

Abstract
The cause of most hypertensive disease is unclear, but inflammation appears critical in disease progression. However, how elevated blood pressure initiates inflammation is unknown, as are the effects of high blood pressure on innate and adaptive immune responses. We now report that hypertensive mice have increased T cell responses to antigenic challenge and develop more severe T cell-mediated immunopathology. A root cause for this is hypertension-induced erythrocyte adenosine 5′-triphosphate (ATP) release, leading to an increase in plasma ATP levels, which begins soon after the onset of hypertension and stimulates P2X7 receptors on antigen-presenting cells (APCs), increasing APC expression of CD86. Hydrolyzing ATP or blocking the P2X7 receptor eliminated hypertension-induced T cell hyperactivation. In addition, pharmacologic or genetic blockade of P2X7 receptor activity suppressed the progression of hypertension. Consistent with the results in mice, we also found that untreated human hypertensive patients have significantly elevated plasma ATP levels compared with treated hypertensive patients or normotensive controls. Thus, a hypertension-induced increase in extracellular ATP triggers augmented APC and T cell function and contributes to the immune-mediated pathologic changes associated with hypertensive disease.
INTRODUCTION
Hypertension is the source of enormous morbidity and mortality throughout the world (1). The World Health Organization estimates that the number of people with uncontrolled hypertension is nearly 1 billion and that this disease causes about 12% of all adult deaths (2). Although hypertension has been studied for many years, the cause of disease in most patients is still not understood. Hypertension is accompanied by low-grade chronic inflammation (3, 4). Recently, evidence suggests that inflammation not only is associated with hypertension but also may represent a major pathologic process driving development and progression of the disease. For example, immune-deficient RAG-1 knockout mice have a reduced blood pressure (BP) response to several models of hypertension (5). In addition, transfer of dendritic cells (DCs) from hypertensive mice to normotensive recipients primed the recipients for CD8+ T cell proliferation and an exaggerated BP response to a mild hypertensive insult (6). These studies, and many others, have suggested that hypertension has some features of an autoimmune disease in which both antigen-presenting cells (APCs) and T cells elicit a higher BP (7, 8). What is not well understood is the cause of the hypertension-associated inflammatory response and the temporal relationship between the elevation of BP and the onset of inflammation. Further, very little is known about the precise effects of hypertension on immune responses, although clinical studies indicate a positive correlation between hypertension and autoimmune diseases (9–11).
Different from pathogen-associated molecular patterns, damage-associated molecular patterns (DAMPs) are host biomolecules that can initiate and perpetuate a noninfectious inflammatory response. Many metabolites can act as DAMPs (12), such as adenosine 5′-triphosphate (ATP), uric acid, and oxidized low-density lipoprotein (oxLDL). When tissue is damaged or under stress, DAMPs may be released or increasingly formed from cells, and the elevated extracellular DAMPs can mobilize and activate immune cells. When serving as a DAMP, ATP exerts its function by binding to and activating purinergic P2 receptors (13). For example, APCs express P2X7 receptors and extracellular ATP has been shown to modulate their response in cancer and in chronic kidney disease (14, 15). P2X7 is a nucleotide-gated ion channel. Activation of P2X7 by extracellular ATP allows for the passage of small cations, including Ca2+, Na+, and K+, across the plasma membrane, which gives rise to a variety of downstream cellular events, such as inflammasome activation, reactive oxygen species (ROS) formation, prostaglandin release, transcription activation [such as through nuclear factor κB (NF-κB) pathway], and phagocytosis (16–18).
In this study, we investigated how hypertension affects the immune response and how the hypertension-associated inflammatory response is triggered. We demonstrate that an increase in plasma ATP is one of the earliest hallmarks of hypertension and is directly responsible for APC-mediated overactivity of T cells in response to immune challenges, thereby predisposing hypertensive mice to immune-mediated diseases. These exaggerated immune responses may also contribute to the progression of hypertension.
RESULTS
Hypertension increases antigen-specific T cell responses
To investigate whether hypertension affects immune responses, we studied the reaction to ovalbumin (OVA) inoculation in C57BL/6 normotensive mice and mice made hypertensive with angiotensin (Ang) II. After a 2-week infusion, when the systolic BP (SBP) was raised to a plateau between 140 and 150 mmHg (fig. S1), mice were immunized subcutaneously with OVA emulsified in complete Freund’s adjuvant (CFA). Seven days later, tetramers were used to measure the quantity of blood CD8+ T cells specific for the OVA epitope SIINFEKL. There were significantly more OVA-specific CD8+ T cells in both absolute number and percentage of total CD8+ T cells in hypertensive mice as compared with normotensive animals (Fig. 1A). To exclude that the heightened immune response was adjuvant or route specific, we also immunized mice intraperitoneally with OVA in combination with lipopolysaccharide (LPS) or alum. For the OVA-alum group, splenocytes were restimulated after 7 days with SIINFEKL peptide and supernatant levels of interleukin-2 (IL-2) and interferon-γ (IFN-γ) were assessed. Again, a heightened immune response was observed in the hypertensive animals (Fig. 1, B and C). The OVA-specific CD4+ T cell response was also examined by restimulating splenocytes from the OVA-CFA and OVA-alum groups with the major histocompatibility complex (MHC) class II peptide OVA 323–339. Hypertensive mice had an elevated CD4+ T cell response in comparison with normotensive mice, as shown by more IL-2 expression upon restimulation (Fig. 1D). In toto, these data indicate that Ang II—induced hypertension elicits increased T cell activation in response to immunization with OVA.
Fig. 1. Antigen-specific immune responses are enhanced in hypertension.
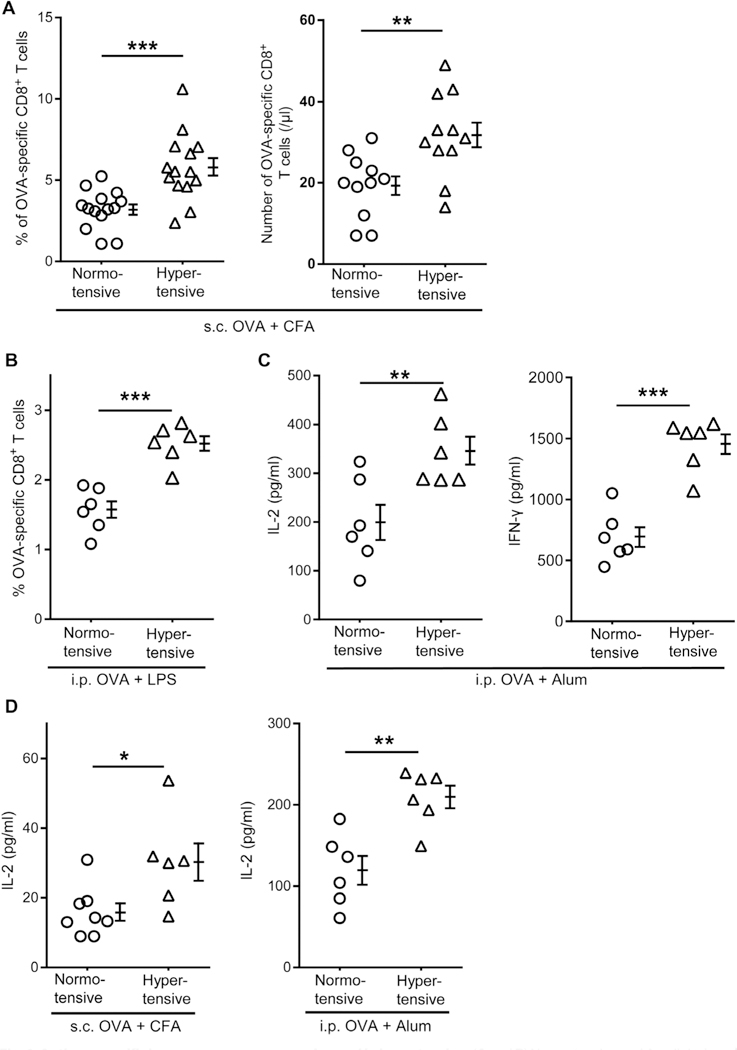
(A and B) Normotensive and Ang II-induced hypertensive C57BL/6 mice were immunized subcutaneously (s.c.) with OVA-CFA (A) or intraperitoneally (i.p.) with OVA-LPS (B). Seven days later, the percentages and numbers of OVA-specific T cells among total CD8+ T cells in blood (A) and spleen (B) were determined by SIINFEKL-H-2Kb tetramer staining. (C) Mice were immunized intraperitoneally with OVA-alum. Seven days later, their splenocytes were restimulated with SIINFEKL peptide. IL-2 and IFN-γ secretion were measured after 24 and 72 hours, respectively, by ELISA. (D) Seven days after the immunization with OVA-CFA or OVA-alum, splenocytes were restimulated with the MHC class II dominant peptide OVA323–339. Secretion of IL-2 was measured at 24 hours by ELISA. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.005.
Hypertension predisposes mice to T cell-mediated immunopathology
To determine whether the enhanced T cell responses in hypertensive mice translate into increased tissue pathology, we first studied a model of autoimmune diabetes. Rat insulin promoter (RIP)-mOVA mice express OVA in insulin-producing pancreatic islet cells (19, 20). Injected OT-I T cells that express transgenic T cell receptors (TCRs) specific for OVA 257–264 are eliminated in the periphery when a low number of such cells are given (20, 21). We found that the adoptive transfer of a high number (5 × 106) of OT-I T cells into normotensive RIP-mOVA mice was insufficient to induce diabetes in the first week. After the second week, mice were on the threshold of diabetes with blood glucose levels that averaged 205 ± 21 mg/dl (Fig. 2A). When the same number of OT-I T cells was transferred into hypertensive RIP-mOVA mice continuously treated with Ang II (fig. S2), 83% of hypertensive mice developed diabetes (blood glucose above 200 mg/dl) during the first week, and by the end of the second week, mice were severely diabetic with an average blood glucose of 361 ± 40 mg/dl (hypertensive versus normotensive, P < 0.001). Nitric oxide synthase inhibitor N(G)-Nitro-L-arginine methyl ester (L-NAME) can trigger hypertension (fig. S1) by a mechanism different from Ang II. The pattern of an accelerated development of diabetes was repeated when hypertension was induced by L-NAME instead of Ang II (Fig. 2A); 2 weeks after OT-I transfer, the hypertensive mice averaged 325 ± 50 mg/dl blood glucose (P = 0.02 versus normotensive mice). The more aggressive diabetes in hypertensive mice was accompanied by increased accumulation/expansion of OT-I T cells in peripancreatic lymph nodes (Fig. 2B). Last, immunohistochemistry documented significantly more CD3+ T cells within the pancreatic islets of hypertensive mice (Fig. 2C).
Fig. 2. Hypertensive mice are predisposed to autoimmune diseases.
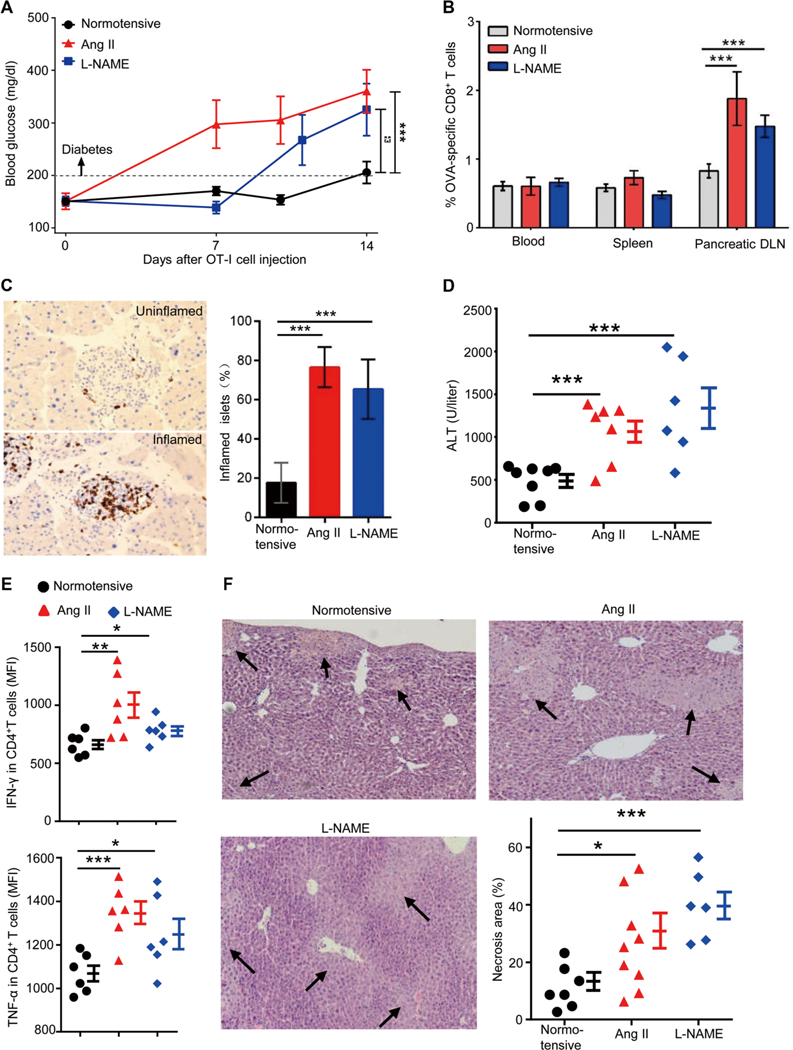
(A to C) RIP-mOVA mice were made hypertensive by treatment with either Ang II or L-NAME. After 2 to 3 weeks, when hypertension was established, 5 × 106 OT-I T cells (CD8+ T cells from OT-I transgenic mice) were transferred intravenously into normotensive and hypertensive RIP-mOVA mice. (A) Blood glucose levels were measured for 2 weeks after OT-I T cell transfer. (B and C) Two weeks after OT-I T cell transfer, the percentages of OT-I T cells among CD8+ T cells in the blood, spleen, and pancreatic draining lymph nodes (DLN) were quantified by tetramer analysis (B), and the numbers of inflamed islets, identified by anti-CD3 staining, were counted in a blinded fashion (C). (D to F) ConA (5 mg/kg) was injected intravenously into normotensive and hypertensive mice. Blood ALT levels were measured after 6 hours. (D) Hepatic inflammatory cells were prepared by tissue enzymatic digestion followed by Percoll centrifugation. (E) Cells were cultured in medium for 6 hours in the presence of brefeldin A, and then intracellular staining was performed to examine the production of IFN-γ and TNF-α by CD4+ T cells. Liver necrosis area was measured after hematoxylin and eosin staining. MFI, mean fluorescence intensity. (F) For each liver, necrosis area represents the average of 10 separate fields. In (A) to (C), n = 17, 6, and 12 for normotensive, Ang II-treated, and L-NAME-treated mice, respectively. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.005.
Concanavalin A (ConA)-induced acute hepatitis is a well-studied mouse model of immune-mediated liver injury, and its pathogenesis is caused by activation of liver-resident T cells including natural killer T (NKT) cells (22–24). The administration of ConA caused acute liver injury, which was followed by the rapid increase of plasma alanine transaminase (ALT) levels (fig. S3A). When mice from both hypertensive models and normotensive mice were compared, plasma ALT levels of hypertensive mice were more than twofold higher than those of normotensive mice at 6 hours after ConA (Fig. 2D). Flow cytometric analysis at 6 hours after ConA showed that the CD4+ T cells in the livers of hypertensive mice produced significantly more of the proinflammatory cytokines TNF-α (tumor necrosis factor-α) and IFN-γ than their counterparts from normotensive mice (Fig. 2E and fig. S3B). This was accompanied by increased necrosis in hypertensive livers as disclosed by histologic analysis (Fig. 2F). Thus, the data from two in vivo models indicate that hypertension predisposes mice to more intense T cell-mediated immunopathology.
APCs from hypertensive mice present antigen more effectively than normotensive cells
To determine whether hypertension induces changes in the sensitivity of TCR-mediated signaling, we used OT-I mice in which CD8+ T cells express a transgenic TCR sensitive to the OVA epitope SIINFEKL. OT-I mice exhibit a normal hypertensive response to infused Ang II (fig. S4A). Two weeks after the start of Ang II infusion, splenocytes were prepared and coincubated with DCs from normotensive C57BL/6 mice previously pulsed with either SIINFEKL (N4), SIITFEKL (T4), SIIGFEKL (G4), or RTYTYEKL (RTY) (25). When presented on MHC class I molecules, these peptides differ in their affinity for the OT-I TCR. Weak agonist peptides and lower doses induced less activation, but the CD8+ T cells derived from hypertensive OT-I mice behaved identically to their counterparts from normotensive OT-I mice, as assessed by both CD69 expression and IFN-γ production (fig. S4B). This indicates that the heightened immune response observed in hypertensive mice was probably not due to changes in the sensitivity of TCR signaling.
To investigate whether APCs are responsible for the enhanced T cell responses in hypertensive animals, we collected splenic DCs and peritoneal macrophages from both normotensive and Ang II-induced hypertensive C57BL/6 mice. Cells were pulsed with OVA, and their ability to activate OT-I T cells was compared. DCs and macrophages from hypertensive mice were both superior to their counterparts from normotensive mice in stimulating OT-I T cells, as indicated by increased T cell expression of CD69 and production of IFN-γ (Fig. 3A). This was not due to a higher efficiency of antigen processing because the hypertensive APCs were still superior to normotensive APCs when both populations were loaded with SIINFEKL peptide (Fig. 3B). In contrast, without antigen feeding, normotensive and hypertensive APCs were equivalent in their effect on OT-I T cells, excluding the possibility of an antigen-independent mechanism. To understand whether this change is Ang II specific, we assessed antigen presentation by splenic DCs from mice made hypertensive with L-NAME (Fig. 3C). Again, the APCs from the L-NAME-induced hypertensive mice were more efficient in presenting and activating T cells ex vivo, suggesting that this change can be induced in multiple mouse models of experimental hypertension.
Fig. 3. APCs from hypertensive mice present antigens more efficiently.
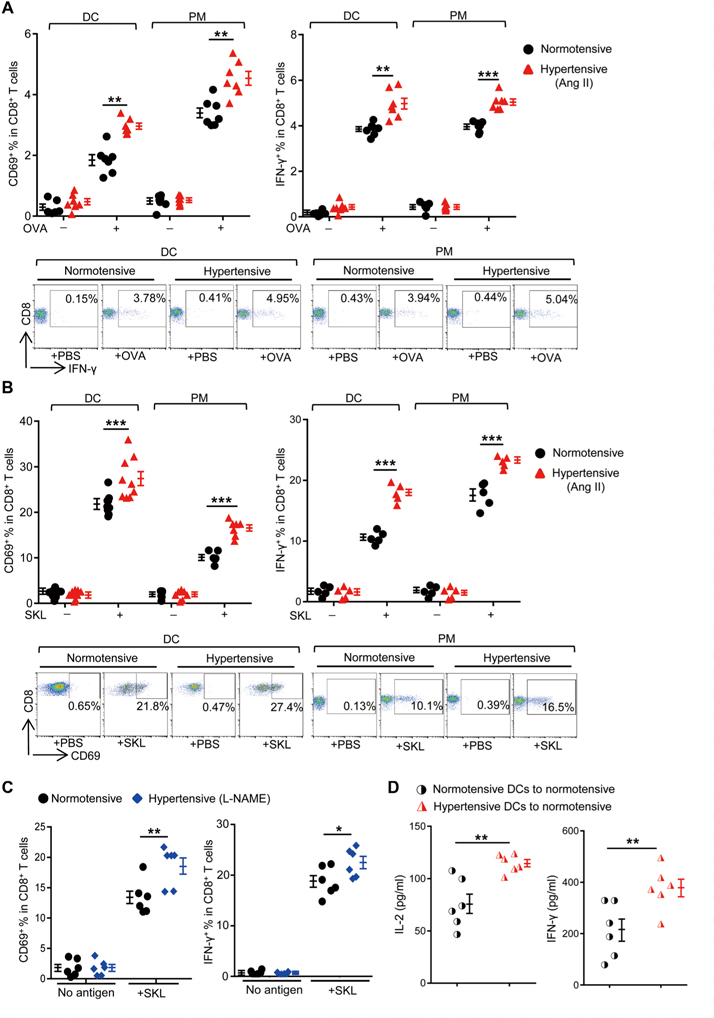
(A and B) Splenic DCs and peritoneal macrophages (PM) from normotensive or Ang Il-induced hypertensive C57BL/6 mice were pulsed with 10 μM OVA (A) or 50 pM SIINFEKL peptide (SKL) (B) in vitro and were then coincubated with OT-I T cells. The percentages of CD69+ (after 4 hours) or IFN-γ+ cells (after 6 hours in the presence of brefeldin A) among OT-I T cells were measured by flow cytometry. Representative flow cytometry dot plots of IFN-γ and CD69 expression are shown. (C) Splenic DCs from normotensive or L-NAME-induced hypertensive mice were pulsed with SIINFEKL and then coincubated with OT-I T cells. The percentages of CD69+ or IFN-γ+ cells among all CD8+ T cells were measured. (D) Splenic DCs from normotensive or Ang II-induced hypertensive mice were loaded with SIINFEKL for 4 hours ex vivo. Cells were then transferred intravenously into naїve C57BL/6 mice. Seven days later, splenocytes from the recipients were restimulated with SIINFEKL and the secretion of IL-2 and IFN-γ was measured. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.005.
To study whether the enhanced immune responses in hypertensive mice can be transferred by APCs, we collected splenic DCs and fed them with SIINFEKL in vitro. The antigen-loaded APCs were then transferred intravenously into naïve (normotensive) C57BL/6 recipients. Eight days after transfer, the SIINFEKL-specific T cell responses of the recipients were compared by restimulating splenocytes and measuring levels of IL-2 and IFN-γ (Fig. 3D). The transfer of hypertensive DCs increased the activation of T cells significantly more than equivalent cells from normotensive mice. These data demonstrate that, in mice, hypertension is accompanied by an enhanced ability of APCs to present antigens to T cells.
Increased CD86 expression on hypertensive APCs is responsible for the enhanced activation of T cells
The activation of T cells by APCs integrates TCR stimulation, costimulatory signals, and cytokine effects, which fine-tune T cell proliferation and differentiation. To understand the cause of enhanced antigen presentation by hypertensive APCs, we first measured the surface levels of MHC molecules on DCs, but neither H-2Kb, H-2Db, nor I-Ab showed any differences between hypertensive and normotensive cells (fig. S5A). We also injected normotensive and hypertensive C57BL/6 mice intravenously with SIINFEKL and, 12 hours later, measured the surface abundance of the SIINFEKL-H-2Kb complex on splenic DCs using the antibody 25-D1.16 (fig. S5B) (26). There was no difference between cells from hypertensive and normotensive mice.
Costimulatory factors on APCs are critical in facilitating T cell activation. We noticed that both splenic DCs and peritoneal macrophages derived from hypertensive mice had higher expression of CD86 but not CD80 as compared with their counterparts from normotensive mice (Fig. 4A). Increased CD86 expression was also observed on Kupffer cells of the liver in hypertensive mice (Fig. 4A). The increase is not due to the pharmacological effects of Ang II on APCs because in vitro stimulation of naïve APCs with Ang II did not increase CD86 expression (Fig. 4B). To determine whether BP affects CD86 expression by APCs, we normalized BP during Ang II infusion by cotreating the mice with hydralazine, a BP-reducing medicine. Lowering BP to normal levels completely abrogated the up-regulation of CD86 on APCs (Fig. 4C).
Fig. 4. CD86 is up-regulated on the APCs of hypertensive mice.
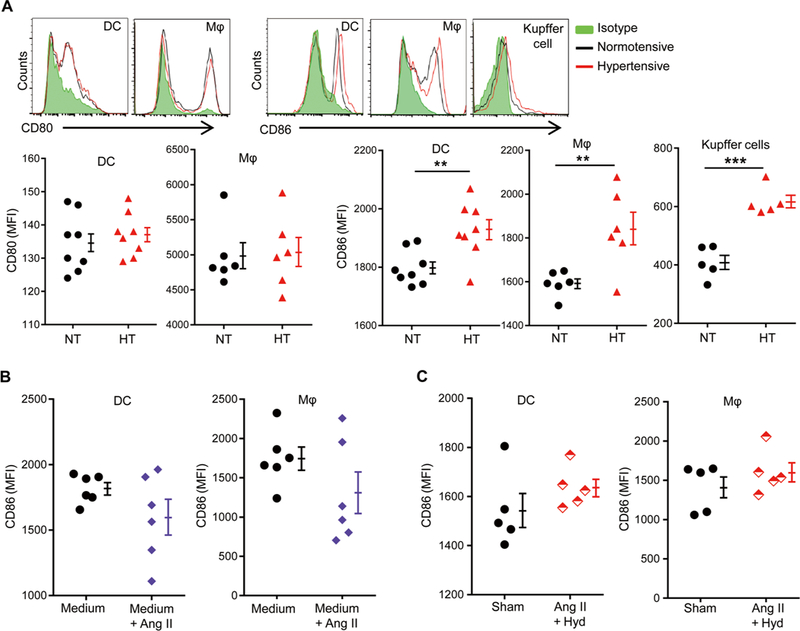
C57BL/6 mice were sham-treated (NT) or Ang Il-treated (HT) for 2 weeks. (A) Surface CD80 and CD86 expression on splenic DCs and peritoneal macrophages (Mφ). Surface expression of CD86 by Kupffer cells was also measured. The representative histograms are shown. (B) DCs and macrophages from naive mice were stimulated in vitro with or without 10 μM Ang II overnight, and surface CD86 expression was measured. (C) Mice were either sham-treated or treated with Ang II and hydralazine (Hyd) for 14 days. Surface CD86 expression was measured in splenic DCs and peritoneal macrophages. Data are means ± SEM. **P < 0.01; ***P < 0.005.
CD40 is another APC maturation marker that functions as a receptor for CD40L-induced DC activation. Indoleamine-pyrrole 2,3-dioxygenase (IDO) has also been implicated in immune modulation through its ability to limit T cell function and engage mechanisms of immune tolerance (27). There was no difference in CD40 expression between hypertensive DCs and normotensive cells (fig. S5C). Although hypertensive DCs did average less expression of IDO, the difference between these cells and normotensive DCs was not significant (P = 0.13). Proinflammatory cytokines secreted by APCs are important in educating naïve T cells and eliciting differentiation. However, there were minor differences in expression of TNF-α, pro-IL-1β/IL-1β, IL-6, or IL-12/p40 between hypertensive and normotensive APCs at baseline and after LPS stimulation (fig. S6). Together, our studies of T cells and APCs indicate that the up-regulation of CD86 on APCs is the most distinguishable difference in the immune systems between hypertensive and normotensive animals.
We wondered whether the elevation of CD86 in hypertensive DCs was important for the observed increase in T cell activation. To study this, we purified DCs from the spleens of hypertensive and normotensive mice and then pretreated them with either anti-CD86 antibody, anti-CD80 antibody, or a combination of both antibodies. DCs were then loaded with SIINFEKL and tested for their ability to activate OT-I cells. We found that anti-CD86, but not anti-CD80, completely eliminated the enhanced ability of hypertensive DCs to activate T cells. Specifically, OT-I T cells incubated with either anti-CD86-treated normotensive or anti-CD86-treated hypertensive DCs showed equivalent expression of CD69 and IFN-γ (Fig. 5A). In contrast, hypertensive DCs treated with anti-CD80 still showed enhanced activation of T cells as compared with equivalently treated DCs from normotensive mice. The treatment of DCs with both anti-CD86 and anti-CD80 antibodies did not reduce T cell activation more than the use of anti-CD86 alone.
Fig. 5. Blocking CD86 eliminates the overactivation of immune responses in hypertensive mice.
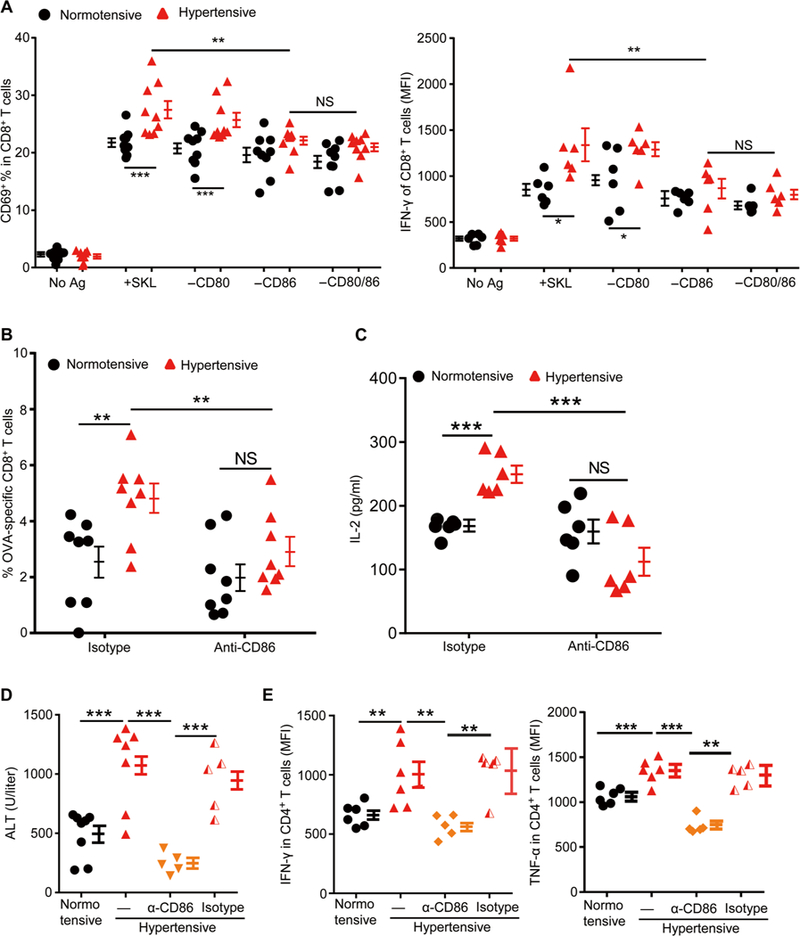
C57BL/6 mice were sham-treated (normotensive) or Ang Il-treated (hypertensive) for 2 weeks. (A) Splenic DCs were purified. Some of the cells were pulsed with SIINFEKL peptide. Some cells were also treated with an anti-CD80 antibody or an anti-CD86 antibody or both. They were then coincubated with OT-I T cells, and the expression of T cell CD69 and IFN-γ was measured. (B and C) Mice were regularly treated with an anti-CD86 antibody or an isotype control antibody from 3 days before being immunized with OVA until sacrifice. Seven days after immunization, the numbers of OVA-specific CD8+ T cells in the blood (B) and splenocyte IL-2 production after SIINFEKL restimulation were measured (C). (D and E) Acute hepatitis was induced by intravenous injection of ConA (5 mg/kg) into normotensive and hypertensive mice. Some hypertensive mice were injected intraperitoneally with either CD86 blocking antibody (α-CD86) or an isotype control antibody (IgG) every other day for three times before ConA administration. (D) Blood ALT levels were measured 6 hours after ConA injection. (E) Leukocytes in the liver were isolated with Percoll gradients. Cells were cultured in medium for 6 hours in the presence of brefeldin A, and then intra-cellular staining was performed to examine the production of IFN-γ and TNF-γ by CD4+ T cells. Data are means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.005. NS, not significant.
To evaluate the role of CD86 in vivo, we administered hypertensive and normotensive mice either anti-CD86 or an isotype control antibody (day 0). Additional antibody was given every 3 days. On day 3, mice were immunized with OVA-CFA, and they were euthanized on day 11. Both the numbers of OVA-specific CD8+ T cells in blood and the IL-2 production by splenocytes were greatly reduced in CD86-blocked hypertensive mice so that they were indistinguishable from those of CD86-blocked normotensive mice (Fig. 5, B and C). When hypertensive mice were treated with the CD86-blocking antibody before challenge with ConA, both plasma ALT levels and the overactivation of hepatic CD4+ T cells could be normalized (Fig. 5, D and E). Thus, both in vitro and in vivo analyses indicate an important role of CD86 in the heightened immune response present in hypertension.
Elevation of plasma ATP is an early event in hypertension development and is responsible for CD86 up-regulation in APCs
If we model hypertension as a sterile inflammatory disease, then an obvious question is what initiates the inflammatory process. We posit that disrupted hemodynamics caused by continuous high BP may lead to the release or formation of DAMPs from otherwise normal tissues. To examine this hypothesis, we evaluated the plasma levels of uric acid, oxLDL, HMGB-1 (high-mobility group box 1 protein), and ATP, a collection of DAMPs involved in a variety of cardiovascular diseases (28–31). Mice were made hypertensive by injection of either Ang II or L-NAME for 6 days, and blood plasma was collected for analysis. At this early stage of hypertension, all the examined DAMPs except ATP were decreased in hypertensive mice as compared with normotensive mice. In contrast, ATP was significantly elevated in both hypertensive models (Fig. 6A). To investigate ATP in more detail, we then measured plasma ATP levels during the course of disease (Fig. 6B). The ATP concentration began to rise as early as 3 days after the induction of hypertension, with a significant rise by day 5 (L-NAME) or day 6 (Ang II) with a peak level of 3 μM after 2 weeks of disease. This is due to the direct effects of BP because a cohort of mice cotreated for 2 weeks with Ang II and hydralazine maintained normal BPs and normal plasma levels of ATP (Fig. 6B). Hypertension also up-regulated CD86 expression by splenic DCs. This began as early as 4 days after initiation of hypertension and was significant at 7 and 14 days (Fig. 6C). The kinetics of CD86 expression exactly parallel the rise of plasma ATP levels.
Fig. 6. ATP is elevated early in hypertension and is responsible for CD86 up-regulation through the P2X7 receptor in APCs.
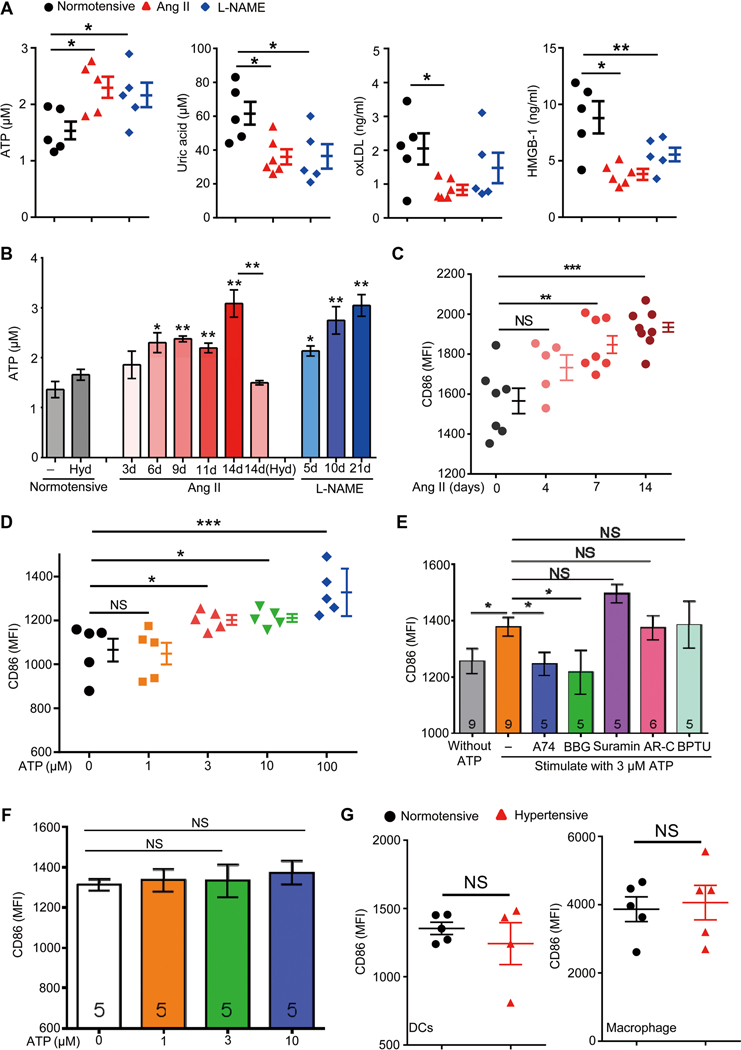
(A) DAMPs including ATP, uric acid, oxLDL, and HMGB-1 were measured in the plasma of normotensive mice, and mice were made hypertensive for 6 days. (B) Blood plasma ATP levels were measured at different time points after hypertension induction. A group of mice were cotreated with Ang II and hydralazine [14d(Hyd)]. n = 5 for normotensive, Ang II-treated, and Ang II and hydralazine cotreated mice; n = 4 for L-NAME-treated mice. (C) Surface CD86 expression on splenic DCs from mice infused with Ang II for 0, 4, 7, or 14 days. (D) Splenic DCs from naïve mice were stimulated with the indicated concentrations of ATP for 12 hours in vitro, and their surface CD86 expression was measured. (E) Splenic DCs were stimulated with medium or 3 μM ATP. Some ATP-treated cells were also incubated with antagonists to P2X7 [A740003 (A74) or BBG], pan-P2Y (suramin), P2Y2 [AR-C 118925XX (AR-C)], or P2Y1 (BPTU). Surface CD86 was measured after 18 hours. (F) Splenic DCs from P2X7-deficient mice were treated with different concentrations of ATP, and surface CD86 was measured. (G) Surface CD86 expression on splenic DCs and peritoneal macrophages was derived from normotensive and hypertensive P2X7-deficient mice.
To investigate the relationship of ATP and CD86 expression, we stimulated naïve DCs in vitro with various concentrations of ATP. The plasma concentration of normotensive mice is about 1 μM. In vitro, this concentration had no effect on DC CD86 expression (Fig. 6D). In contrast, as little as 3 μM ATP increased DC CD86 expression. ATP levels did up-regulate CD80 but in a relatively in-efficient manner, with a significant effect only at 100 μM (fig. S7A). In hypertensive mice, we did not detect an increase of CD86 on blood monocytes even after 2 weeks of Ang II administration. This may be due to a difference in sensitivity of monocytes and DCs to ATP because the up-regulation of CD86 by monocytes was observed at 30 μM but not at 3 μM ATP (fig. S7B). Considering that myeloid cells express several different ATP P2 receptors (32–34), we next investigated which receptor mediates the effect of ATP. Naïve DCs derived from spleen were stimulated in vitro with 3 μM ATP in the presence of the inhibitors A740003 (P2X7), Brilliant Blue G (BBG) (P2X7), suramin (pan-P2Y), AR-C 118925XX (P2Y2), or BPTU (P2Y1). The P2X7 blockers completely abrogated CD86 up-regulation, whereas other blockers showed no effect (Fig. 6E). To substantiate that the P2X7 receptor is critical, we made use of DCs derived from mice in which the P2rx7 gene was deleted. ATP (3 μM) had no effect on CD86 expression in P2X7-deficient DCs (Fig. 6F). In addition, CD86 overexpression was not observed on either DCs or macrophages derived from hypertensive P2X7 knockout mice (Fig. 6G).
Activation of the P2X7 receptor on APCs by 3 μM ATP
The P2X7 receptor is a nucleotidegated ion channel. Activation by extracellular ATP allows for the passage of small cations, including Ca2+, across the plasma membrane (34). It is generally thought that the P2X7 receptor has low affinity for ATP and requires more than 100 μM ATP for activation (35, 36). However, these studies were mostly performed in P2X7-transfected kidney human embryonic kidney (HEK)-293 cells or in astrocytoma 1321N1 cells and may not reflect the biological activity of P2X7 in APCs. When APCs were preincubated with the calcium indicator Fluo-4 AM and then treated with 3 μM ATP, this triggered an acute Ca2+ influx that, although less than the response to 500 μM ATP, could still be readily detected by both flow cytometry and confocal microscopy (Fig. 7A and fig. S8). T cells also express P2X7 (34, 37). When the same stimuli were applied to splenic T cells, 500 μM ATP induced a similar Ca2+ influx as that in APCs. However, 3 μM ATP did not trigger Ca2+ influx in T cells (Fig. 7A). The specificity of P2X7 in mediating these effects was verified by testing P2X7-deficient cells, which exhibited no Ca2+ influx when stimulated with either dose of ATP (Fig. 7A). These data suggest that P2X7 activity is cell specific and that this receptor in APCs is sensitive to 3 μM ATP.
Fig. 7. Activation of the P2X7 receptor on APCs by 3 μM ATP initiates the heightened immune responses associated with hypertension.
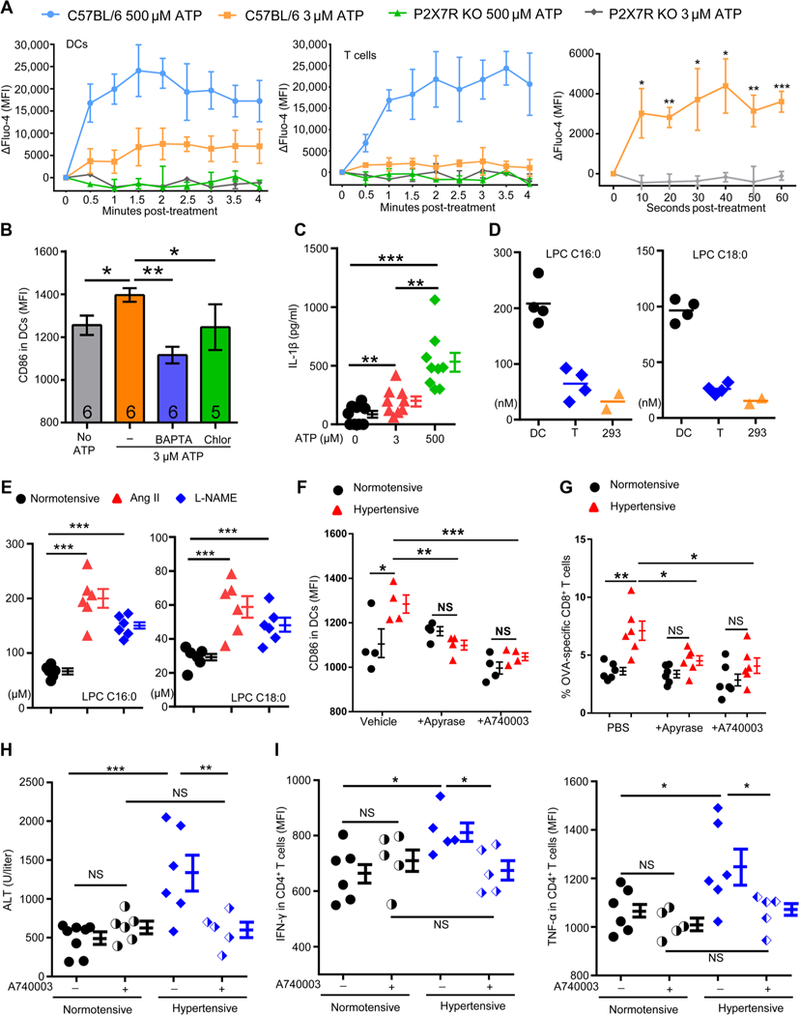
(A) Splenocytes from either C57BL/6 or P2X7 knockout mice were labeled with the Ca2+ probe Fluo-4 AM in vitro. The cells were then treated with either 3 or 500 μM ATP, and Ca2+ influx into DCs (left) and T cells (middle) was monitored continuously for 4 min by flow cytometry (right). The first minute kinetics of Ca2+ influx into DCs in the presence of 3 μM ATP is also shown. (B) Splenic DCs were stimulated with or without 3 μM ATP. Some of the ATP-treated cells were pretreated with the calcium chelator BAPTA or the calmodulin inhibitor chlorpromazine (Chlor). Surface CD86 was measured after 18 hours. (C) Peritoneal macrophages were treated with 3 or 500 μM ATP for 18 hours ex vivo, and IL-1β in the medium was measured by ELISA. Data were analyzed with paired Student′s t test. (D) Splenic DCs and T cells were purified from the spleen of naїve mice. DCs (2 × 106), T cells, and HEK-293 cells were pelleted and then dissolved in 100 μl of acetonitrile. The concentrations of LPCs C16:0 and C18:0 were determined by LC-MS/MS. (E) Concentrations of LPCs C16:0 and C18:0 in the plasma of normotensive and hypertensive mice. (F) Splenic DCs from normotensive and hypertensive mice were treated with vehicle, apyrase, or A740003 for 18 hours. Surface expression of CD86 was then measured. (G) Normotensive and hypertensive mice were pretreated intraperitoneally with apyrase (0.2 U/g weight) or A740003 (50 nmol/mouse) every other day for 6 days, and they were then immunized with OVA-CFA. Apyrase or A740003 was continued after immunization. The percentages of OVA-specific CD8+ T cells in the blood were evaluated 7 days after immunization. (H and I) Normotensive and hypertensive mice were treated with ConA to induce hepatitis with or without the P2X7 receptor antagonist A740003. Plasma ALT levels (H) and CD4+ T cell cytokines (I) were measured.
To understand the effect of Ca2+ influx on CD86 up-regulation, we stimulated DCs with 3 μM ATP in the presence of either BAPTA [1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid], a Ca2+ chelator, or chlorpromazine, a calmodulin inhibitor (Fig. 7B). CD86 up-regulation was completely inhibited by either BAPTA or chlorpromazine, suggesting that P2X7-gated Ca2+ influx mediates CD86 expression.
The downstream effects of P2X7 activation are multifaceted, including inflammasome activation, production of ROS, and release of prostaglandin E2 (PGE2) (34). ATP (3 μM) stimulation did induce a mild but significant release of IL-1β by APCs (Fig. 7C), but it did not trigger the production of ROS or PGE2 (fig. S9). We did not detect elevated IL-1β in the plasma of hypertensive mice probably due to the far lower concentration of ATP compared with the millimolar range upon tissue damage; we note that 500 μM ATP caused a three-fold higher IL-1 b secretion than 3 μM ATP (Fig. 7C). An important feature of the P2X7 receptor is that, in addition to the usual rapid opening of the cation-selective ion channel, it becomes permeable to larger molecules with prolonged exposure to ATP, often leading to cell death (34). Our results show that, unlike 500 μM ATP, 3 μM ATP did not induce YO-PRO-1 entrance into DCs or cell death (fig. S10). Thus, the effects of 3 μM ATP on DCs are dose specific.
Next, we asked why the P2X7 receptors on DCs but not those on T cells or HEK-293 cells respond to low concentration of ATP. Lysophosphatidylcholine (LPC) has been reported to increase agonist potency at the P2X7 receptor possibly due to changes in membrane properties (38, 39). Among the various forms of LPC, the greatest effects on P2X7 were observed with C16:0 and C18:0 (39). We studied C16:0 and C18:0 in DCs, T cells, and HEK-293 cells by mass spectrometry and found that DCs have the highest levels of both LPCs (C16:0: 3.2- and 6.3-fold of those in T cells and in HEK-293 cells, respectively; C18:0: 3.7- and 6.3-fold of those in T cells and in HEK-293 cells, respectively) (Fig. 7D). These differences may reflect the requirements of professional phagocytes. We also measured LPC in the plasma of hypertensive mice. There were consistently higher concentrations of C16:0 and C18:0 in both Ang II and L-NAME hypertensive mice than in normotensive mice (Fig. 7E). It is reported that supplementation of 80 to 100 μM LPC can enhance P2X7 activity (38, 39). The sum of the concentrations of C16:0 and C18:0 is about 200 to 250 μM in hypertensive mice. Thus, besides a high content of LPC in the membrane of DCs, high concentrations of free LPC may give an additional boost to the activation of P2X7 in hypertension.
Increased plasma ATP initiates the heightened immune responses associated with hypertension
To further examine the role of ATP, we purified splenic DCs from hypertensive mice and treated them with either apyrase (an ATP diphosphohydrolase), A740003, or vehicle ex vivo. Both apyrase and A740003 reduced CD86 expression to normotensive levels (Fig. 7F). Similarly, when mice were treated with either apyrase or A740003, the difference in CD86 levels on DCs and macrophages between normotensive and hypertensive mice was abrogated (fig. S11). In hypertensive mice, apyrase or A740003 also reverted the level of OVA-specific CD8+ T cells after OVA immunization to that of normotensive mice (Fig. 7G). When DCs derived from P2X7-deficient mice were loaded with SIINFEKL ex vivo and then infused into naïve C57BL/6 mice, the P2X7-deficient hypertensive DCs did not induce an enhanced T cell response (fig. S12). This contrasts with the observation that recipients infused with hypertensive DCs having P2X7 showed an enhanced immune response (Fig. 3D). Thus, P2X7 appears central to the increased ability of hypertensive DCs to initiate T cell immunity. Blocking the P2X7 receptor also significantly alleviated the severity of hepatitis induced by ConA in hypertensive mice (Fig. 7, H and I). Treating mice with A740003 did not alter the BP increase in the first week of induced hypertension, but, compared with vehicle, it reduced the BP in the second week when the elevated ATP-P2X7-CD86 axis would be expected to be active (fig. S13). This was also true when P2X7-deficient mice were examined for the development of hypertension (Fig. 8A), implying that the intensified immune responses associated with the ATP-P2X7-CD86 axis play an important role in the pathobiology of hypertension.
Fig. 8. The correlation of plasma ATP levels and P2X7 receptor expression with BP in mice and humans.
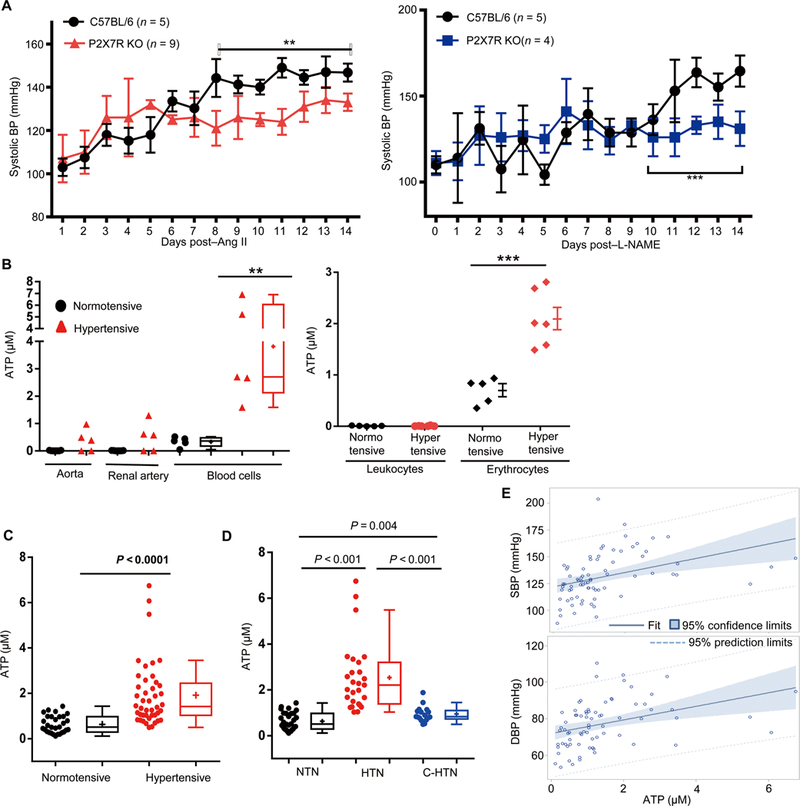
(A) BP of P2X7 knockout mice was made hypertensive with Ang II (left) or L-NAME (right). Two-way ANOVA with Bonferroni’s correction was used for BP data analysis. (B) Aorta, renal arteries, and total blood cells were collected from normotensive mice and hypertensive mice treated with Ang II for 2 weeks. Some blood cells were further separated to isolate leukocytes and erythrocytes with Percoll gradients. The arteries and cells were placed in serum-free medium for 3 hours, and the ATP levels in the medium were measured. (C) Distribution of plasma ATP in hypertensive patients and normotensive controls. The Kruskal-Wallis nonparametric test was used for analysis. (D) Distribution of plasma ATP in normotensive people (NTN), hypertensive patients with BPs not well controlled (HTN), and hypertensive patients with BP controlled to normal levels (C-HTN). Wilcoxon rank sum test (LSD test adjusted) was used for analysis. (E) Fit plots between ATP and SBP (left) and between ATP and DBP (right) among all subjects. *P < 0.05; **P < 0.01; ***P < 0.005.
To determine the source of elevated ATP in hypertension, we measured ATP release from aorta, renal artery, and total blood cells based on the hypothesis that the disrupted hemodynamics associated with hypertension directly affects blood flow and blood vessels. None of the blood vessels from the normotensive mice released ATP (Fig. 8B). However, both aorta and renal arteries in three of five hypertensive mice released ATP. There was also a significant elevation of ATP levels in the blood cells of hypertensive mice compared with normotensive animals (Fig. 8B). We further examined ATP release from erythrocytes and white blood cells, respectively, and found that the erythrocytes are the major source of plasma ATP in hypertension (Fig. 8B). We conclude that in hypertension, both erythrocytes and blood vessels are prone to release ATP, which potentially functions as an “alarmin.”
Plasma ATP correlates with BP in humans
To determine the relevance of plasma ATP in human hypertension, we analyzed plasma ATP levels of hypertensive patients (n = 44) and normotensive controls (n = 30). Subject enrollment criteria are described in Materials and Methods. Demographics and clinical characteristics of the studied groups are shown in table S1. ATP levels were significantly higher in hypertensive patients compared with controls (median ATP, 1.42 μM versus 0.51 μM; P < 0.0001) (Fig. 8C).
In a second analysis, the same 44 hypertensive patients were assigned into either a hypertensive group (HTN, n = 27) with BP not well controlled or a controlled group (C-HTN, n = 17) with BP within normal levels due to treatment with anti-hypertensive medications. Demographics and clinical characteristics of these groups are shown in table S2. These two groups were individually compared against normotensive controls. As expected, both SBP and DBP (diastolic BP) are highest in the patients of the HTN group. In contrast, there was no difference in BP between the normotensive (NTN) and C-HTN groups. The ATP levels were significantly higher in the HTN group than in the NTN group [median ATP, 2.21 μM versus 0.51 μM; P < 0.001, Wilcoxon test and least significant difference (LSD) test adjusted] (Fig. 8D). ATP levels were significantly reduced in the C-HTN group (median ATP, 0.83 μM in C-HTN versus 2.21 μM in HTN; P < 0.001, Wilcoxon test and LSD test adjusted).
As shown in Fig. 8E, a linear trend was observed between ATP levels and SBP or DBP among all subjects. The results of multivariate regressions between ATP and SBP or DBP are summarized in tables S3 and S4. ATP was positively correlated with SBP (β = 5.62, P = 0.0021) and DBP (β = 3.80, P = 0.0004), respectively. Thus, consistent with animal models, plasma ATP elevation in humans appears to be a biomarker for hypertension.
DISCUSSION
Hypertension is both extremely common and highly deleterious to human health. Hence, the control of hypertension has been identified as exceedingly advantageous and cost-effective (40). Although it is recognized that, in both animals and humans, excessive inflammation can raise BP, mechanistically there is much less known about how high BP affects inflammation and the generalized immune response. This question is important because an initial rise in BP may initiate a destructive cycle whereby inflammation and a further increase of BP cause clinically detectable disease.
Our initial experiments explored the impact of hypertension on the immune system. These studies showed that high BP was associated with an elevated antigen-specific T cell response. This was true for both CD8+ and CD4+ T cells. This conclusion was supported by experiments testing the effects of hypertension in two separate models of T cell-mediated immunopathology. Both diabetes in RIP-mOVA mice and acute hepatitis induced by ConA were significantly worsened by hypertension as compared with the pathology observed in normotensive mice. Thus, in three separate mouse models of immune challenge, there was a consistent pattern of an increased T cell-mediated immune response associated with an elevation of BP, which is due to aberrant immune activation in hypertensive mice. Although many clinical studies indicate that hypertension is one of the most common comorbidities of many autoimmune diseases (41,42), our animal studies provide evidence and mechanistic insight into why there is an association between hypertension and high prevalence of autoimmune disease, even in humans (9–11).
Our studies led to the realization that more effective APC presentation of antigen, but not differences in intrinsic T cell signaling, appears to be a major instigator of the increased T cell response in hypertension. Further examination of APCs revealed that hypertension induces increased CD86 expression, which is consistent with the report of Vinh et al. (43) that the percentage of CD86+ DCs is elevated in Ang II-treated mice. Our data highlight that ATP-driven P2X7-mediated CD86 up-regulation is the hallmark of hypertension-associated inflammation and is central to the heightened immune response observed after further immune challenge. In this setting, it is very possible that some autoantigens will be efficiently presented by APCs to T cells, eventually leading to antigen-specific T cell-mediated pathology. T cells have been shown to play a major role in furthering hypertension development and progression (5, 8). Our data demonstrate that in both P2X7-deficient mice and wild-type mice treated with a P2X7 blocker, BP rises in a pattern similar to that of control mice in the first week but that the BP rise significantly slowed in the second week after hypertension induction, indicating that activation of the ATP-P2X7-CD86 axis follows an initial BP increase and eventually leads to T cell activation.
This study also revealed some of the earliest biochemical events that provoke high BP-associated immune activation. We examined plasma concentrations of a plethora of DAMPs during the early stage of hypertension and found that only the ATP level was significantly elevated. Our data indicate that a short elevation of BP might alter red blood cells, resulting in the release of ATP that, acting as a DAMP, initiates activation of APCs. It is known that shear stress can induce ATP release from erythrocytes (44). However, it is still unclear whether ATP is passively released due to erythrocyte damage or ATP is actively pumped out by erythrocytes in hypertension. The lack of clinical evidence of anemia in hypertensive patients supports the latter. Future studies should address the mechanism of ATP release. Interference with ATP release could be an attractive potential means to treat hypertensive patients.
Pharmacological and genetic evidence shows that up-regulation of CD86 is mediated by P2X7 receptors on APCs. However, hypertension is initially accompanied by an ATP increase only at micromolar levels, which challenges the paradigm that P2X7 is a low-affinity receptor of ATP (34). Many of the studies on P2X7 affinity were performed on cell lines (35), whereas primary APCs have not been studied. Here, we found that 3 μM ATP did induce Ca2+ inflow, CD86 up-regulation, and inflammasome activation as indicated by IL-1β release, all of which were mediated by P2X7 receptors on APCs. Consistent with our findings, Wilhelm et al. (45) observed that a low concentration of ATP (10 μM in their case) could up-regulate CD86 in DCs. LPC may act as a cofactor in sensitizing the P2X7 receptor to ATP (38, 39). Our data show that DCs contain more LPC than T cells and HEK-293 cells, suggesting that cellular differences in P2X7 activity may result from different levels of LPC inside cells. P2X7 is mainly expressed in inflammatory cells. Most previous studies of P2X7-mediated effects on APCs usually evaluated high concentrations of ATP as the stimulus (46). This dose results in the formation of large membrane pores and eventually cell death. The activation of APC P2X7 by low-dose (micromolar) ATP is different, and its biological effects, particularly on APC-mediated antigen presentation, require reevaluation of the current P2X7 paradigm.
Last, different from the immediate reaction to ATP by endothelial cells resulting in vasodilation (47,48), a long-term ATP increase appears to instigate the immune system and promote inflammation. Our human study shows that hypertensive patients present with significantly elevated plasma levels of ATP compared with normotensives. The positive correlation between both SBP and DBP with plasma ATP levels suggests that ATP can serve as a surrogate marker for the degree of high BP. Moreover, our animal studies also found elevation of LPC in the plasma of hypertensive mice. Thus, further clinical cohort studies are required to evaluate whether ATP alone, or together with LPC, can also serve as a predictor for the degree of organ damage associated with hypertension.
MATERIALS AND METHODS
Study design
The aims of this study were to characterize the changes in APCs after hypertension, identify the molecular trigger of the changes, and evaluate the consequences of these changes for immunopathogenesis. Age- and gender-matched C57BL/6, RIP-mOVA, and P2X7 purinergic receptor-deficient mice were used for this study. We performed flow cytometric experiments to analyze the activation markers of APCs in two mechanistically different mouse hypertension models. Pharmacological and genetic approaches were used to assess the roles of the P2X7 receptor. T cell-mediated diabetes and hepatitis models along with antigen immunization were used to assess the effects of hypertension in mice on in vivo T cell responses. In addition, we measured the plasma levels of ATP in hypertensive patients and normotensive controls.
Mice and hypertension models
C57BL/6 mice were purchased from the Jackson Laboratory and Shanghai Research Center for Model Organisms. OT-I mice [C57BL/6-Tg(TcraTcrb)1100Mjb/J] and RIP-mOVA mice [C57BL/6-Tg(Ins2-TFRC/OVA)296Wehi/WehiJ] were purchased from the Jackson Laboratory. P2X7-deficient mice (B6.129P2-P2rx7tm1Gab/J) were originally obtained from the Jackson Laboratory. Because there are significant gender and age differences in hypertension development, all mice used in this study were 8- to 12-week-old males. Hypertension was induced by subcutaneous infusion of Ang II (490 ng/kg per min) (Phoenix Pharmaceuticals) via osmotic minipump (ALZET) or by L-NAME treatment (1.5 mg/ml in drinking water) (Bachem). BP was monitored in conscious mice with a radio telemetry device (model PA-C10, Data Sciences International). Mice were anesthetized with isoflurane, and a catheter connected to a radio telemetry device was inserted in the left carotid artery. After a 14-day recovery phase, baseline levels were established before hypertension induction. Data were collected, stored, and analyzed using Dataquest A.R.T. 4.0 software (Data Sciences International). In some experiments, hydralazine (320 mg/liter; Sigma-Aldrich) was administered in the drinking water to normalize BP. Mice were anesthetized by inhalation of 2 to 3% isoflurane/O2 mixture. All animal experiments were approved by the Institutional Animal Care and Use Committee at Zhejiang University and by the Institutional Animal Care and Use Committee at Cedars-Sinai Medical Center.
Antibodies and reagents
The following fluorophore-conjugated antibodies were purchased from BioLegend or Thermo Fisher Scientific: anti-CD3e (145–2C11), anti-CD4 (GK1.5), anti-CD8 (53–6.7), anti-CD11a (M17/4), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD25 (3C7), anti-CD44 (IM7), anti-CD49b (DX5), anti-CD62L (MEL-14), anti-CD69 (H1.2F3), anti-CD80 (16–10A1), anti-CD86 (GL-1), anti-CD122 (TM-β1), anti-F4/80 (BM8), anti-IDO (2E21/IDO1), anti–IFN-γ (XMG1.2), anti–IL-1β (NJTEN3), anti–IL-6 (MP5–20F3), anti–IL-7R (A7R34), anti–IL-12p40 (C15.6), anti-TCRγδ (GL3), anti–TNF-α (MP6-XT22), anti–H-2Kb-SIINFEKL (25-D1.16), anti-H-2Kb (AF6–88.5), anti-H-2Db (KH95), anti–I-Ab (AF6–120.1), CX3CR1 (SA011F11), and Ly6C (HK1.4). Annexin V and 7-aminoactinomycin D (7-AAD) were purchased from BioLegend and used for live/dead staining. Alexa Fluor 488-conjugated H-2Kb-SIINFEKL was obtained from the National Institutes of Health (NIH) tetramer core facility. For staining of intracellular proteins, fixation and permeabilization buffers were purchased from Thermo Scientific. Apyrase, BBG, and suramin were purchased from Sigma-Aldrich. A740003 and AR-C 118925XX were purchased from Tocris Bioscience. BAPTA was purchased from MedchemExpress. N-[2-[2-(1,1-dimethylethyl)phenoxy]-3-pyridinyl]-N′-[4-(trifluoromethoxy) phenyl]urea (BPTU) and chlorpromazine were purchased from Target Molecule. Uric acid was measured with a Beckman Coulter chemistry analyzer (AU5421). Plasma PGE-2, oxLDL, and HMGB-1 were analyzed with enzyme-linked immunosorbent assay (ELISA) kits (Elabscience Biotechnology). Fluo-4 AM, NAD assay kit, and lactic dehydrogenase cytotoxicity assay kits were purchased from Beyotime Biotech.
Splenic DC and CD8+ T cell isolation
Spleens were minced with scissors and digested with collagenase IV (Worthington) and deoxyribonuclease I (Sigma-Aldrich) for 30 min at 37°C. Cells were passed through a 70-μm strainer (BD Biosciences) and then selected for either CD11c+ DCs with a CD11c positive selection magnetic kit (Miltenyi Biotec) or CD8+ T cells with a negative selection magnetic kit (Miltenyi Biotec).
Immunization and immune response assays
OVA (100 μg) was dissolved in phosphate-buffered saline (PBS; 50 ml) and emulsified with an equal volume of CFA (0.05% Mycobacterium tuberculosis; MP Biomedicals), and a total volume of 100 ml was injected subcutaneously into the dorsal flank of a mouse. In another group, a 100-μg OVA/PBS solution was mixed with 2 μg of LPS for 1 hour and injected intraperitoneally to mice. After 7 days, blood (CFA immunized) and spleen (LPS immunized) cells were harvested and stained with an H-2Kb-SIINFEKL tetramer in combination with anti-CD3ε and anti-CD8 staining. For the experiments using alum as adjuvant, mice were immunized intraperitoneally with a mixture of 100 μg of OVA and 50 μl (2 mg) of Imject Alum (Pierce Biochemicals). Seven days after the immunization, splenocytes were collected and restimulated with SIINFEKL (40 μg/ml) or OVA323–339 (10 μg/ml). After 24 hours (IL-2) or 72 hours (IFN-γ), the cytokines were measured in the supernatants using ELISA kits (BioLegend). For in vivo CD86 blockade, anti-CD86 antibody was purified from the culture supernatant of GL-1 hybridomas (American Type Culture Collection) with the Nab Protein G Spin Kit (Thermo Scientific). The control isotype immunoglobulin G (IgG) was purchased from Abcam. Anti-CD86/IgG (100 μg), apyrase (0.2 U/g weight), or A740003 (50 nM) was administered intraperitoneally 3 days before OVA-CFA immunization and every 3 days afterward.
In vitro antigen presentation
Splenic DCs (1 × 105) or peritoneal macrophages were fed with either no antigen,1 μM OVA (Invivogen), or 50 pM SIINFEKL in Iscove’s modified DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% fetal bovine serum (FBS). After 3 hours, cells were washed and 1 × 106 splenocytes from OT-I mice were coincubated. Brefeldin A (BioLegend) was included in some of the cultures. OT-I T cell activation was detected by surface staining with anti-CD69 after 4 hours or by intracellular IFN-γ staining after 6 hours in the presence of brefeldin A. In some experiments, APCs were pretreated with anti-CD86 (1 μg/ml; GL1, BioLegend) and/or anti-CD80 (1 μg/ml; 16–10A1, BioLegend) before coincubation with T cells.
DC adoptive transfer
Splenic DCs (1.5 × 106) were purified from normotensive or Ang II-induced hypertensive C57BL/6 or P2X7-deficient mice. DCs were cultured with 1 nM SIINFEKL for 3 hours and then washed and adoptively transferred to normotensive C57BL/6 mice by intravenous injection. After 8 days, splenocytes were collected from the recipients and restimulated with SIINFEKL. The supernatants were collected for IFN-γ and IL-2 ELISA.
Autoimmune type 1 diabetes model
Two to 3 weeks after hypertension induction by either Ang II or L-NAME, hypertensive RIP-mOVA mice, together with their normotensive counterparts, were injected intravenously with 5 × 106 CD8+ T cells isolated from OT-I mice. Blood from the tail vain was read on a Contour blood glucose monitoring system (Bayer HealthCare). Two weeks after injection, mice were euthanized. Cells from blood, spleen, and pancreatic draining lymph nodes were assessed with H-2Kb–SIINFEKL tetramers. Formalin-fixed pancreatic sections were stained with an anti-CD3 antibody (BioLegend). Islets infiltrated with ≥10 CD3+ T cells were defined as “inflamed.”
ConA-induced acute hepatitis model
Normotensive and Ang II–induced hypertensive mice were intravenously injected with ConA (5 mg/kg). Blood plasma ALT levels were monitored by colorimetric ALT enzyme assay (Nanjing Jiancheng Bioengineering Institute, China). Twelve hours later, inflammatory cells were purified from the livers as previously described (49) and cultured in the presence of brefeldin A for 6 hours. Then, cells were examined for TNF-α and IFN-γ production by intracellular staining and flow cytometry in combination with surface anti-CD3 and anti-CD4 staining.
In vitro ATP stimulation and P2X7 receptor inhibition
Splenocytes (3 × 105) were cultured in Iscove’s modified Dulbecco’s medium containing 2% FBS, with 0, 1, 3, 10, 30, 100, or 300 μM ATP (Sigma-Aldrich) for 18 hours. CD80 and CD86 were measured on DCs (CD11c+) or monocytes (Ly6C+ CD11b+ CX3CR1+) by surface staining and flow cytometry. In some experiments, splenic DCs were stimulated with 3 μM ATP in the presence of the P2X7 receptor antagonist A740003 (25 μM), BBG (1 μM), the P2Y receptor antagonist suramin (15 μM), the P2Y1 receptor antagonist BPTU (1 μM), the P2Y12 receptor antagonist AR-C 118925XX (10 μM), the calcium chelator BAPTA (20 μM), or the calmodulin inhibitor chlorpromazine (1 μM). Surface CD86 expression was measured after 18 hours by flow cytometry.
ATP assays
Whole blood from normotensive and hypertensive mice was collected at different time points. Plasma was prepared by spinning the whole blood at 1500g for 15 min and collecting the supernatant. ATP levels were determined with an ATP assay bioluminescence detection kit (Beyotime). For testing tissue ATP release, aorta and renal artery were removed from PBS-perfused mice and cultured in 30 μl of RPMI 1640 for 3 hours. To measure ATP production by blood cells, 50 μl of whole blood was centrifuged at 300g for 5 min to remove plasma. Blood cells were resuspended in 50 μl of RPMI 1640 and transferred into a fresh well for a 3-hour incubation. After 3 hours, the culture plates containing blood cells or tissue were centrifuged at 300g for 5 min and supernatants were then collected for ATP measurement.
Liquid chromatography-tandem mass spectrometry analysis
Ninety-microliter plasma samples were mixed with 10 μl of internal standard working solution (2 μg/ml), 5 μl of10 mM acetic acid, and 200 μl of acetonitrile for 1 min. Two million cells were dissolved in 100 μl of acetonitrile and then subjected to ultrasonic processing. Samples were then centrifuged at 13,000 rpm at 4°C for 10 min. The supernatant was processed by vacuum drying and reconstituted in 200 ml of mobile phase consisting of (A) ammonium formate buffer (pH 3.5; 10 mM) and (B) acetonitrile (90% B). Ten microliters of solution was used each time for liquid chromatography-mass spectrometry (LC-MS) analysis. Quantitative analysis was performed with an Agilent 1260 high-performance liquid chromatographic system (Agilent Technologies, USA) coupled to an API4000 mass spectrometer (AB Sciex, USA) operated in negative electrospray ionization through multiple reaction monitoring (MRM) mode. Data were analyzed with Analyst 1.5.2 software Hotfixes (AB SCIEX, Ontario, Canada). Chromatographic separations were achieved with Poroshell 120 SB-C18 (2.1 × 100 mm, 2.7 μm; Agilent, USA) at 30°C. The mobile phase was pumped with a gradient elution program that consists of (A) ammonium format buffer (pH 3.5; 10 mM) and (B) acetonitrile and was delivered at a flow rate of 0.25 ml min−1 according to the following programs: 0.0 to 0.5 min (20% B), 0.5 to 1.5 min (90% B), 1.5 to 4.0 min (90% B), 4.0 to 4.5 min (20% B), and 4.5 to 5.0 min (20% B). Mass spectrometer and ionization conditions were optimized as follows: heated gas pressure, 60 psi; nebulizer gas pressure, 60 psi; curtain gas pressure,30 psi; collision gas pressure, 7 U (N2); heated gas temperature, 400°C; ion spray voltage, −5500 V; decluttering potential, −55 V [lysophosphatidic acid (LPA), 18:0], −50 V (LPA, 16:0), and −35 V (salicylic acid; internal standard); collision energy was −30 V (LPA 18:0), −25 V (LPA, 16:0), and −15 V (internal standard), respectively. Detection of ions from the analytes was carried out in the MRM by monitoring transition of mass/charge ratio (m/z) 437.1 → 152.9 (LPA, 18:0), m/z 409.1 → 152.9 (LPA, 16:0), and m/z 136.0 → 92.0 for the IS as precursor ion and product ion, respectively.
Human plasma ATP analysis
Zhejiang Hospital and the Fourth Affiliated Hospital of Zhejiang University reviewed and approved the human studies. All the participants signed a written informed consent form before being enrolled in the study. Patients with a history of hypertension between 40 and 70 years of age and normotensive controls were recruited. Their BP was measured on two consecutive days. Patients were assigned to the HTN group when either average SBP was ≥140 mmHg or average DBP was ≥90 mmHg. Hypertensive patients (by history) who were taking anti-hypertensive medications and had both average SBP below 140 mmHg and average DBP below 90 mmHg were assigned to the C-HTN group. The NTN group is normotensive controls with normal measured BP and no previous history of hypertension. Patients with histories of autoimmune diseases, lung diseases, asthma, stroke, AIDS, cancer, and transplantation and those having symptoms or history of infection in the last month were excluded. Whole blood of selected subjects was collected into heparin-treated anticoagulation tubes. Plasma was isolated by centrifuging the whole blood at 1000g for 15 min. Hemolyzed samples were not used. All blood samples used were not subjected to a freeze-thaw cycle at any step in the process. Plasma ATP levels were assessed with an ATP assay bioluminescence detection kit (Beyotime).
Ca2+ influx measurement
Splenocytes from normotensive mice were incubated with the calcium indicator Fluo-4 AM (1 μM) at 37°C for 30 min. Unloaded Fluo-4 AM was removed by three washes with PBS. DCs (CD11c+) and T cells (CD3+) were distinguished by surface staining with relevant antibodies for 15 min at 37°C. After surface staining, cells were resuspended in Hank’s balanced salt solution (HBSS) buffer containing Ca2+ and Mg2+. Flow cytometry analysis was started immediately after ATP addition and performed for 4 min.
Ca2+ imaging
Peritoneal exudate cells were cultured on glass-bottom cell culture dish (NEST) in RPMI 1640 containing 2% FBS. After 2 hours, unattached cells were washed out with PBS, and the attached macrophages were loaded with 2 μM Fluo-4 AM in PBS for 30 min at 37°C. Those macrophages were stimulated with 3 or 500 μM ATP in HBSS buffer containing Ca2+ and Mg2+. Images were acquired every 75 ms for 30 s (400 pictures per sample) with an Andor spinning disc confocal microscope (Nikon). Fluo-4 AM fluorescence intensity was measured with ImageJ.
YO-PRO-1 uptake and ATP-induced apoptosis
Splenic cells were treated with 3 or 500 μM ATP in RPMI 1640/1% bovine serum albumin for 3 hours at 37°C. For the YO-PRO-1 uptake assay, cells were stained with 1 μM YO-PRO-1 (Thermo Scientific) and analyzed by flow cytometry. For apoptosis evaluation, cells were stained with 7-AAD and annexin V to distinguish apoptotic cells from viable and necrotic cells.
Statistics
Data from mice are expressed as means ± SEM, and values of P < 0.05 were considered statistically significant. Two-way analysis of variance (ANOVA) with Bonferroni’s post-test was used when analyzing changes in data collected over time. Paired one-way ANOVA and two-tailed unpaired Student’s t test were used to compare groups. For the human ATP study, categorical variables were presented as numbers and relative frequencies (percentages); continuous variables were presented as either mean ± SD or median with interquartile range according to their distributions, which were checked by using the Kolmogorov-Smirnov and Levene tests. Normally distributed continuous variables were compared using Student’s t test or one-way ANOVA. Either Kruskal-Wallis test or Wilcoxon rank sum test was used for the data that were not normally distributed. Differences between groups in categorical variables were analyzed by the χ2 test or Fisher’s exact test. The LSD test was used for multiple comparisons of measurement variables, and a Bonferroni correction was used for multiple comparisons of categorical variables. A generalized linear model was performed to explore the linear correlation between ATP, SBP, and DBP. All statistical tests were two tailed, and P values of <0.05 were considered significant. OriginPro 2016 was used to plot the distribution of ATP. All statistical analyses were performed by using SAS 9.1 (SAS Institute) and SPSS 19.0 (SPSS Inc., IL, USA).
Supplementary Material
Acknowledgments:
We acknowledge the technical support by the Core Facilities, Zhejiang University School of Medicine. We are grateful to Y. Zhang (Institutes of Brain Science, Fudan University) for sharing P2X7 knockout mice, H. Xu (University of Florida) for assistance in biostatistics, and H. Wang and Y. Wang (Zhejiang University) for assistance in confocal microscopy analyses.
Funding: This work was supported by the National Natural Science Foundation of China (81670378 to X.Z.S. and 81700365 and 31771266 to P.S.), China Postdoctoral Science Foundation (2018M632486 to T.V.Z.), Science and Technology Agency Foundation of Zhejiang Province (2015C33118 to S.X.), Chinese Traditional Medicine of Zhejiang Province (2013ZA009 to X.L.), NIH (P01 HL129941, R21 AI114965, and R01 AI143599 to K.E.B. and R01 HL142672 to J.F.G.), American Heart Association (16SDG30130015 to J.F.G. and Grant-in-Aid 17GRNT33661206 to K.E.B.), and UCSD/UCLA Dream Resource Center (P&F P30DK063491 to J.F.G.).
Footnotes
Competing interests: The authors declare that they have no competing financial interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Luscher TF, Prevention: Some important steps forward, but many unmet needs in a world with cardiovascular disease as the leading cause of death. Eur. Heart J. 37, 3179–3181 (2016). [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization, A Global Brief on Hypertension (World Health Organization, 2013); www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/. [Google Scholar]
- 3.Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, Ganio EA, Fragiadakis GK, Spitzer MH, Douchet I, Daburon S, Moreau J-F, Nolan GP, Blanco P, Dechanet-Merville J, Dekker CL, Jojic V, Kuo CJ, Davis MM, Faustin B, Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat.Med. 23, 174–184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savoia C, Schiffrin EL, Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 15, 152–158 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG, Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 204, 2449–2460 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen S.-c, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts II J, Harrison DG, DC isoketal-modified proteins activate T cells and promote hypertension. J. Clin. Invest. 124, 4642–4656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG, Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55, 500–507 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Marko L, Vvedenskaya O, Kleiner FH, Tsvetkov D, Klug L, Costea PI, Sunagawa S, Maier L, Rakova N, Schatz V, Neubert P, Frätzer C, Krannich A, Gollasch M, Grohme DA, Côrte-Real BF, Gerlach RG, Basic M, Typas A, Wu C, Titze JM, Jantsch J, Boschmann M, Dechend R, Kleinewietfeld M, Kempa S, Bork P, Linker RA, Alm EJ, Muller DN, Salt-responsive gut commensal modulates TH17 axis and disease. Nature 551, 585–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerola AM, Kerola T, Kauppi MJ, Kautiainen H, Virta LJ, Puolakka K, Nieminen TVM, Cardiovascular comorbidities antedating the diagnosis of rheumatoid arthritis. Ann. Rheum. Dis. 72, 1826–1829 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Norton S, Koduri G, Nikiphorou E, Dixey J, Williams P, Young A, A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology 52, 99–110 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Lin L-T, Wang P-H, Tsui K-H, Cheng J-T, Cheng J-S, Huang W-C, Tang P-L, Hu L-Y, Increased risk of systemic lupus erythematosus in pregnancy-induced hypertension: A nationwide population-based retrospective cohort study. Medicine 95, e4407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin C, Flavell RA, Innate sensors of pathogen and stress: Linking inflammation to obesity. J. Allergy Clin. Immunol. 132, 287–294 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Burnstock G, Purinergic signaling in the cardiovascular system. Circ. Res. 120, 207–228 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Burnstock G, Di Virgilio F, Purinergic signalling and cancer. Purinergic Signal. 9, 491–540 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lajdova I, Oksa A, Chorvat D Jr., P. Topor, V. Spustova, Purinergic P2X7 receptors participate in disturbed intracellular calcium homeostasis in peripheral blood mononuclear cells of patients with chronic kidney disease. Kidney Blood Press. Res. 35, 48–57 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Savio LEB, de Andrade Mello P, da Silva CG, Coutinho-Silva R, The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 9, 52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P, The NLRP3 and pyrin inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front. Immunol. 8, 43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S, The P2X7 receptor in infection and inflammation. Immunity 47, 15–31 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H, Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 184, 923–930 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurts C, Kosaka H, Carbone FR, Miller JFAP, Heath WR, Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp.Med. 186, 239–245 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, von Herrath MG, Suppression of diabetes by accumulation of non-islet-specific CD8+effector T cells in pancreatic islets. Sci. Immunol. 3, eaam6533 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Tiegs G, Hentschel J, Wendel A, A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90, 196–203 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heymann F, Hamesch K, Weiskirchen R, Tacke F, The concanavalin A model of acute hepatitis in mice. Lab. Anim. 49, 12–20 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K, Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc. Natl. Acad. Sci. U.S.A. 97, 5498–5503 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salmond RJ, Brownlie RJ, Morrison VL, Zamoyska R, The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat. Immunol. 15, 875–883 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN, Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 6, 715–726 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Mellor AL, Munn DH, IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4, 762–774 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Bangert A, Andrassy M, Muller A-M, Bockstahler M, Fischer A, Volz CH, Leib C, Göser S, Korkmaz-Icöz S, Zittrich S, Jungmann A, Lasitschka F, Pfitzer G, Müller OJ, Katus GA, Kaya Z, Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. U.S.A. 113, E155–E164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stachon P, Geis S, Peikert A, Heidenreich A, Michel NA, Ünal F, Hoppe N, Dufner B, Schulte L, Marchini T, Cicko S, Ayata K, Zech A, Wolf D, Hilgendorf I, Willecke F, Reinohl J, von Zur Muhlen C, Bode C, Idzko M, Zirlik A, Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arterioscler. Thromb. Vasc. Biol. 36, 1577–1586 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D, Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 84, 1086–1095 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feig DI, Kang D-H, Johnson RJ, Uric acid and cardiovascular risk. N. Engl. J. Med. 359, 1811–1821 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baron L, Gombault A, Fanny M, Villeret B, Savigny F, Guillou N, Panek C, Le Bert M, Lagente V, Rassendren F, Riteau N, Couillin I, The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 6, e1629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamson SE, Montgomery G, Seaman SA, Peirce-Cottler SM, Leitinger N, Myeloid P2Y2 receptor promotes acute inflammation but is dispensable for chronic high-fat diet-induced metabolic dysfunction. Purinergic Signal 14, 19–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartlett R, Stokes L, Sluyter R, The P2X7 receptor channel: Recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Chessell IP, Simon J, Hibell AD, Michel AD, Barnard EA, Humphrey PPA, Cloning and functional characterisation of the mouse P2X7 receptor. FEBS Lett. 439, 26–30 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF, Mammalian P2X7 receptor pharmacology: Comparison of recombinant mouse, rat and human P2X7 receptors. Br. J. Pharmacol. 157, 1203–1214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheuplein F, Schwarz N, Adriouch S, Krebs C, Bannas P, Rissiek B, Seman M, Haag F, Koch-Nolte F, NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J. Immunol. 182, 2898–2908 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Takenouchi T, Sato M, Kitani H, Lysophosphatidylcholine potentiates Ca2+ influx, pore formation and p44/42 MAP kinase phosphorylation mediated by P2X7 receptor activation in mouse microglial cells. J. Neurochem. 102, 1518–1532 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Michel AD, Fonfria E, Agonist potency at P2X7 receptors is modulated by structurally diverse lipids. Br. J. Pharmacol. 152, 523–537 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moran AE, Odden MC, Thanataveerat A, Tzong KY, Rasmussen PW, Guzman D, Williams L, Bibbins-Domingo K, Coxson PG, Goldman L, Cost-effectiveness of hypertension therapy according to 2014 guidelines. N. Engl. J. Med. 372, 447–455 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ, Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension 64, 792–800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackey RH, Kuller LH, Deane KD, Walitt BT, Chang Y-F, Holers VM, Robinson WH, Tracy RP, Hlatky MA, Eaton CB, Liu S, Freiberg MS, Talabi MB, Schelbert EB, Moreland LW, Rheumatoid arthritis, anti-cyclic citrullinated peptide positivity, and cardiovascular disease risk in the women’s health initiative. Arthritis Rheum. 67, 2311–2322 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ, Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122, 2529–2537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan J, Ristenpart WD, Stone HA, Dynamics of shear-induced ATP release from red blood cells. Proc.Natl. Acad. Sci. U.S.A. 105, 16432–16437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilhelm K, Ganesan J, Müller T, Dürr C, Grimm M, Beilhack A, Krempl CD, Sorichter S, Gerlach UV, Jüttner E, Zerweck A, Gärtner F, Pellegatti P, Di Virgilio F, Ferrari D, Kambham N, Fisch P, Finke J, Idzko M, Zeiser R, Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 16, 1434–1438 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, Mittal M, Hong Z, Kanneganti T-D, Rehman J, Malik AB, The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49, 56–65.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellsworth ML, Forrester T, Ellis CG, Dietrich HH, The erythrocyte as a regulator of vascular tone. Am. J. Physiol. 269, H2155–H2161 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ, ATP: The red blood cell link to NO and local control of the pulmonary circulation. Am. J. Physiol. 271, H2717–H2722 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Xu F, Zhen P, Zheng Y, LIjuan F, Aiting Y, Min C, Hong Y, Jidong J, Preparation of Kupffer cell enriched non-parenchymal liver cells with high yield and reduced damage of surface markers by a modified method for flow cytometry. Cell Biol. Int. 37, 284–291 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.