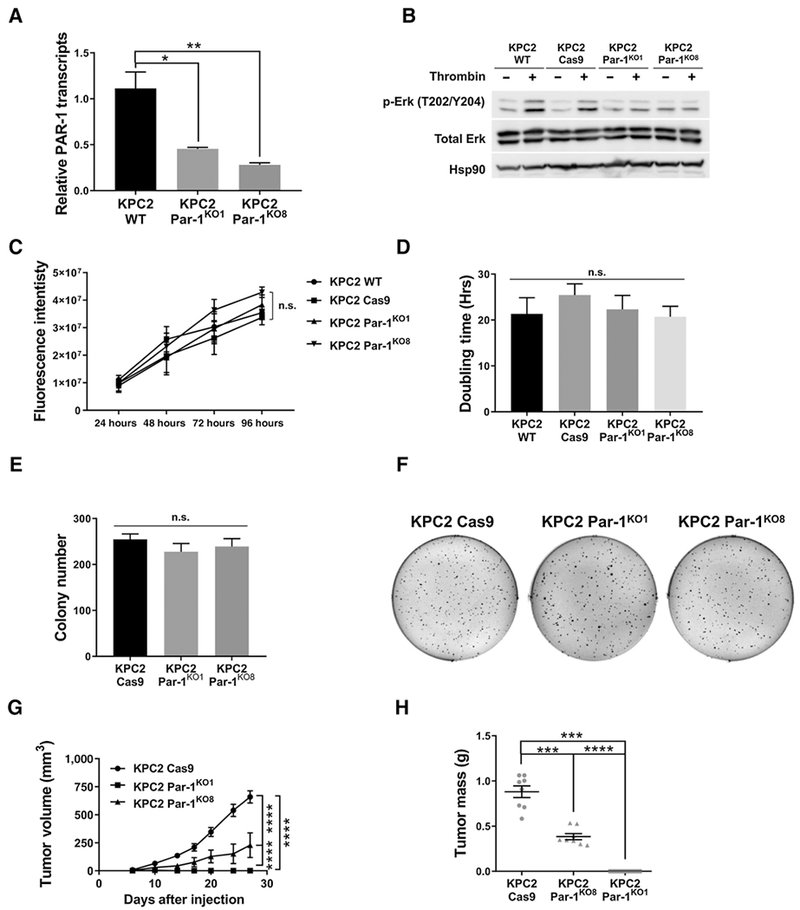
Elimination of PAR-1 activity in KPC2 cells does not alter cell proliferation or anchorage-independent growth in vitro, but significantly reduces tumor growth in vivo. A, qRT-PCR of Par-1 transcripts from KPC2 WT, KPC2 Par-1KO1, and KPC2 Par-1KO8 cells (N = 3 per group). B, Immunoblots for MAPK pathway activation of KPC2 WT, KPC2 Cas9 control, KPC2 Par-1KO1, and Par-1KO8 lines ± thrombin. Hsp90 served as a loading control. C, Growth curves for KPC2 WT, KPC2Cas9 control, KPC2 Par-1KO1, and KPC2 Par-1KO8 cells (N = 3 per group). D, Cell doubling times for the KPC2 WT, KPC2 Cas9 control, KPC2 Par-1KO1, and KPC2 Par-1KO8 lines (N = 3 per group). E and F, Soft agar colony formation for the KPC2 Cas9 Control, Par-1KO1, and Par-1KO8 lines (N = 3 per group). G and H, Analysis of tumor growth and final tumor mass following subcutaneous injection of KPC2 Cas9 Control, KPC2 Par-1KO1, and KPC2 Par-1KO8 lines into C57Bl/6 mice (N = 8 mice per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant.
KPC2 Par-1KO1 and KPC2 Par-1KO8 cells exhibited near identical growth rates and doubling times compared with WT KPC2 and control Cas9 cells, demonstrating that inactivating PAR-1 does not affect cell proliferation in vitro (Fig. 4C and D). KPC2 Par-1KO1 and KPC2 Par-1KO8 lines also produced similar numbers and sizes of distinct colonies in soft agar assays to that of KPC2-Cas9 cells, indicating that PAR-1 is dispensable for anchorage-independent growth in vitro (Fig. 4E and F). However, in allograft tumor studies, KPC2 Par-1KO8 cells exhibited a dramatic reduction in tumor growth, which translated to a significant reduction in final tumor mass compared with the Cas9 line (Fig. 4G and H). Par-1KO1 cells exhibited an even more impressive phenotype as these cells produced little to no tumor tissue 4 weeks postinjection. Intriguingly, elimination of PAR-1 activity in the microenvironment using Par-1−/− mice only reduced KPC tumor growth when Par-1 expression was also decreased in the tumor cells (i.e., for KPC2-shPAR-1 cells), but had no effect on tumor growth when tumor cell Par-1 expression was high (i.e., for KPC2-shControl; Supplementary Fig. S6). These results support the concept that tumor cell PAR-1 signaling is a potent determinant of PDAC tumor growth in vivo despite robust proliferative activity in vitro.
The TF-thrombin-PAR-1 pathway promotes metastasis of PDAC cells
To determine if the TF–thrombin axis contributes to PDAC dissemination, KPC cells were tested in tail vein injection metastasis assays. KPC2 shControl cells generated substantial metastatic burden in lung tissues 3 weeks postinjection such that individual metastatic lesions were too numerous to quantify (Supplementary Fig. S7A). In contrast, KPC2 shTF cells produced markedly fewer metastatic foci (Supplementary Fig. S7A). Lung weights revealed a significantly higher value for mice injected with KPC2 shControl cells compared with those injected with shTF cells (Supplementary Fig. S7B). Histology confirmed KPC2 shControl tumor tissue throughout the organ, effectively replacing lung epithelial cells and parenchyma. In contrast, mice injected with the shTF cells exhibited distinct metastatic foci with a large portion of lung architecture remaining unperturbed (Supplementary Fig. S7C). A similar reduction in metastatic burden was observed when KPC2 WT cells were tested in flllow mice versus WT (Supplementary Fig. S7D–S7F). Interestingly, the limited lung metastasis in flllow mice represented a reduction in initial adhesion and/or survival of tumor cells. In contrast to the substantial accumulation of KPC2GFP micro-metastases in WT mice, KPC2GFP cells failed to significantly accumulate in lungs of fIIlow mice just 24 hours postinjection (Supplementary Fig. S7G and S7H). Finally, KPC2 shPar-1 cells exhibited a similar significantly reduced metastatic potential (Supplementary Fig. S7I–S7K). Collectively, these results demonstrate that cancer cell–derived TF, prothrombin, and tumor cell–derived PAR-1 promote PDAC hematogenous metastatic potential.
Ectopic PAR-1 rescues the tumor growth potential of Par-1KO cells
To confirm that the failure of tumor growth potential in KPC PAR-1KO cells was indeed due to loss of PAR-1 activity and not to an off-target effect, we introduced a Dox-inducible Par-1Myc transgene (Tg) into KPC2 Par-1KO1 cells (resulting in the derivative line KPC2 Par-1KO/Tg). As expected, PAR-1Myc protein was not detected in KPC WT cells or in the KPC2 Par-1KO/Tg line maintained in the absence of Dox (Fig. 5A). However, treatment of KPC2 Par-1KO/Tg cells with Dox led to rapid induction of PAR-1Myc that localized predominantly to the cell surface (Fig. 5A). Importantly, KPC2 Par-1KO/Tg cells expressed Par-1Myc transcripts at levels comparable to that observed for the endogenous Par-1 gene in KPC2 WT cells (Fig. 5B). The functionality of the PAR-1Myc protein was then confirmed by treating KPC2 Par-1KO/Tg cells with thrombin following Dox induction. Under these conditions, thrombin addition led to the expected PAR-1Myc-dependent activation of phospho-ERK, whereas no increase in phospho-ERK was observed for KPC2 Par-1KO/Tg cells treated with thrombin in the absence of simultaneous Dox treatment (Fig. 5C).
Figure 5.
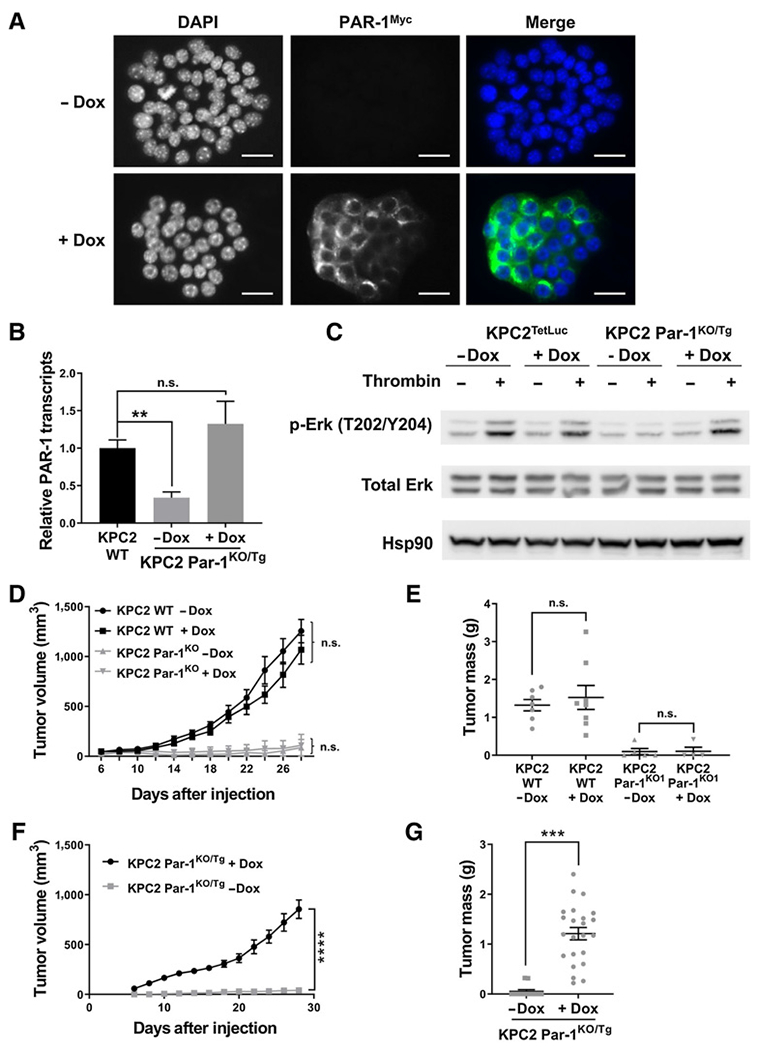
Ectopic PAR-1 activity restores KPC2 Par-1KO cell tumor growth. A, Anti-Myc immunofluorescence of KPC2 Par-1KO/Tg cells treated ± Dox. B, qRT-PCR for endogenous and Tg Par-1 transcripts comparing KPC2 WT and KPC2 Par-1KO/Tg cells ± Dox stimulation (N = 3 per group). C, Immunoblot for phospho-ERK in KPC2 cells containing a Tet-inducible luciferase expression vector (KPC2TetLuc) and KPC2 Par-1KO/Tg cells stimulated ± Dox and ± thrombin. D and E, Analysis of tumor volume and final tumor mass following subcutaneous injection of KPC2 WT cells or KPC2 Par-1KO1 cells ± Dox (N = 8 mice per group). F and G, Analysis of tumor volume and final tumor mass following subcutaneous injection of KPC2 Par-1KO/Tg cells into cohorts of mice treated ± Dox (N = 12 mice per group). Scale bars, 25 μm. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant.
The potential of KPC2 Par-1KO/Tg cells to form tumors in mice was next evaluated. Dox treatment in and of itself did not alter tumor progression as WT KPC2 cells formed large tumors following subcutaneous injection, whereas KPC2 Par-1KO1 cells generated only small tumor nodules with or without Dox (Fig. 5D and E). In comparative tumor growth studies, KPC2 Par-1KO/Tg cells failed to produce significant tumors when mice were not administered Dox (Fig. 5F and G). However, when the same KPC2 Par-1KO/Tg cells were tested in mice maintained on Dox, large tumors developed (Fig. 5F and G). Importantly, the growth rate of the KPC2 Par-1KO/Tg cells in mice with Dox was similar to that observed with KPC2 WT cells (Supplementary Fig. S8A and S8B). To test PAR-1–dependent tumor growth in a more clinically relevant setting, we next performed orthotopic injections of KPC2 WT cells or KPC2 Par-1KO/Tg cells into the mouse pancreas. The orthotopic model mimicked results from the subcutaneous tumor model where mice injected with KPC2 Par-1KO/Tg cells in the absence of Dox treatment exhibited minimal tumor formation (Fig. 6A and B). In contrast, a robust tumor growth response was obtained in Dox-treated mice tested with KPC2 Par-1KO/Tg cells. Again, tumor growth was equivalent to that observed following orthotopic injection of KPC2 WT cells (Fig. 6A and B). These results show that the limited tumor growth of KPC2 Par-1KO1 cells is fully restored by the reintroduction of PAR-1.
Figure 6.
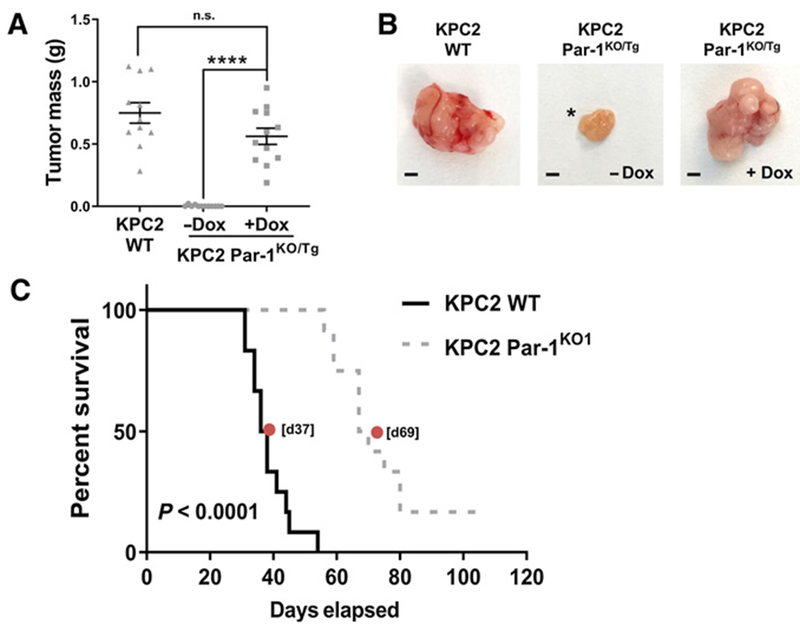
Expression of PAR-1 drives tumor growth in the pancreas microenvironment, resulting in poor host survival. A, KPC2 WT or KPC2 Par-1KO/Tg cells were injected into the pancreas and mice were treated ± Dox (N = 12 mice per group). Pancreas tumors were harvested at 21 days postsurgery.B, Representative examples of isolated tumors from the pancreas of mice 21 days postsurgery. Asterisk indicates one of only two tumors generated out of a cohort of 12 mice from the Par-1KO/Tg cells in the absence of Dox. The remaining 10 animals failed to develop visible tumors within the 21-day period. C, Survival study following orthotopic pancreas injection of KPC2 WT vs. KPC2 Par-1KO1 cells (N = 12 mice per group). Median survival times are indicated for each study (red dot). Scale bar, 2 mm. ****, P < 0.0001; n.s., not significant.
We next evaluated the contribution of tumor cell PAR-1 expression to PDAC-induced host mortality. Mice injected with KPC2 WT cells uniformly succumbed to the PDAC challenge with a median survival time of 37 days (Fig. 6C). In contrast, mice tested with KPC2 Par-1KO1 cells displayed a significantly prolonged lifespan with a median survival of 69 days (Fig. 6C). Indeed, 2 of 12 mice injected with KPC2 Par-1KO1 cells had no tumors and survived through the entire evaluation period of 106 days (Fig. 6C). Collectively, these findings confirm that reduced tumor growth displayed by PDAC cells in which PAR-1 signaling has been eliminated leads to significantly improved survival for tumor-bearing hosts.
Tumor cell thrombin-PAR-1 signaling supports tumor growth by modulating immune pathways
To begin to determine the mechanism by which tumor cell thrombin-PAR-1 signaling promotes tumor growth, we investigated in vivo changes in cell proliferation and apoptosis. Despite significant changes in overall tumor growth, no genotype-dependent differences were detected in the amount of IHC staining for the proliferation marker phospho-histone 3 (pH3) in sections of KPC2 WT and KPC2-Par-1KO1 or KPC2-Par-1KO8 tumors grown in WT mice for 1 week following subcutaneous injection (Fig. 7A). Similar findings were observed for KPC2 WT and KPC2-shPar-1 following subcutaneous injection. A significantly higher percentage of apoptotic cells as identified by cleaved caspase-3 (CC3+) were observed in tumor sections of KPC2-Par1KO1 or KPC2-Par1KO8 subcutaneous tumors compared with control KPC2-Cas9 cells (Fig. 7B). Similar findings were again observed for KPC2 WT and KPC2-shPar-1 following subcutaneous injection. To begin to understand potential differences in the TME, IHC analyses were performed on KPC2 and KPC2-shPar-1 tumors grown for 2 weeks following subcutaneous injection. Although tumors grown from KPC2 WT cells had significant numbers of KPC2 cells as shown by cytokeratin 19 (K19) staining, tumors grown from KPC2-shPar-1 cells had very few K19+ KPC2-shPar-1 cells within the tumor area (Supplementary Fig. S9A and S9B). Vessel density as determined by the number of CD31+ vessels per area of healthy tumor tissue was similar (Supplementary Fig. S9A and S9B), but the total number of vessels was diminished in KPC2-shPar-1 tumors as a result of those tumors being smaller overall and having a larger area of central necrosis. A key difference between tissues was the presence of immune cells. In KPC2 WT tumors, activated macrophages (Iba-1+; Supplementary Fig. S9A and S9B) were generally found at the perimeter of the tumor tissue, whereas in KPC2-shPar-1 tumors these cells were observed throughout the tumor. Analysis of macrophage subtype markers revealed very little iNOS (M1 macrophage) staining in KPC2 shControl tumors with an incremental increased number of iNOS+ cells in the KPC2 shPar-1 tumors (Supplementary Fig. S9A and S9B). Cells positive for the Arg1 marker (M2 macrophage) appeared dispersed throughout the tumors of both genotypes with a trend towards increased staining within KPC2 shPar-1 tumors (Supplementary Fig. S9A). Perhaps the most-striking feature was the distribution of T cells. In KPC2 WT tumors, CD3+ T cells (Supplementary Fig. S9A and S9B) were restricted to the perimeter of the tumor tissue, whereas these cells were found throughout the tumor tissue in KPC2 shPar-1 samples (Supplementary Fig. S9A). Collectively, these studies suggest a possible mechanism whereby loss or reduction of PAR-1 in the tumor cells results in a failure of tumor growth coupled to enhanced immunemediated clearance of tumor cells.
Figure 7.
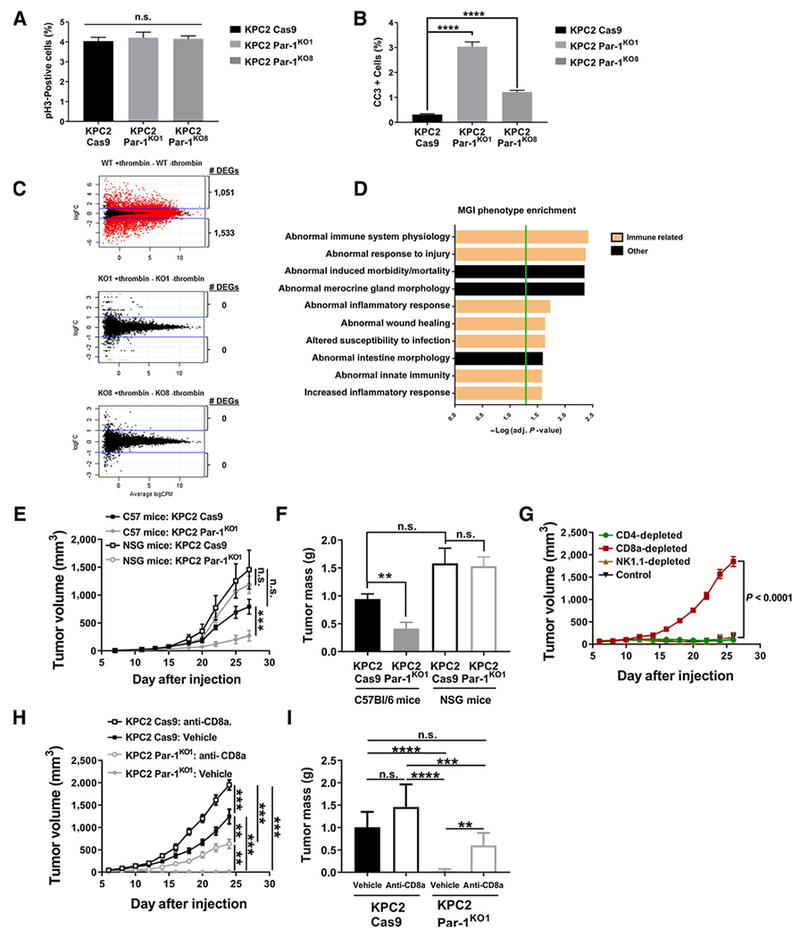
Reduction of Par-1 in KPC cells leads to induced apoptosis and susceptibility to immune cell invasion and antitumor immunity. A and B, pH3 (A) and cleaved caspase-3 (CC3; B) staining analysis of tissue sections of KPC2-Cas9, KPC2-Par-1KO1, and KPC2-Par-1KO8 (n = 3-4 mice/group) tumor tissue 1 week following subcutaneous injection into WT C57Bl/6 mice. C, MA plots showing the contrast in gene expression between samples treated with vehicle or thrombin based on RNA-seq. Red dots represent genes with an FDR ≤ 0.01, black dots represent genes with no significant change in expression, orange dots represent genes flagged for showing expression only in one sample group. The number of DEGs with two-fold change in expression is written beside each graph. D, The top 10 significantly enriched MGI phenotypes based on analysis of the DEGs from the KPC2 WT ± thrombin RNA-seq. Immune-related pathways have been highlighted. Green bar represents a significance cut-off of P ≤ 0.05. E and F, Analysis of tumor volume and final tumor mass following subcutaneous injection of KPC2 Cas9 control and KPC2 Par-1KO1 cells into cohorts of C57Bl/6and immune-deficient NSG mice (N = 7 mice per group). G, Analysis of tumor volume following subcutaneous injection of KPC2 Par-1KO1 cells into C57Bl/6 mice and immunodepletion of either CD4+, CD8a+, or NK cells (N = 4 mice per group). H and I, Analysis of tumor volume and final tumor mass following subcutaneous injection of KPC2 Cas9 control and KPC2 Par-1KO1 cells into cohorts of C57Bl/6 mice treated with anti-CD8a antibody or vehicle control (N = 7 mice per group). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; n.s., not significant.
To identify pathways by which thrombin-PAR-1 signaling may be driving tumor growth, RNA sequencing (RNA-seq) analysis was performed on KPC2 WT and KPC2-PAR-1KO cells in culture with and without thrombin stimulation. Numerous differentially expressed genes (DEG) were detected for thrombin-stimulated KPC2 WT cells relative to unstimulated KPC2 WT cells with 1,051 genes upregulated and 1,533 genes downregulated when applying a two-fold cutoff with an FDR ≤ 0.01 (Fig. 7C; Supplemental Excel File; Data deposited to GEO database accession no. GSE120370). In contrast, 0 DEGs were detected for thrombin-stimulated KPC2-PAR-1KO cells relative to unstimulated cells, a finding observed for the 2 independent KPC-PAR-1KO clones (KO1, KO8). Mouse genome informatics (MGI) phenotype enrichment indicated that the most profoundly thrombin–PAR-1 impacted categories were linked to immune regulation (Fig. 7D), a finding replicated by pathway analysis using other algorithms (e.g., Gene Ontology).
To directly test the hypothesis that thrombin–PAR-1 signaling in the PDAC tumor cells promotes PDAC tumor growth by suppressing antitumor immunity in the TME, parallel tumor growth studies were performed with KPC2-Cas9 cells and KPC2-PAR1KO1 cells in immune competent C57Bl/6 mice and immune-deficient NSG mice. Similar to earlier findings, KPC2-Cas9 cells formed large tumors in C57Bl/6 mice whereas minimal tumor growth of KPC2-PAR-1KO1 in C57Bl/6 mice was observed (Fig. 7E and F). In stark contrast, both KPC2-Cas9 and KPC2-PAR-1KO1 formed large tumors (equivalent in overall size) in immune-deficient NSG mice (Fig. 7E and F). NSG mice are known to be deficient in mature B cells, T cells, NK cells, and have significantly defective macrophages and dendritic cells. To determine whether the suppression of KPC2-PAR1KO1 tumor growth could be linked to a specific immune cell type compromised in NSG mice, an immune cell depletion screen was performed. Elimination of CD8a+ cells, but not CD4 T cells or NK cells (Supplementary Fig. S10) rescued tumor growth of KPC2-PAR-1KO1 cells in C57Bl/6 mice (Fig. 7G). Additional comparative studies revealed that depletion of CD8a+ (Fig. 7H and I) or CD8b+ resulted in a significant, but partial, restoration of tumor growth in C57Bl/6 mice. Collectively, these findings suggest that the failure of KPC2-PAR-1KO cell tumor growth in immune-competent C57Bl/6 mice is not due to cell intrinsic alterations but rather is linked to aberrant interactions with the TME to suppress antitumor immunity, in part driven by CD8 T cells.
Discussion
These studies emphasize that TF expression by pancreatic epithelial tumor cells is not merely an epiphenomenon but represents a leading factor in PDAC disease progression. Indeed, TF has long been proposed to influence tumor biology. For example, TF can mediate cell signaling events through TF-Factor VIIa complex formation and subsequent activation of PAR-2 (19). This receptor complex has been linked to numerous signaling pathways that drive tumor progression, including ERK1/2, JNK, PI3K, and PKCα (5, 20, 21). TF can also exert cell effects by engaging β1 integrins where modulation of integrin binding function and integrin-driven signaling is controlled in part by phosphorylation of the short TF-cytoplasmic tail (22). Beyond cell surface-associated TF, an alternatively spliced soluble form of TF (termed asTF) has been shown to play a critical role in cancer. Elevated asTF expression by cancer cells, including PDAC tumor cells, correlates with advanced histologic grade and poor patient prognosis (8, 23, 24) and in pancreatic cancer models asTF can drive inflammatory cell infiltration and increased metastatic potential (25, 26). Our current study implicating the TF-thrombin axis in PDAC tumor progression adds yet another layer of complexity to the role of TF within the microenvironment and tumor cells (Supplementary Fig. S11). Despite these diverse pathways, the different proposed mechanisms are not mutually exclusive and may function in concert. Additional studies will be required to determine if pathways are additive or synergistic and whether any are uniquely functional at distinct stages of PDAC disease.
Thrombin has also been linked to cancer progression; however, precise mechanisms by which thrombin activation contributes to lethal pancreatic cancer progression have not been elucidated. We show that genetic or pharmacologic reduction of prothrombin suppresses the growth of KPC PDAC tumor cells in vivo. Likewise, our studies revealed that tumor cell–derived PAR-1 is a key target of thrombin in that Par-1 shRNA and CRISPR knockout strategies confirmed that tumor cell PAR-1 is a primary factor in PDAC tumor growth. The reduction in tumor growth in PAR-1–deficient cells was not simply a function of cell-intrinsic defects because in vitro proliferation and colony formation were equivalent in KPC cells with and without PAR-1. Importantly, induction of Dox-inducible PAR-1Myc rescued full tumor growth in PAR-1KO cells, confirming a direct dependency of PAR-1 activity by PDAC cells. Tumor PAR-1 was also required for metastasis as PAR-1–deficient cells exhibited a greatly reduced metastatic tumor burden. Together, our studies support a model where PAR-1 is critical for primary tumor growth and formation of metastatic lesions.
The role of PAR-1 in stromal cells has also been shown to contribute to pancreatic cancer progression (27). Our studies suggest that even in the context of a normal microenvironment with PAR-1–expressing stromal cells of multiple origins (i.e., fibroblasts, inflammatory cells), reduction of thrombin generation in the microenvironment, or elimination of PAR-1 in the tumor cells, is a powerful negative determinant of tumor growth. Interestingly, WT KPC cells readily formed tumors in Par1−/− mice, indicating tumor growth was not dependent on the presence of PAR1 in the TME, a finding that is inconsistent with another study that reported that stromal PAR-1 expression promotes PDAC progression (27). The different observations between studies may link to tumor cell heterogeneity where the murine cell line Panc02 in their study has a SMAD4 driver mutation but retains WT KRAS and P53 (28). However, we also observed an even greater reduction in growth of KPC shPar-1 cells in Par-1−/− mice (Supplementary Fig. S6), suggesting the existence of a complex synergistic network between PDAC tumor cells and PAR-1 expressing cells within the stromal microenvironment. These findings highlight the need to better understand the relative contribution of PAR-1 signaling to the various stages of tumor growth and PDAC disease progression by individual cell types other than tumor cells.
PAR-1 may not be the only TF–thrombin target important in PDAC disease progression. Analyses of multiple cancer cell types (e.g., Lewis Lung carcinoma, B16 melanoma, MC38 colon cancer) have indicated that elimination or blockade of TF, prothrombin, or thrombin activity results in diminution of metastasis (29–31). Separate studies have demonstrated that exacerbation of thrombin procoagulant function through imposition of a thrombomodulin mutation (i.e., TmPro/Pro mice) leads to increased, metastasis (32). Our studies now add KPC PDAC cells to the list of tumor cell types whose metastatic potential is sensitive to TF–thrombin activity, suggesting that the TF–thrombin axis is a point of commonality in metastatic pathways. In support of this idea, fibrinogen, fXIII, and platelets are known to function as down-stream thrombin targets that are critical for mediating a prometastatic potential (33). Multiple mechanistic features have been elucidated regarding the thrombin–fibrinogen pathway and metastasis including that these factors are critical for the initial adhesion and or survival of tumor cells to the endothelium (29), a functional element shared by KPC tumor cells. Further, the mechanism by which fibrinogen and platelets support metastasis is linked to protection from NK-cell antitumor activity (34). Here, we show that tumor cell PAR-1 is a downstream target of thrombin that significantly enhances KPC metastatic potential. Future studies will focus on examining if the molecular pathways linking tumor cell PAR-1 to metastasis are also related to modulation of immune function.
The mechanism by which PAR-1 promotes tumor growth and formation of metastatic lesions has been studied primarily focusing on intrinsic cell signaling in vitro, such as promoting cell proliferation, migration, and adhesion to the cellular matrix (35–37) or by in vivo analyses using implanted human cells in immune-deficient mice (38, 39). Such strategies do not allow for identifying potential roles of PAR-1 in modulating antitumor immune responses. Our strategy using the KPC system in C57Bl/6 mice alleviated this significant limitation. The identification of the TF–thrombin–PAR-1 axis in immune suppression is consistent with studies showing PDAC has been known to have poor immunogenicity. Recent advances in immune therapies using single-agent immune checkpoint inhibitors targeting cytotoxic T-lymphocyte–associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) have little to no effect on patients with PDAC (40, 41), indicating that other mechanisms dominantly contribute to PDAC immune suppression. It stands to reason that immune evasion mechanisms for PDAC are associated with the driver mutation genes (i.e., mutations in KRAS, TP53) because PDAC has a relatively low mutation rate during cancer progression (42). Accordingly, the driver mutation genes would confer upon the PDAC cancer cell the immune escape capacity at the earliest stages of PDAC disease. The identification of TF and PAR-1 genes as downstream targets of KrasG12D and Trp53R172H are consistent with this working model (43, 44). In addition, PAR-1 has been shown to alter immune responses in other contexts, including inflammatory disease and response to viral infection (45, 46). The identification of CD8 T cells as being part of the mechanism by which the immune system eliminates KPC cells that lack PAR-1 signaling is consistent with other reports that document CD8 cytotoxic T cells as a primary effector mediating the elimination of transformed cells or foreign tissues and that the adaptive immune response occurs within 7 to 10 days following tumor cell inoculation (47–50). Although our current data strongly support a role for tumor cell–derived PAR-1 as an immune modulator in the PDAC TME, critical future experiments will focus on identifying the additional components of the immune system beyond CD8 T cells impacted (e.g., additional immune effector cell types) and the specific gene products and immune mediators downstream of PAR-1 that connect to the immune system.
In summary, this study provides clear evidence that the tumor cell TF–thrombin–PAR-1 axis promotes PDAC tumor growth and metastasis in vivo. In addition, one critical mechanistic feature is tumor cell PAR-1–mediated suppression of antitumor immunity. Studies are currently underway to elucidate the specific molecular pathways and immune-related mechanisms downstream of PAR-1 that are driving the documented PDAC phenotypes observed here. Determining whether the TF–thrombin–PAR-1 pathway plays a fundamental role early in PDAC disease (i.e., initial events such as acinar-ductal metaplasia or early development) remain critical open questions for investigation. Nonetheless, our results offer the proof-of-principle that targeting thrombin and/or tumor cell PAR-1 may offer substantial therapeutic benefits to patients with PDAC.
Supplementary Material
Significance:
The tissue factor-thrombin-PAR-1 signaling axis in tumor cells promotes PDAC growth and disease progression with one key mechanism being suppression of antitumor immunity in the microenvironment.
Acknowledgments
This work was supported by grants to M.J. Flick (NIH R01CA211098: NIH U01HL143403), S.F. Konieczny (NIH R01CA211098, NIH R01CA124586), and Y. Yang (Purdue University Center for Cancer Research SIRG Graduate Research Assistantship and Purdue University Cancer Prevention Internship Program Graduate Fellowship). The authors gratefully acknowledge support from the Purdue University Center for Cancer Research, NIH Grant No. P30 CA023168.
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
A.S. Revenko has ownership interest (including stock, patents, etc.) in Ionis Pharmaceuticals. E.S. Mullins is a consultant/advisory board member at Takeda and OctaPharma. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel) 2017; 9:42–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer 2007;110:2339–46. [DOI] [PubMed] [Google Scholar]
- 4.Mandalá M, Reni M, Cascinu S, Barni S, Floriani I, Cereda S, et al. Venous thromboembolism predicts poor prognosis in irresectable pancreatic cancer patients. Ann Oncol 2007;18:1660–5. [DOI] [PubMed] [Google Scholar]
- 5.Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Ying CH, et al. Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin Cancer Res 2007;13:2870–5 [DOI] [PubMed] [Google Scholar]
- 6.Nitori N, Ino Y, Nakanishi Y, Yamada T, Honda K, Yanagihara K, et al. Prognostic significance of tissue factor in pancreatic ductal adenocarcinoma. Clin Cancer Res 2005;11:2531–9. [DOI] [PubMed] [Google Scholar]
- 7.Rak J, Klement P, Yu J. Genetic determinants of cancer coagulopathy, angiogenesis and disease progression. Vnitr Lek 2006;52:135–8. [PubMed] [Google Scholar]
- 8.van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood 2012;119:924–32. [DOI] [PubMed] [Google Scholar]
- 9.Palumbo JS. Mechanisms linking tumor cell–associated procoagulant function to tumor dissemination. Semin Thromb Hemost 2008;34: 154–60. [DOI] [PubMed] [Google Scholar]
- 10.Kakkar AK, Lemoine NR, Scully MF, Tebbutt S, Williamson RC. Tissue factor expression correlates with histological grade in human pancreatic cancer. Br J Surg 1995;82:1101–4. [DOI] [PubMed] [Google Scholar]
- 11.Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004;58:213–9. [DOI] [PubMed] [Google Scholar]
- 12.Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, et al. Colon cancer growth and dissemination relies upon thrombin, stromal PAR-1, and fibrinogen. Cancer Res 2015;75:4235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu C, Konieczny SF. Pancreatic acinar cell 3-dimensional culture. Bio-protocol 2013;3:e930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014;32:569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 16.Shi G, Zhu L, Sun Y, Bettencourt R, Damsz B, Hruban RH, et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 2009;136:1368–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullins ES, Kombrinck KW, Talmage KE, Shaw MA, Witte DP, Ullman JM, et al. Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood 2009;113:696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crosby JR, Zhao C, Zhang H, MacLeod AR, Guo S, Monia BP. Reversing antisense oligonucleotide activity with a sense oligonucleotide antidote: proof of concept targeting prothrombin. Nucleic Acid Ther 2015;25: 297–305. [DOI] [PubMed] [Google Scholar]
- 19.Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost 2004;2:93–101. [DOI] [PubMed] [Google Scholar]
- 20.Schaffner F, Ruf W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler Thromb Vasc Biol 2009;29:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bluff JE, Brown NJ, Reed MWR, Staton CA. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res 2008;10:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, et al. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med 2004;10:502–9. [DOI] [PubMed] [Google Scholar]
- 23.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cisregulatory elements required for macrophage and B cell identities. Mol Cell 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unruh D, Sagin F, Adam M, Van Dreden P, Woodhams BJ, Hart K, et al. Levels of alternatively spliced tissue factor in the plasma of patients with pancreatic cancer may help predict aggressive tumor phenotype. Ann Surg Oncol 2015;22:1206–11. [DOI] [PubMed] [Google Scholar]
- 25.Signaevsky M, Hobbs J, Doll J, Liu N, Soff GA. Role of alternatively spliced tissue factor in pancreatic cancer growth and angiogenesis. Semin Thromb Hemost 2008;34:161–9. [DOI] [PubMed] [Google Scholar]
- 26.Unruh D, Turner K, Srinivasan R, Kocatüurk B, Qi X, Chu Z, et al. Alternatively spliced tissue factor contributes to tumor spread and activation of coagulation in pancreatic ductal adenocarcinoma. Int J Cancer 2014;134: 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Queiroz KCS, Shi K, Duitman J, Aberson HL, Wilmink JW, van Noesel CJM, et al. Protease-activated receptor-1 drives pancreatic cancer progression and chemoresistance. Int J Cancer 2014;135:2294–304. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Zhang Y, Yang J, Ni X, Liu S, Li Z, et al. Genomic sequencing of key genes in mouse pancreatic cancer cells. Curr Mol Med 2012;12:331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palumbo JS, Talmage KE, Massari J V, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 2007;110:133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milsom C, Yu J, May L, Meehan B, Magnus N, Al-Nedawi K, et al. The role of tumor-and host-related tissue factor pools in oncogene-driven tumor progression. Thromb Res 2007;120:S82–91. [DOI] [PubMed] [Google Scholar]
- 31.Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci U S A 1992;89:11832–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horowitz NA, Blevins EA, Miller WM, Perry AR, Talmage KE, Mullins ES, et al. Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood 2011;118:2889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell mediated elimination of tumor cells. Blood 2005;105:178–85. [DOI] [PubMed] [Google Scholar]
- 34.Palumbo JS, Barney KA, Blevins EA, Shaw MA, Mishra A, Flick MJ, et al. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost 2008;6:812–9. [DOI] [PubMed] [Google Scholar]
- 35.Darmoul D, Gratio V, Devaud H, Peiretti F, Laburthe M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol Cancer Res 2004;2:514–22. [PubMed] [Google Scholar]
- 36.Rudroff C, Striegler S, Schilli M, Scheele J. Thrombin enhances adhesion in pancreatic cancer in vitro through the activation of the thrombin receptor PAR 1. Eur J Surg Oncol 2001;27:472–6. [DOI] [PubMed] [Google Scholar]
- 37.Even-Ram SC, Maoz M, Pokroy E, Reich R, Katz BZ,Gutwein P, et al. Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the alpha vbeta 5 integrin. J Biol Chem 2001;276: 10952–62. [DOI] [PubMed] [Google Scholar]
- 38.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005;120:303–13. [DOI] [PubMed] [Google Scholar]
- 39.Agarwal A, Covic L, Sevigny LM, Kaneider NC, Lazarides K, Azabdaftari G, et al. Targeting a metalloprotease-PAR1 signaling system with cell-penetrating pepducins inhibits angiogenesis, ascites, and progression of ovarian cancer. Mol Cancer Ther 2008;7:2746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467:1114–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellis CA, Malik AB, Gilchrist A,Hamm H, Sandoval R, Voyno-Yasenetskaya T, et al. Thrombin induces proteinase-activated receptor-1 gene expression in endothelial cells via activation of Gi-linked Ras/mitogen-activated protein kinase pathway. J Biol Chem 1999;274:13718–27. [DOI] [PubMed] [Google Scholar]
- 44.Salah Z, Haupt S, Maoz M, Baraz L, Rotter V, Peretz T, et al. p53 controls hPar1 function and expression. Oncogene 2008;27:6866–74. [DOI] [PubMed] [Google Scholar]
- 45.Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, et al. PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 2013;123:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saban R, D’Andrea MR, Andrade-Gordon P, Derian CK, Dozmorov I, Ihnat MA, et al. Mandatory role of proteinase-activated receptor 1 in experimental bladder inflammation. BMC Physiol 2007;7:4. doi: 10.1186/1472-6793-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.North RJ, Bursuker I.Generation and decay of the immune response to a progressive fibrosarcoma. I. Ly-1+2- suppressor T cells down-regulate the generation of Ly-1–2+ effector T cells. J Exp Med 1984;159: 1295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ibe S, Qin Z, Schuler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med 2001;194: 1549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammond-McKibben D, Lake P, Zhang J, Tart-Risher N, Hugo R, Weetall M. A high-capacity quantitative mouse model of drug-mediated immunosuppression based on rejection of an allogeneic subcutaneous tumor. J Pharmacol Exp Ther 2001;297:1144–51. [PubMed] [Google Scholar]
- 50.Celli S, Albert ML, Bousso P. Visualizing the innate and adaptive immune responses underlying allograft rejection by two-photon microscopy. Nat Med 2011;17:744–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.