

Abstract
Background
Triatoma rubrofasciata is a widespread pathogen vector for Chagas disease, an illness that affects approximately 7 million people worldwide. Despite its importance to human health, its evolutionary origin has not been conclusively determined. A reference genome for T. rubrofasciata is not yet available.
Finding
We have sequenced the genome of a female individual with T. rubrofasciatausing a single molecular DNA sequencing technology (i.e., PacBio Sequel platform) and have successfully reconstructed a whole-genome (680-Mb) assembly that covers 90% of the nuclear genome (757 Mb). Through Hi-C analysis, we have reconstructed full-length chromosomes of this female individual that has 13 unique chromosomes (2n = 24 = 22 + X1 + X2) with a contig N50 of 2.72 Mb and a scaffold N50 of 50.7 Mb. This genome has achieved a high base-level accuracy of 99.99%. This platinum-grade genome assembly has 12,691 annotated protein-coding genes. More than 95.1% of BUSCO genes were single-copy completed, indicating a high level of completeness of the genome.
Conclusion
The platinum-grade genome assembly and its annotation provide valuable information for future in-depth comparative genomics studies, including sexual determination analysis in T. rubrofasciata and the pathogenesis of Chagas disease.
Keywords: Triatoma rubrofasciata, PacBio Sequel platform, Hi-C, chromosomal-level assembly, comparative genomics, RNA-Seq, Iso-Seq
Data Description
Introduction
The insect Triatoma rubrofasciata (De Geer) (Hemiptera, Triatominae) is the first Triatominae species formally described, initially with the name Cimex rubrofasciatus De Geer, 1773 [1]. This insect presents anthropogenic habits with its dispersion favored by the interaction between residential settlement and human activities [2]. It is considered of global epidemiological importance because it has a pantropical widespread distribution that is found in approximately 45 countries from the Old World to the New World [3]. It is one of the 151 species of Triatominae that has 18 genera currently described worldwide that can transmit American trypanosomiasis, known as Chagas disease [4]. This condition has a great impact on public health, with 7–8 million people estimated to be infected worldwide, mostly in Latin America. It has become a global health issue in this century with the spread to nonendemic countries due to growing population movements [5].
Due to growing population movements, important epidemiological changes have occurred in recent decades, and the disease has now spread to many nonendemic countries [6]. The widespread of T. rubrofasciata emerges as a potential risk of outbreaks in these regions, which demands urgent studies through comprehensive sampling and comparative studies. The lack of a high-quality reference genome represents a major hurdle for such efforts. Here, we present a platinum-grade reference genome for T. rubrofasciata, which will be valuable for developing vector control programs.
Sample description and DNA sequencing
An adult female insect T. rubrofasciata (Fig. 1) was used for reference genome construction in this study. This insect was the second-generation offspring of a population that was established from the eggs of single female adult collected in Shunde County, Foshan City, Guangdong Province (22°42′44.63″N, 113°08′45.34″E), China, in 2016 [7]. DNA was extracted from this individual using the traditional phenol/chloroform extraction method and was quality checked using agarose gel electrophoresis. A single band was observed, indicating high integrity of DNA molecules for library construction for the Illumina X Ten (Illumina, Inc., San Diego, CA, USA) and the PacBio Sequel (Pacific Biosciences of California, Menlo Park, CA, USA) sequencing platforms.
Figure 1:

Dorsal (left) and ventral (right) views of a female Triatoma rubrofasciata.
Using DNA preparation, a library with an insertion length of 350 bp was constructed for the Illumina sequencing platform according to the manufacturer's protocol. In total, 46.75-Gb short reads were obtained from the Illumina X Ten DNA sequencing platform (Table 1), and 39.32-Gb filtered reads were used for the following genome survey analysis and for final-stage base-level genome sequence polishing. Meanwhile, 20-kb libraries were constructed for PacBio Sequel sequencing. Using 14 SMRT cells, 8.23 million reads were generated, with the total length of 69.38 Gb (Table 1). The mean length of these subreads was 8.43 kb, and the plot of the read length distribution/ratio is shown in Fig. 2.
Table 1:
Sequencing data generated for Triatoma rubrofasciata genome assembly and annotation
| Library type | Platform | Library size (bp) | Data size (Gb) | Application |
|---|---|---|---|---|
| Short reads | HiSeq X Ten | 350 | 46.75 | Genome survey and genomic base correction |
| Long reads | PacBio Sequel | 20,000 | 69.38 | Genome assembly |
| Hi-C | HiSeq X Ten | 300–500 | 103.61 | Chromosome construction |
Figure 2:
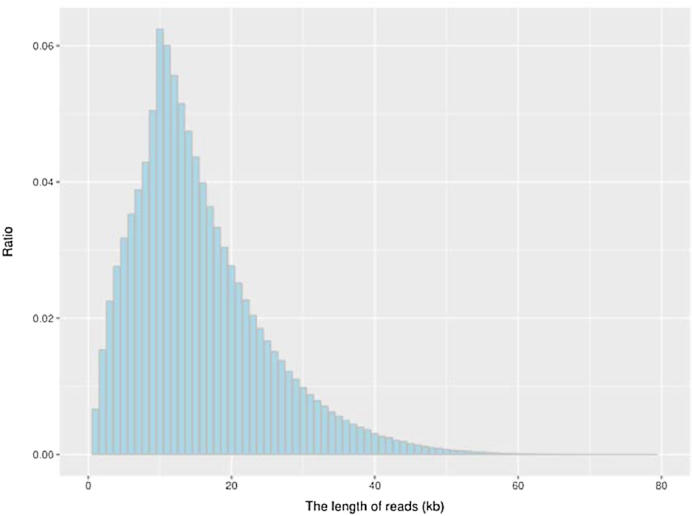
The plot of the read length distribution/ratio of the subreads.
Genome features estimation through k-mer analysis
With sequencing data from the Illumina HiSeq X Ten DNA sequencing platform, several genome features were evaluated for the genome of T. rubrofasciata. To ensure the quality of the analysis, ambiguous bases and low-quality reads were first trimmed and filtered using the HTQC package [8]. First, the quality of bases at 2 read ends was checked. Bases in sliding 5-bp windows were deleted if the average quality of the window was below 20. Second, reads were filtered if the average quality was smaller than 20 or the read length was shorter than 75 bp. Third, the mate reads were also removed if the corresponding reads were filtered.
The processed reads were used for genome assessment. We calculated the number of each 17-mer from the sequencing data using the jellyfish software (v2.1.3) [9], and the distribution was analyzed with GCE software [10]. We estimated the genome size of 757 Mb with the heterozygosity of 1.01% and repeat content of 55.49% in the genome. K-mer analysis was used to estimate the genome size, which showed the PacBio assembly was of good quality (Fig. 3). The genome size of T. rubrofasciata is similar to that of Rhodnius prolixus, another insect vector of Chagas disease, which has a predicted 733-Mb genome size [11].
Figure 3:
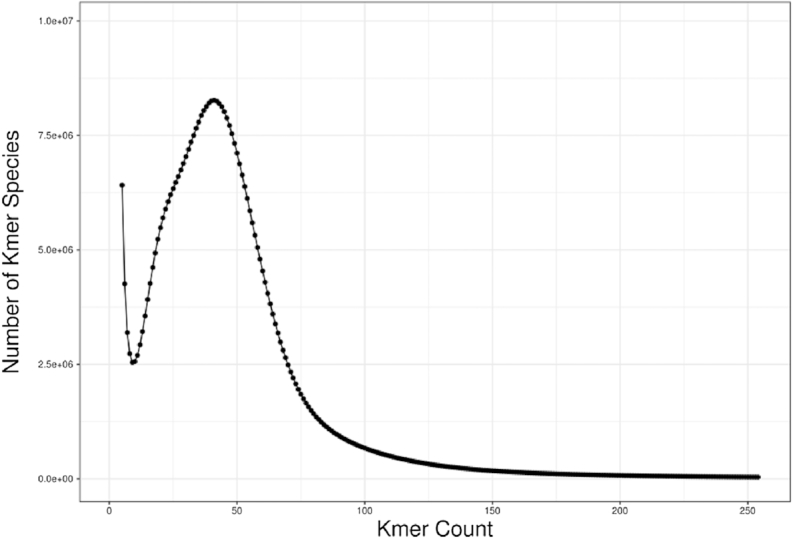
17-mer depth distribution for genome size estimation analysis of Triatoma rubrofasciata.
Genome assembly using PacBio long reads
FALCON [12] was employed using the length_cut_off and length_cutoff_pr parameters of 3 kb and 3 kb, respectively. We first obtained a 677.72-Mb genome with a contig N50 of 2.71 Mb. The genome sequences were subsequently polished using PacBio long reads using arrow [13] and Illumina short reads by pilon [14] to correct base errors.
In situ Hi-C library construction and chromosome assembly using Hi-C data
A separate female individual T. rubrofasciata was used for library construction for Hi-C analysis as described previously [15, 16]. The library was sequenced with a 150-bp paired-end mode on the Illumina HiSeq X Ten platform.
From the Illumina HiSeq X Ten platform, 103.61-Gb reads were obtained for the Hi-C library and 99.28-Gb filtered reads were used for the following Hi-C analysis. The reads were mapped to the above T. rubrofasciata genome with Bowtie [17], with both ends of paired reads being mapped to the genome separately. To increase the interactive Hi-C reads ratio, an iterative mapping strategy was performed as in previous studies, and only read pairs that both ends uniquely mapped were used for the following analysis. From the alignment of the paired ends, self-ligation, nonligation, and other sorts of invalid reads, including StartNearRsite, PCR amplification, random break, LargeSmallFragments, and ExtremeFragments, were filtered out by Hi-C library, with the method described in a previous study [15]. Through the recognition of restriction sites in sequences, contact counts among contigs were calculated and normalized.
By clustering the contigs using the contig contact frequency matrix, we were able to correct some minor errors in the FALCON assembly results. Contigs with errors were corrected by breaking into shorter contigs, and we obtained a chromosome-level genome assembly of 680.73 Mb with 2126 contigs and a contig N50 of 2.72 Mb. The longest contig was 10.27 Mb in size (Table 2). Among these 2126 contigs, 626 contigs were mounted to 13 chromosomes with Lachesis [18] using the agglomerative hierarchical clustering method. Lachesis was further applied to order and orient the clustered contigs according to the contact matrix. Contigs anchored to chromosomes accounted for 92.51% of the total genome bases (Fig. 4). The number of chromosomes matched nicely to a previously published karyotype of a female T. rubrofasciata individual (2n = 24 = 11 * 2 + X1 + X2) [1]. Taken together, we have successfully reconstructed the first chromosomal-level assembly of T. rubrofasciata of 680.73 Mb, with 2126 contigs, a contig N50 of 2.72 Mb, and a scaffold N50 of 50.70 Mb (Table 2).
Table 2:
Statistics for genome assembly of Triatoma rubrofasciata
| Sample | Length | Number | ||
|---|---|---|---|---|
| Contig (bp) | Scaffold (bp) | Contig | Scaffold | |
| Total | 680,314,598 | 680,726,098 | 2126 | 1303 |
| Max | 10,270,547 | 97,329,580 | – | – |
| N50 | 2,722,109 | 50,700,875 | 76 | 6 |
| N60 | 2,121,675 | 50,415,845 | 104 | 7 |
| N70 | 1,587,961 | 46,556,423 | 140 | 8 |
| N80 | 1,038,484 | 37,928,883 | 193 | 10 |
| N90 | 338,786 | 20,341,594 | 301 | 12 |
Figure 4:
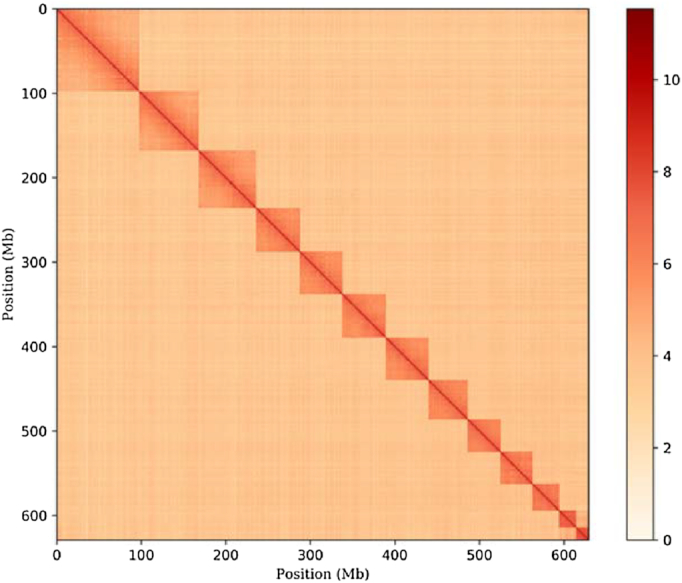
DNA interaction heatmap generated in Hi-C analysis (resolution: 500 Kb).
Genome quality evaluation
We assessed the quality of genome of T. rubrofasciata in 3 aspects: sequence continuity, genome completeness, and base-level accuracy.
First, we compared the contig/scaffold number and N50 length of contig of T. rubrofasciata with insect species with sequenced genomes and found that our assembly has much improved quality over other insects (Fig. 5). We attributed the improvement to the application of the PacBio long reads for genome assembly. With Hi-C data analysis, we successfully assembled the T. rubrofasciata genome to chromosome level with just 1 individual. Like previous studies, insect genome heterozygosity was one of the biggest challenges for genome assembly, both in terms of contig and scaffold assembly. Traditional chromosomal genome assembly requires physical maps and genetic maps, which is enormously time- and labor-consuming. Our work illustrated that the genome assembly using PacBio long sequencing data was not only affordable but also effective for overcoming the difficulties presented by insect genome assembly.
Figure 5:
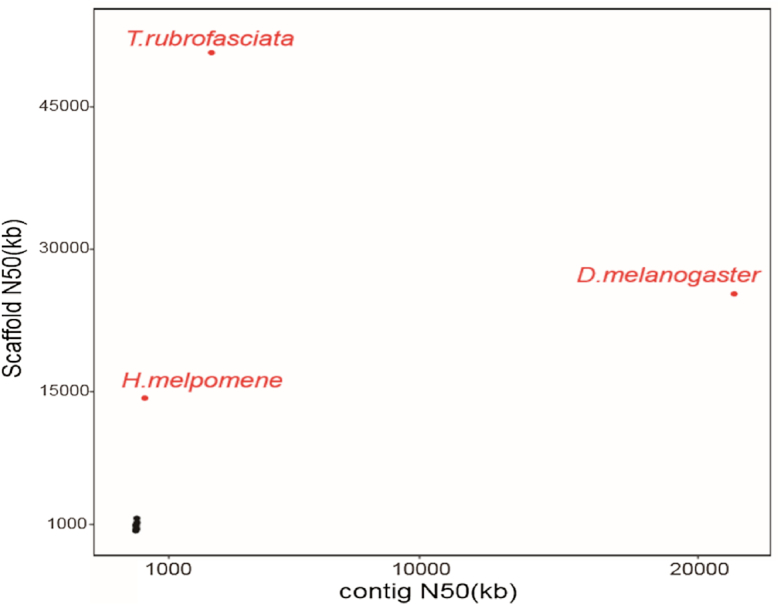
Genome assembly comparison of Triatoma rubrofasciata with other sequenced insect genomes (Apis mellifera, Acyrthosiphon pisum, Cimex lectularius, Culex quinquefasciatus, Drosophila melanogaster, Gerris buenoi, Glossina palpalis, Halyomorpha halys, Heliconius melpomene, Homalodisca vitripennis, Oncopeltus fasciatus, Rhodnius prolixus). The x- and y-axes represent the contig and scaffold N50s, respectively. The genomes with both contig and scaffold N50s less than 2M are hignlighted in black.
Second, the assembled genome was subjected to the BUSCO v.3.0.2 (RRID:SCR_015008) [19] to assess the completeness of the genome assembly. We used the “insect_obd9” gene set, and 98.2% of the BUSCO genes were identified in theT. rubrofasciata genome. More than 95.1% of BUSCO genes were single-copy completed in our genome, illuminating a high level of completeness of the genome.
Third, NGS short reads were aligned to the genome using BWA [20]. About 98.1% of reads were aligned to the genome, of which 98.0% were reads paired aligned. The insertion length distribution of read pairs exhibited a single peak around 300 bp, which was consistent with the design for the Illumina sequencing library construction. Note that the NGS data, which were used for error correction, were not used in the contig assembly. Therefore, the insertion length distribution of NGS data illustrated the high quality of our assembly at the contig level. From the NGS reads alignment, we detected 8478 homologous SNP loci using GATK [21], demonstrating the high base-level accuracy of 99.99%.
Repeat element and gene annotation
TRF [22] was used for repetitive element identification in theT. rubrofasciata genome. A de novo method applying RepeatModuler [23] was used to detect TEs. The resulting de novo data, combined with a known repeat library from Repbase [24], were used to identify TEs in the T. rubrofasciata genome by RepeatMasker [25].
Protein-coding genes in the T. rubrofasciata genome were annotated using the de novo program Augustus (RRID:SCR_008417) [26]. Protein sequences of the closely related species, including Rhodnius prolixus (from VectorBase), Halyomorpha halys (from NCBI), Oncopeltus fasciatus (from USDA), Cimex lectularius (from NCBI), and Drosophila melanogaster (from NCBI), were aligned to the T. rubrofasciata genome with tblastn. Full-length transcripts obtained using Iso-Seq were mapped to the genome using Gmap [27]. Finally, gene models predicted from all above methods were combined by MAKER [28], resulting in 12,691 protein-coding genes. The gene number, gene length, CDS length, exon length, and intron length distribution were all comparable with the related insects (Fig. 6).
Figure 6:

Length distribution comparison on total gene, CDS, exon, and intron of annotated gene models of Triatoma rubrofasciata with other closely related insect species. Length distribution of total gene (A), CDS (B), exon (C), and intron (D) was compared to those of Rhodnius prolixus, Halyomorpha halys, Oncopeltus fasciatus, Cimex lectularius, and Drosophila melanogaster.
To functionally annotate protein-coding genes in the T. rubrofasciata genome, we searched all predicted gene sequences to NCBI nonredundant protein (NR), InterPro (InterProScan, RRID:SCR_005829) [29], GO, KEGG (RRID:SCR_012773) [30], Swissprot [31], and TrEMBL databases [31] by BLASTN [32] and BLASTX [33]. A threshold e-value of 1e-5 was used for all BLAST applications. Finally, 12,063 genes were functionally annotated (Table 3).
Table 3:
Statistics for genome annotation of Triatoma rubrofasciata
| Database | Number | Percent |
|---|---|---|
| NR | 11,451 | 90.23 |
| InterPro | 9625 | 75.84 |
| GO | 7180 | 56.58 |
| KEGG ALL | 10,867 | 85.63 |
| KEGG KO | 6112 | 48.16 |
| Swissprot | 9448 | 74.45 |
| TrEMBL | 11,989 | 94.47 |
| Total | 12,063 | 95.05 |
Phylogenetic analysis of T. rubrofasciata with other insects
OrthMCL was used to cluster gene families. First, proteins from T. rubrofasciata and the closely related insects, including Rhodnius prolixus, Oncopeltus fasciatus, Halyomorpha halys, Cimex lectularius, Drosophila melanogaster, Gerris buenoi, Homalodisca vitripennis, Acyrthosiphon pisum, Culex quinquefasciatus, Glossina palpalis, Apis mellifera, and Heliconius melpomene, were all-to-all blasted by BLASTP [33] utility with an e-value threshold of 1e-5. Only proteins from the longest transcript were used for genes with alternative splices. We identified 21,850 gene families for T. rubrofasciata and the related species, among them 330 single-copy ortholog families.
Using single-copy orthologs, we probed the phylogenetic relationships for T. rubrofasciata and other insects. To this end, protein sequences of single-copy genes were aligned using MUSCLE [34]. Guided by the protein multisequence alignment, the alignment of the CDS for those genes was generated and concatenated for the following analysis. The phylogenetic relationships were constructed using PhyML [35] using the concatenated nucleotide alignment with the JTT+G+F model. We first obtained divergent times for all pairs with the phylogenetic tree using r8s [36], which were used as input, together with molecular clock data from the divergence time from the TimeTree database [37], to estimate species divergence time for all pairs of species in the phylogenetic tree using the MCMCtree program (from PAML) [38]. We found that T. rubrofasciata was most closely related to R. prolixus, and the 2 species diverged from their common ancestor around 60.00–95.00 MYA (Fig. 7).
Figure 7:

Phylogenetic analysis of T. rubrofasciata with other insect species. The estimated species divergence time (million years ago) and the 95% confidential intervals are labeled at each branch site. The divergence used for time recalibration is illuminated as red dots in the tree.
Conclusion
We reconstructed the first high-quality, chromosome-level assembly of T. rubrofasciata using an integrated strategy of PacBio, Illumina, and Hi-C technologies. Using the long reads from the PacBio Sequel platform and short reads from the Illumina HiSeq X Ten platform, we successfully constructed contig assembly for Triatoma. Leveraging contact information among contigs from Hi-C technology, we further improved the assembly to the chromosome-level quality. We annotated 12,691 protein-coding genes in the T. rubrofasciata genome, 12,063 of which were functionally annotated. With 330 single-copy orthologs from T. rubrofasciata and other related insects, we constructed the phylogenetic relationship of these insects and found that T. rubrofasciata might have diverged from its common ancestor of R. prolixus around 60.00–95.00 MYA. Given the increasing interests in insect genome evolution and the biological importance of T. rubrofasciata as the vector for Chagas disease, our genomic and transcriptome data provide a valuable genetic resource for the following functional genomics investigations for the research community.
Availability of supporting data
The raw data from our genome project was deposited in the NCBI Sequence database with Bioproject IDs PRJNA516044. The Illumina, PacBio, and Hi-C sequencing data are available from NCBI via accession numbers SRR8466736, SRR8466737, and SRR8466756, respectively. The Illumina transcriptome sequencing data were deposited to NCBI via accession numbers SRR8468315 and SRR8468316. Other data further supporting this work are available in the GigaSciencerepository, GigaDB [39].
Ethics Statement
This study was approved by the Animal Care and Use Committee of the National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention. All participants consented to the study under the "Ethics, Consent and Permissions" heading. All participants consented to publish the work under the "Consent to Publish" heading.
Abbreviations
BLAST, Basic Local Alignment Search Tool; bp, base pairs; BUSCO, Benchmarking Universal Single-Copy Orthologs; CDS, coding DNA sequences; Gb, gigabase pairs; GO, Gene Ontology; Hi-C, High-throughput/resolution chromosome conformation capture; kb, kilobase pairs; KEGG, Kyoto Encyclopedia of Genes and Genomes; Mb, megabase pairs; MYA, million years ago; NCBI, National Center for Biotechnology Information; NGS, Next Generation Sequencing; NR, nonredundant protein; PCR, polymerase chain reaction; SMRT, Single Molecule Real Time; SNP, single-nucleotide polymorphism; TE, transposable element; TRF, Tandem Repeat Finder; USDA, US Department of Agriculture.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFC1202000), the National Science and Technology Project (No. 2018ZX10101002), and the CAS Pioneer Hundred Talents Program (to N.S.C.) and Taishan Scholar Project Special Fund (to N.S.C.).
Author Contributions
Z.X.N., L.Q., Z.Y., and H.W. conceived the project. L.Q., G.Y.H., Z.Y., Z.D., L.Y.Y., W.J.T., and Z.Z.B. collected the samples and extracted the DNA and RNA. L.Q, G.Y.H., and Z.Y. performed the genome assembly and data analysis. C.N.S. performed the data analysis. L.Q. and C.N.S. wrote the paper. Z.X.N. revised the manuscript. All authors read, edited, and approved the final version of the manuscript.
Supplementary Material
Ben Mans -- 2/25/2019 Reviewed
Ben Mans -- 5/13/2019 Reviewed
Ting-Fung Chan, PhD -- 3/9/2019 Reviewed
Ting-Fung Chan, PhD -- 5/18/2019 Reviewed
ACKNOWLEDGEMENTS
We thank Frasergen Bioinformatics for providing technical support for this work.
References
- 1. Alevi KCC, Nascimento JGO, Moreira FFF et al.. Cytogenetic characterisation of Triatoma rubrofasciata (De Geer) (Hemiptera, Triatominae) spermatocytes and its cytotaxonomic application. African Entomology. 2016;24(1):4. [Google Scholar]
- 2. Hypsa V, Tietz DF, Zrzavý J, et al.. Phylogeny and biogeography of Triatominae (Hemiptera: Reduviidae): molecular evidence of a New World origin of the Asiatic clade. Mol Phylogenet Evol. 2002;23(3):447–57. [DOI] [PubMed] [Google Scholar]
- 3. Galvão C, Carcavallo R, Rocha DS, et al.. A checklist of the current valid species of the subfamily Triatominae Jeannel, 1919 (Hemiptera, Reduviidae) and their geographical distribution, with nomenclatural and taxonomic notes. Zootaxa. 2003;202:36. [Google Scholar]
- 4. Justi SA, Galvão C. The evolutionary origin of diversity in Chagas disease vectors. Trends Parasitol. 2017;33(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carod-Artal FJ. American trypanosomiasis. Handbook of Clinical Neurology. 2013;114:103–123. [DOI] [PubMed] [Google Scholar]
- 6. Coura JR, Vinas PA. Chagas disease: a new worldwide challenge. Nature. 2010;465:2. [DOI] [PubMed] [Google Scholar]
- 7. Liu Q, Guo YH, Zhang Y, et al.. First records of Triatoma rubrofasciata (De Geer, 1773) (Hemiptera, Reduviidae) in Foshan, Guangdong Province, Southern China. Infect Dis Poverty. 2017;6(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neff KL, Argue DP, Ma AC, et al.. Mojo Hand, a TALEN design tool for genome editing applications. BMC Bioinformatics. 2013;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu B, Shi Y, Yuan J et al.. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biol. 2013;35:3. [Google Scholar]
- 11. Mesquita RD, Vionette-Amaral RJ, Lowenberger C et al.. Genome of Rhodnius prolixus, an insect vector of Chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc Natl Acad Sci U S A. 2015;112(48):14936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chin CS, Peluso P, Sedlazeck FJ et al.. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 2016;13(12):1050–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chin CS, Alexander DH, Marks P, et al.. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9. [DOI] [PubMed] [Google Scholar]
- 14. Walker BJ, Abeel T, Shea T, et al.. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong G, Dan C, Xiao S, et al.. Chromosomal-level assembly of yellow catfish genome using third-generation DNA sequencing and Hi-C analysis. Gigascience. 2018;7(11):1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu S, Xiao S, Zhu S et al.. A draft genome assembly of the Chinese sillago (Sillago sinica), the first reference genome for Sillaginidae fishes. Gigascience. 2018;7(9):1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Langmead B, Trapnell C, Pop M et al.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Near TJ, Dornburg A, Eytan RI, et al.. Phylogeny and tempo of diversification in the superradiation of spiny-rayed fishes. Proc Natl Acad Sci U S A. 2013;110(31):12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Simao FA, Waterhouse RM, Ioannidis P, et al.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McKenna A, Hanna M, Banks E, et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. http://www.repeatmasker.org/RepeatModeler.html. [Google Scholar]
- 24. Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2004; 4:1-14. [DOI] [PubMed] [Google Scholar]
- 26. Stanke M, Keller O, Gunduz I, et al.. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(Web Server issue):W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu TD, Reeder J, Lawrence M, et al.. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol Biol. 2016;1418:283–334. [DOI] [PubMed] [Google Scholar]
- 28. Campbell MS, Holt C, Moore B, et al.. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics. 2014;48:1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quevillon E, Silventoinen V, Pillai S, et al.. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(Web Server issue):W116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boeckmann B, Bairoch A, Apweiler R, et al.. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31(1):365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gertz EM, Yu YK, Agarwala R, et al.. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Camacho C, Coulouris G, Avagyan V, et al.. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. [DOI] [PubMed] [Google Scholar]
- 36. Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–2. [DOI] [PubMed] [Google Scholar]
- 37. Kumar S, Stecher G, Suleski M, et al.. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 2017;34(7):1812–9. [DOI] [PubMed] [Google Scholar]
- 38. Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13(5):2. [DOI] [PubMed] [Google Scholar]
- 39. Liu Q, Guo YH, Zhang Y et al.. Supporting data for "A chromosomal-level genome assembly for the insect vector for Chagas disease, Triatoma rubrofasciata". GigaScience Database. 2019. 10.5524/100614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ben Mans -- 2/25/2019 Reviewed
Ben Mans -- 5/13/2019 Reviewed
Ting-Fung Chan, PhD -- 3/9/2019 Reviewed
Ting-Fung Chan, PhD -- 5/18/2019 Reviewed
Data Availability Statement
The raw data from our genome project was deposited in the NCBI Sequence database with Bioproject IDs PRJNA516044. The Illumina, PacBio, and Hi-C sequencing data are available from NCBI via accession numbers SRR8466736, SRR8466737, and SRR8466756, respectively. The Illumina transcriptome sequencing data were deposited to NCBI via accession numbers SRR8468315 and SRR8468316. Other data further supporting this work are available in the GigaSciencerepository, GigaDB [39].