

Abstract
Somatic mutations in cytosolic or mitochondrial isoforms of isocitrate dehydrogenase (IDH1 or IDH2, respectively) contribute to oncogenesis via production of the metabolite 2-hydroxyglutarate (2HG). Isoform-selective IDH inhibitors suppress 2HG production and induce clinical responses in patients with IDH1- and IDH2-mutant malignancies. Despite the promising activity of IDH inhibitors, the mechanisms that mediate resistance to IDH inhibition are poorly understood. Here, we describe four clinical cases that identify mutant IDH isoform switching, either from mutant IDH1 to mutant IDH2 or vice versa, as a mechanism of acquired clinical resistance to IDH inhibition in solid and liquid tumors.
INTRODUCTION
Somatic mutations in isocitrate dehydrogenase (IDH) enzymes are observed in a wide spectrum of human cancers, most commonly gliomas (1, 2), myeloid malignancies (3), chondrosarcomas (4), and intrahepatic cholangiocarcinomas (ICC; ref. 5). IDH enzymes normally function as components of the tricarboxylic acid cycle, catalyzing the interconversion of isocitrate and alpha-ketoglutarate (αKG; ref. 6). Cancer-associated IDH mutations occur at catalytic-site arginine residues in cytoplasmic IDH1 (R132) and mitochondrial IDH2 (R140 and R172), altering the enzymatic activity to promote efficient reduction of αKG to the “oncometabolite” 2-hydroxyglutarate (2HG; refs. 7, 8). Cellular accumulation of 2HG inhibits αKG-dependent dioxygenases that mediate histone and DNA demethylation, leading to a repressive chromatin landscape that disrupts normal cellular differentiation and contributes to oncogenesis (9–12).
Importantly, the effects of 2HG on chromatin and cell differentiation are largely reversible (11); therefore, drugs that inhibit 2HG production by mutant IDH represent potential therapies for IDH-mutant malignancies (13). Small-molecule inhibitors of IDH1 or IDH2 inhibit 2HG production by IDH-mutant enzymes, induce cellular differentiation, and inhibit cancer growth in preclinical models (6, 13). In phase I/II trials, the mutant IDH1 inhibitor ivosidenib (AG-120) and the mutant IDH2 inhibitor enasidenib (AG-221) induced clinical responses in patients with relapsed/refractory IDH-mutant myeloid malignancies (14, 15). Clinical trials with ivosidenib are under way for the treatment of relapsed/refractory IDH 1-mutant solid tumors, including glioma, ICC, and chondrosarcoma (16).
The mechanisms that mediate response and resistance to small-molecule IDH inhibition are not fully understood (17–19). Here, we identify mutant IDH isoform switching, either from cytoplasmic mutant IDH1 to mitochondrial mutant IDH2 or vice versa, as a mechanism of acquired clinical resistance to IDH inhibition. The first two cases describe patients with relapsed/refractory IDH1 R132C-mutant acute myeloid leukemia (AML) who achieved durable remissions in response to the mutant IDH1 inhibitor ivosidenib followed by leukemic progression on therapy, rise of blood 2HG levels, and emergence of IDH2 R140Q mutations. The third case describes a patient with treatment-refractory IDH1 R132C-mutant ICC who attained a sustained partial response to ivosidenib followed by progression of disease associated with acquisition of a new IDH2 R172V mutation. The fourth case describes a patient with relapsed/refractory IDH2 R140Q-mutant AML who achieved a durable remission to the mutant IDH2 inhibitor enasidenib, followed by progression on therapy associated with emergence of a new IDH1 R132C mutation that was sensitive to combined IDH1/2 blockade with AG-881. These findings provide evidence of selective pressure to maintain 2HG production in IDH-mutant malignancies, as well as suggest potential strategies for disease monitoring and therapies that might overcome acquired resistance to IDH inhibition.
RESULTS
Case Reports 1 and 2
A 54-year-old man with normal karyotype AML relapsed 100 days after allogeneic bone marrow transplantation with progressive pancytopenia and a bone marrow biopsy showing 30% leukemic blasts (Fig. 1A and B). Targeted next-generation sequencing (NGS) of bone marrow cells using a microdroplet-PCR assay (20) demonstrated the presence of an IDH1 R132C mutation and two DNMT3A mutations (Fig. 1C; Supplementary Tables S1 and S2). The patient began treatment with the mutant IDH1 inhibitor ivosidenib 500 mg orally daily, with a complete remission evident after three 28-day cycles of therapy. After completing twelve 28-day cycles of ivosidenib, the blast count and blood 2HG levels began to rise and a new IDH2 R140Q mutation was detected (Fig. 1A, C and D). Droplet digital PCR (ddPCR) analysis of DNA from bone marrow cells demonstrated that the IDH2 R140Q mutation was not detectable prior to treatment but was present at low levels early in the course of ivosidenib treatment (Supplementary Fig. S1A; Supplementary Table S3). Ivosidenib was discontinued, and the mutant IDH2 inhibitor enasidenib was started. After several days of treatment with enasidenib, the patient developed fevers and hypoxia suspected to be secondary to IDH inhibitor differentiation syndrome (21); enasidenib was discontinued.
Figure 1.
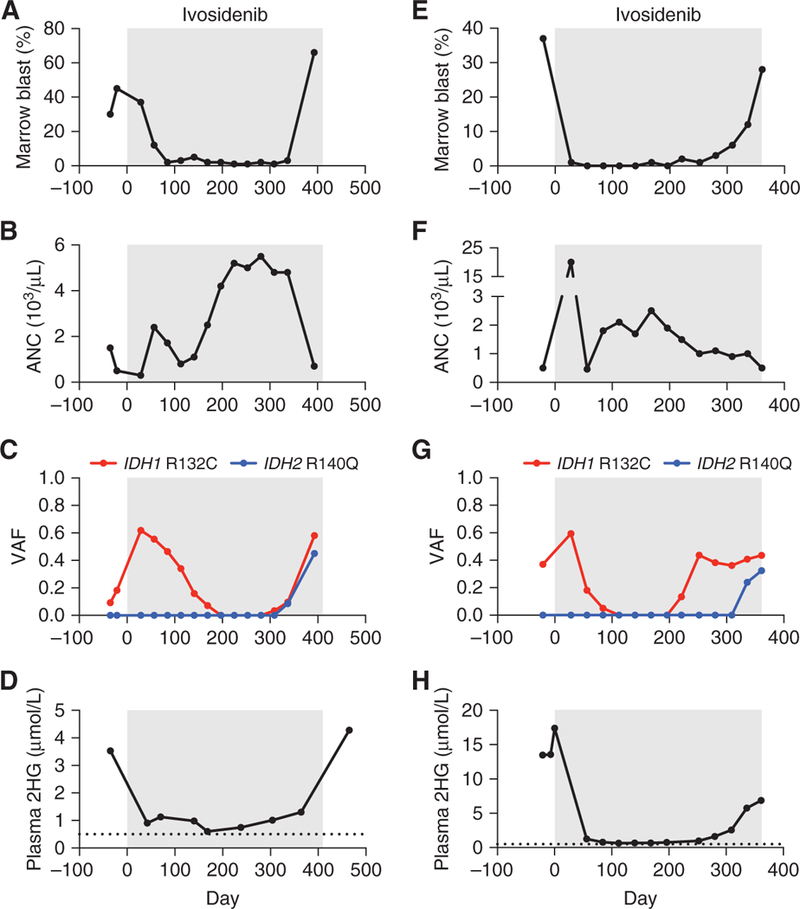
Acquired resistance to mutant IDH1 inhibition associated with emergence of oncogenic IDH2 mutations in AML. Clinical and laboratory features of two patients (case 1 = A–D; case 2 = E–H) with IDH1 R132C-mutant AML treated with the mutant IDH1 inhibitor ivosidenib (gray boxes), including A, E, bone marrow blast percentage; B, F, absolute neutrophil count (ANC); C, G, variant allele frequency (VAF) for IDH1 and IDH2 mutations identified by targeted NGS of bone marrow cells; and D, H, plasma 2-hydroxyglutarate (2HG) concentration measured by gas chromatography–mass spectrometry. Dotted line indicates limit of detection.
A 72-year-old man presented with AML arising from pre-existing JAK2 V617F-mutant myelofibrosis. For several years prior, the myelofibrosis had been treated successfully with single-agent ruxolitinib and then combination therapy with ruxolitinib plus decitabine. However, at the time of presentation with secondary AML, there were 37% blasts in the bone marrow, and the patient was neutropenic (Fig. 1E and F). Targeted NGS of bone marrow mononuclear cells using a microdroplet-PCR assay (20) identified JAK2 V617F and IDH1 R132C mutations (Fig. 1G), as well as mutations in ASXL1, EZH2, and RUNX1 (Supplementary Tables S1 and S4). The patient began treatment with ivosidenib 500 mg orally daily, and a complete response was evident after one 28-day cycle of therapy. The IDH1 mutation became undetectable after four 28-day cycles of ivosidenib, but then reappeared after the 11th 28-day cycle (Fig. 1G). The patient remained in complete morphologic remission until the start of the 12th cycle when the bone marrow blasts increased to 12%, then 28% four weeks later (Fig. 1E). The increase in AML blasts was associated with the emergence of a new IDH2 R140Q mutation and a rise in the serum 2HG levels (Fig. 1G and H). ddPCR analysis of DNA from bone marrow cells demonstrated that the IDH2 R140Q mutation was detectable at low levels both before treatment and early during the course of therapy with ivosidenib, well before overt clinical resistance developed (Supplementary Fig. S1B; Supplementary Table S5). Ivosidenib was discontinued. The patient subsequently pursued treatment elsewhere with low-dose cytarabine and venetoclax (22), but was lost to follow-up.
Case Report 3
A 79-year-old woman with American Joint Committee on Cancer stage IV (T3N1M1) ICC presented for evaluation. One month prior to presentation, she had developed anorexia, unintentional weight loss, and abdominal distention. Cross-sectional imaging with computed tomography revealed an 8 × 5 × 7.5 cm hypoattenuating mass with peripheral enhancement and capsular retraction in the right hepatic lobe, multiple hepatic satellite tumors, and extensive retroperitoneal lymphadenopathy. Core biopsy of the dominant right hepatic mass revealed a poorly differentiated adenocarcinoma with IHC markers consistent with a primary biliary tumor. An analysis of genomic DNA from the biopsy specimen using targeted NGS revealed an IDH1 R132C mutation and no other detectable mutations (Supplementary Tables S6 and S7).
After documented radiographic progression on multiple standard and investigational agents, the patient began treatment with ivosidenib 500 mg orally daily. Computed tomographic scans prior to treatment and then serially over the course of treatment demonstrated a partial response to treatment with a 50% decrease in disease burden (Fig. 2A and B). An escape lesion was identified at day 392, and this lesion continued to enlarge on a follow-up scan at day 446, despite a further decrease in size of the original tumor (Fig. 2B). Ivosidenib was discontinued and a postprogression biopsy of the enlarging lesion was performed. Sequencing of the resistant tumor confirmed the presence of the initial IDH1 R132C mutation in addition to a new IDH2 R172V mutation, CDKN2A/B loss, and several other alterations of uncertain significance (Fig. 2C; Supplementary Tables S7 and S8). Subsequently, the patient was treated with pembrolizumab, a monoclonal antibody to PD-1, with slow progression of disease on serial imaging and clinical progression with worsening ascites and diminution of Eastern Cooperative Oncology Group performance status. She then began treatment with the mutant IDH2 inhibitor enasidenib, but within 8 weeks underwent clinical deterioration and expired.
Figure 2.
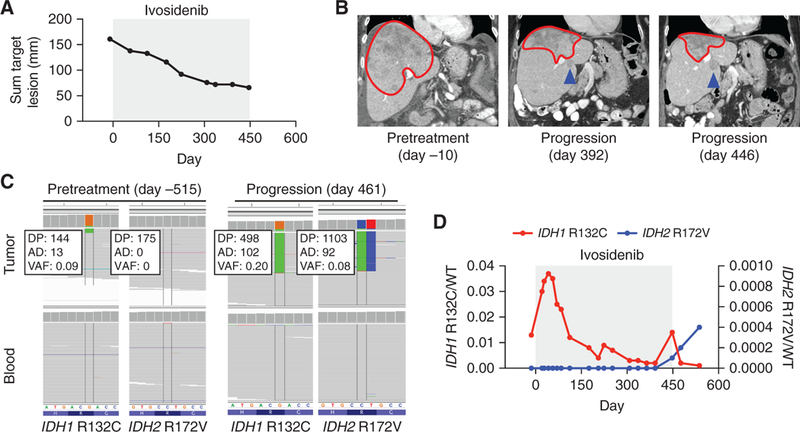
Acquired resistance to mutant IDH1 inhibition associated with emergence of an oncogenic IDH2 mutation in cholangiocarcinoma. Radiographic and laboratory features of a patient (case 3) with IDH1 R132C-mutant ICC treated with the mutant IDH1 inhibitor ivosidenib (gray box), including (A) relative change in cumulative tumor size over time, (B) serial cross-sectional imaging of the tumor mass in the liver, (C) Integrative Genomics Viewer (IGV) images highlighting relevant regions of IDH1 and IDH2 in pretreatment (day −515) and progression biopsy (day 461) sequenced by MSK-IMPACT, and (D) ratios of sequencing reads for IDH1 R132C/WT (red, left axis) or IDH2 r172V/Wt (blue, right axis) as assessed by ddPCR of cell-free DNA isolated from peripheral blood. For B, the red line demarks the primary tumor and the blue arrow indicates the escape lesion. DP, total depth; AD, depth of alternative allele; VAF, variant allele frequency.
Serial monitoring of cell-free DNA (cfDNA) by ddPCR for IDH1 R132C and IDH2 R172V demonstrated the presence of the IDH1 R132C-mutant allele prior to ivosidenib, with an allelic fraction that decreased over the course of ivosidenib therapy (Fig. 2D). In contrast, the IDH2 R172V mutation first became detectable at day 448, coinciding with radiographic disease progression (Fig. 2D). Biopsy of the escape lesion shortly after discontinuation of ivosidenib demonstrated increased 2HG levels by LC/MS (Supplementary Fig. S2A). Possibly due to the small size of the escape lesion, there was no detectable increase in plasma 2HG at the time of progression on ivosidenib (Supplementary Fig. S2B).
Case Report 4
A previously healthy 70-year-old man was found to be pancytopenic on routine bloodwork. Bone marrow aspiration and core biopsy demonstrated a hypercellular marrow with dysplastic neutrophils, erythroid precursors, and megakaryocytes. Myeloblasts were increased and comprised 15% of the marrow cellularity. These morphologic findings were consistent with myelodysplastic syndrome (MDS), subtype refractory anemia with excess blasts 2 (RAEB-2). Cytogenetics and FISH were normal. Clinical amplicon-based genotyping of bone marrow cells demonstrated the presence of an IDH2 R140Q mutation without mutation of NPM1, CEBPA, FLT3, or KIT (not shown). After 4 cycles of azacytidine, pancytopenia persisted (Supplementary Fig. S3A–S3C). Repeat bone marrow evaluation demonstrated a hypercellular marrow with persistent trilineage dysplasia (Supplementary Fig. S3D) and increased myeloid blasts comprising up to 20% of the marrow cellularity (Fig. 3A). Targeted NGS of bone marrow cells using a microdroplet-PCR assay (20) confirmed persistence of the IDH2 R140Q mutation (Fig. 3B; Supplementary Tables S1 and S9). On the basis of these findings, the patient met the diagnostic criteria for IDH2-mutant secondary AML evolved from MDS.
Figure 3.
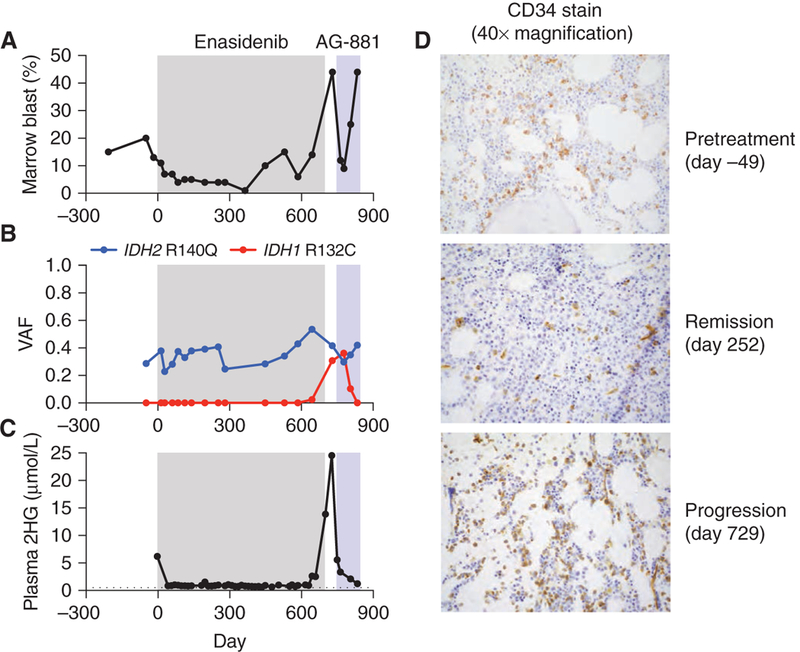
Acquired resistance to mutant IDH2 inhibition associated with emergence of an oncogenic IDH1 mutation in AML. Clinical, laboratory, and pathologic features of a patient (case 4) with IDH2 R140Q-mutant AML treated with the mutant IDH2 inhibitor enasidenib (gray box) and the dual IDH1/2 inhibitor AG-881 (blue box), including (A) bone marrow blast percentage, (B) VAF for IDH1 and IDH2 mutations identified by targeted NGS of bone marrow cells, (C) plasma 2HG concentration measured by gas chromatography–mass spectrometry (day −3 by liquid chromatography–mass spectrometry), and (D) CD34 IHC staining (identifies leukemic blasts) of bone marrow cells at indicated points in the disease course. For C, dotted line indicates limit of detection.
The patient began treatment with the mutant IDH2 inhibitor enasidenib 100 mg orally twice daily. Monthly bone marrow evaluation demonstrated a progressive decrease in the myeloid blast percentage with a complete response (blasts <5%) evident after completing three 28-day cycles of therapy (Fig. 3A; Supplementary Fig. S3D). Although the pancytopenia persisted, there was a modest improvement in the absolute neutrophil count (Supplementary Fig. S3A–S3C). The variant allele frequency (VAF) for IDH2 R140Q remained in the ~20% to 40% range (Fig. 3B), consistent with the observation that enasidenib promotes differentiation of IDH2-mutant blasts rather than cytotoxicity (14, 17). Serial measurements of plasma 2HG concentration demonstrated a decrease from the pretreatment level of 6.2 μmol/L to levels ranging from 0.5 to 1.5 μmol/L while on therapy with enasidenib (Fig. 3C).
The patient remained in complete remission until the start of cycle 17 of enasidenib (day 448), at which time the bone marrow blast percentage increased to 10% (Fig. 3A), leukemic blasts appeared in the blood (not shown), and two CEBPA mutations became detectable by NGS of bone marrow cells (Supplementary Table S9). On day 497, the dose of enasidenib was increased to 200 mg orally twice daily, resulting in a transient decrease in bone marrow blasts (Fig. 3A). However, 8 weeks later there was an increase in bone marrow blasts accompanied by a progressive rise in plasma 2HG concentration (Fig. 3A, C, and D). Targeted NGS of bone marrow cells identified a new IDH1 R132C mutation (Fig. 3B) coinciding with the leukemic progression and rise in plasma 2HG levels. ddPCR analysis of DNA from bone marrow cells demonstrated that the IDH1 R132C mutation was not detectable prior to treatment but was present at low levels relatively early in the course of enasidenib treatment, well before the development of overt clinical resistance (Supplementary Fig. S3E; Supplementary Table S10).
Enasidenib was discontinued on day 700, and the patient began therapy on a clinical study of the dual IDH1/2 inhibitor AG-881 at 400 mg orally daily. There were rapid decreases in the plasma 2HG concentration, bone marrow blast percentage, and VAF for IDH1 R132C (Fig. 3A–C). These effects were transient, as the patient underwent a prolonged hospitalization for Clostridium difficile infection and gastrointestinal bleeding and ultimately expired approximately 3 months after starting therapy with AG-881. In the interval prior to expiration, there was progression of leukemic blasts despite ongoing suppression of blood 2HG levels.
IDH Inhibition in Cells Expressing Both Mutant IDH Isoforms
As the IDH2 R172V mutation identified in case 3 has not been previously reported in patients, we examined the effect of introducing this mutation into IDH1-mutant cancer cells. Expression of the IDH2 R172V mutation in IDH1 R132G-mutant chondrosarcoma cells resulted in 2HG production that was resistant to ivosidenib but susceptible to combined treatment with ivosidenib and enasidenib (Fig. 4A). Likewise, we examined the effect of introducing a neomorphic IDH1 R132H mutation into chondrosarcoma cells harboring an endogenous IDH2 R172S mutation. Although the IDH2-mutant chondrosarcoma cells did not tolerate transient transfection of the IDH1 R132H mutation, we were able to achieve low-level expression of the IDH1 R132H mutation via a doxycycline-inducible retroviral system (Fig. 4B). Expression of the IDH1 R132H mutation conferred enasidenib-resistant 2HG production (albeit less than the uninhibited endogenous IDH2 mutation), which was suppressed by combined inhibition of both IDH isoforms (Fig. 4B). IDH inhibition impaired cellular fitness in both experimental systems as evidenced by decreased cellular biomass (Supplementary Fig. S4A and S4B) and modestly induced apoptosis in the IDH2 R172S-mutant cells as evidenced by cleavage of caspase-3 (Supplementary Fig. S4B). Taken together, these findings demonstrate that bidirectional mutant IDH isoform switching can restore 2HG production in the face of isoform-selective IDH inhibition and that this effect can be overcome by combined inhibition of both isoforms of mutant IDH.
Figure 4.
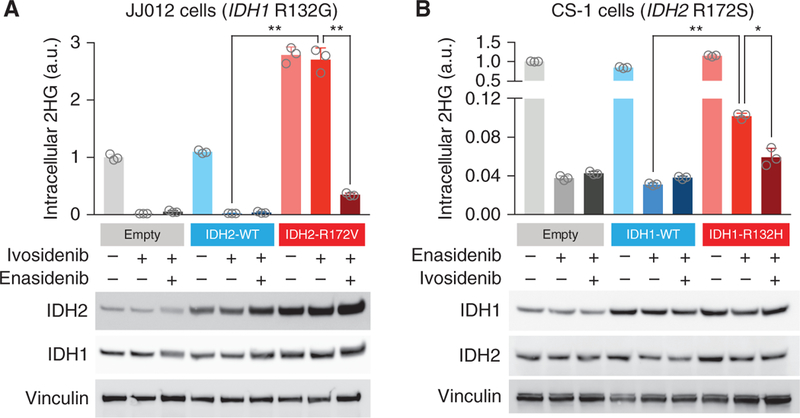
Response to IDH inhibition in cells expressing both mutant IDH isoforms. A, IDH1 R132G-mutant chondrosarcoma cells (JJ012) were transfected with empty vector (empty), IDH2 wifigd-type (IDH2-WT), or IDH2-R172V. Cells were treated for 24 hours with vehicle, ivosidenib (50 μmol/L), or ivosidenib (50 μmol/L) plus enasidenib (50 μmol/L). B, IDH2 R172S-mutant chondrosarcoma cells (CS-1) were stably transduced with retrovirus expressing doxycycline-inducible empty vector (empty), IDH1 wild-type (IDH1-WT), or IDH1-R132H. Cells in doxycycline were treated for 24 hours with vehicle, enasidenib (50 μmol/L), or enasidenib (50 μmol/L) plus ivosidenib (50 μmol/L). For both A and B, intracellular 2HG levels were measured by gas chromatography–mass spectrometry. Error bars, mean ± SEM for triplicate cultures. *, P = 0.0043; **, P < 0.0001 by one-way ANOVA with Tukey multiple comparisons test. Western blot showing IDH2 and IDH1 protein levels. Vinculin serves as a loading control. Results are representative of ≥2 independent experiments. a.u., arbitrary units.
DISCUSSION
We have identified mutant IDH isoform switching as a mechanism of acquired resistance to IDH-targeted therapy. Both IDH1 and IDH2 mutations are observed in liquid tumors, whereas in solid tumors IDH1 mutations vastly outnumber IDH2 mutations (6). The biological underpinning for the preferential selection of cytosolic IDH1 mutation versus mitochondrial IDH2 mutation in different cancer types remains poorly understood. Experimental evidence suggests that the level of 2HG production can vary based on the subcellular location of the IDH mutation (23). Our findings suggest that the selective pressure of inhibiting mutant IDH activity in one subcellular compartment might provide a growth advantage for malignant subclones with unimpeded mutant IDH activity in another subcellular compartment.
Resistance alleles assessed using ddPCR were detectable prior to and/or early during treatment. This suggests that rare subclones harboring neomorphic mutations in both IDH1 and IDH2 can emerge under the selective pressure of isoform-selective IDH inhibition. Indeed, the VAFs of the IDH mutations in cases 1, 2, and 4 allow us to speculate that the IDH1 and IDH2 mutations are present within the same subclone. However, it remains possible that resistance to isoform-selective IDH inhibitors might also occur through parallel acquisition of IDH1 and IDH2 mutations in distinct clones. Single-cell analyses will be required to formally delineate the clonal architecture of IDH inhibitor resistance, particularly for solid tumors such as case 3. Likewise, it will be important to determine whether additional co-occurring genomic alterations contribute to acquired resistance to IDH inhibition.
Mutant IDH isoform switching represents a mechanism of acquired resistance to isoform-selective IDH-targeted therapy with potential therapeutic import. Experimental evidence demonstrated that cancer cells expressing both mutant IDH1 and mutant IDH2 exhibit 2HG production that is resistant to monotherapy yet sensitive to combined inhibition of both isoforms (Fig. 4). The patient in case 4 exhibited potent inhibition of 2HG production and a transient clinical response to the dual IDH1/2 inhibitor AG-881, although there was subsequent 2HG-independent disease progression resulting from unknown molecular mechanisms. Alternatively, a strategy of cotargeting of mutant IDH1 and IDH2 from the outset might delay or prevent resistance and should be explored in clinical trials. These efforts could be focused on patients with low levels of the alternative mutant IDH isoform detectable by sensitive sequencing assays (e.g., ddPCR) prior to or early during therapy with an isoform-selective IDH inhibitor.
The frequency of mutant IDH isoform switching as a mechanism of resistance to IDH inhibition remains uncertain. Prior studies in liquid tumors have begun to elucidate potential mechanisms of resistance to mutant IDH2 inhibitors. We previously reported IDH2 dimer–interface mutations in 2 of 9 patients with IDH2-mutant AML and acquired resistance to enasidenib therapy (18). The current study identifies isoform switching to mutant IDH1 in an additional case (case 4) from this cohort (1 of 9; 11%). Similarly, a recent report using single-cell sequencing to study clonal dynamics of IDH2-mutant AML in response to enasidenib found that 2 of 12 (17%) cases developed resistance associated with emergence of new IDH1 mutations (19). Here, we establish bidirectional mutant IDH isoform switching, either from cytoplasmic mutant IDH1 to mitochondrial mutant IDH2 or vice versa, as a generalized mechanism of acquired clinical resistance to IDH inhibition in both liquid and solid tumors. As larger numbers of patients with IDH-mutant malignancies are treated with isoform-selective IDH inhibitors, it will be important to determine the precise frequency of isoform switching and to elucidate other mechanisms of de novo and acquired resistance (17–19). These cases underscore the potential utility of serial DNA sequencing and 2HG measurement in patients treated with IDH inhibitors as means to monitor response, define resistance mechanisms, and inform treatment recommendations.
METHODS
Clinical Specimens
The patients described were enrolled on the phase I/II studies NCT01915498 (14), NCT02073994, or NCT02074839 (15). Enrollment was open to all patients with relapsed or refractory AML with a mutation in IDH1 (NCT02074839) or IDH2 (NCT01915498) or patients with advanced solid tumors with a mutation in IDH1 (NCT02073994). IDH mutations were identified locally and confirmed centrally. Patients were required to be 18 years or older at the time of study entry. Both men and women were enrolled on the studies. Patients were required to have acceptable performance status and adequate organ function as defined in the study protocols. Clinical data, blood, bone marrow, or tumor biopsy specimens were obtained after receiving written informed consent from patients. Studies were conducted in accordance with the Belmont Report and the U.S. Common Rule. Approval was obtained from the Institutional Review Board at each institution participating in these clinical trials. Additional consent was obtained from participants at Memorial Sloan Kettering Cancer Center with analyses performed on the institutional biobanking protocol approved by the Institutional Review Board. Patient biospecimens were anonymized by creating unique identifiers with no associated PHI and keeping the key on a password-protected server. Data collection and research was performed in compliance with all relevant ethical regulations for human research participants. Absolute neutrophil count, hemoglobin concentration, platelet count, and blast percentage were determined by standard clinical assays. Targeted NGS and determination of VAF were performed by Raindance microdroplet PCR (20) for AML or MSK-IMPACT hybrid capture (24) for cholangiocarcinoma as previously described (see Supplementary Tables S1, S6, and S8 for gene lists). Sequencing data are deposited in the European Nucleotide Archive under accession number PRJEB29344.
cfDNA Isolation and Sequencing
Whole blood was collected in 10-mL Cell-Free DNA BCT tubes (STRECK) and centrifuged at 800 × g for 10 minutes at ambient temperature. Plasma was separated from red blood cells and then subjected to an additional centrifugation at 18,000 × g for 10 minutes at ambient temperature. Cell-free plasma was aliquoted and stored at −80°C. Extraction of cfDNA was performed using a fully automated QIAGEN platform, QIAsymphony SP, and QIAsymphony DSP Virus/Pathogen Midi Kit (QIAGEN). This is a bead-based custom protocol, optimized to work with 3 mL of plasma as starting material. The extraction process includes lysis, binding, wash, and elution steps. The final product was a 60 μL elution of cfDNA with an average size of ~170 to 200 bp. Quality and quantity of cfDNA were evaluated with automated electrophoresis using a fragment analyzer with a high-sensitivity genomic DNA analysis kit (Advanced Analytical). A mean 122 ng of cfDNA was isolated at each time point and analyzed for wild-type and mutant IDH1 and IDH2 alleles by ddPCR
ddPCR
An assay specific for each wild-type and mutant allele was designed and ordered through Bio-Rad (IDH1 R132C: dHsaMDV2010053; IDH2 R140Q: dHsaCP2000057; IDH2 R172V: dHsaMDS874523342). Cycling conditions were tested to ensure optimal annealing/extension temperature as well as optimal separation of positive from empty droplets. All reactions were performed on a QX200 ddPCR System (Bio-Rad) and evaluated in technical duplicates. Reactions were partitioned into a median of ~16,000 droplets per well using the QX200 droplet generator and run on a 96-well thermal cycler. Plates were then analyzed with the QuantaSoft v1.7 to assess the number of droplets positive for mutant or wild-type DNA.
Cell Culture
The chondrosarcoma cell lines JJ012 with an endogenous IDH1 R132G mutation and CS-1 with an endogenous IDH2 R172S mutation were obtained from Dr. Justin Cross (Memorial Sloan Kettering) and previously validated by sequencing the IDH1 and IDH2 genes as described (23, 25). Cells were maintained at low passage number (<10 passages after thawing) in high-glucose DMEM with 10% FBS, glucose 25 mmol/L, and glutamine 2 mmol/L and split every 2 to 3 days before reaching confluence. Cultured cells tested negative for Mycoplasma at least monthly throughout the experimental period.
Statistical Analyses
Statistical analyses for 2HG levels in Fig. 4 were performed with Prism 7.0 (GraphPad), using one-way ANOVA with Tukey multiple comparisons test.
DNA Constructs
Please refer to the Supplementary Methods.
Gel Electrophoresis and Western Blotting
Please refer to the Supplementary Methods.
Metabolite Extraction and Analysis
Please refer to the Supplementary Methods.
Supplementary Material
SIGNIFICANCE:
IDH-mutant cancers can develop resistance to isoform-selective IDH inhibition by “isoform switching” from mutant IDH1 to mutant IDH2 or vice versa, thereby restoring 2HG production by the tumor. These findings underscore a role for continued 2HG production in tumor progression and suggest therapeutic strategies to prevent or overcome resistance.
Acknowledgments
We thank members of the Finley, Intlekofer, Levine, Papaemmanuil, and Reznik laboratories for helpful discussions. We thank Walid K. Chatila and Nikolaus D. Schultz for assistance with data deposition. A. Shih is supported by the NIH/NCI (K08 CA181507) and the Leukemia and Lymphoma Society. J. Schvartzman is a Hope Funds for Cancer Research Postdoctoral Fellow. A. Intlekofer is supported by the NIH/NCI (K08 CA201483), Burroughs Wellcome Fund (CAMS 1015584), Damon Runyon Cancer Research Foundation (CI 95–18), Leukemia and Lymphoma Society (Special Fellow 3356–16), Susan and Peter Solomon Divisional Genomics Program, Steven A. Greenberg Fund, and Cycle for Survival. The research was supported, in part, by the Leukemia and Lymphoma Society Specialized Center of Research Program (7011–16; A. Intlekofer), a Translational and Integrative Medicine Research Fund (TIMRF) grant (A. Shih and E. Stein), and grants from the NIH, including R35 CA197594–01A1 (R. Levine), U54 0D020355 (R. Levine), and the Memorial Sloan Kettering Cancer Center Support Grant (NIHP30 CA008748). The research was also supported, in part, by a Stand Up To Cancer Colorectal Cancer Dream Team Translational Research Grant (grant number: SU2C-AACR-DT22–17) to L. Diaz. Stand Up To Cancer (SU2C) is a division of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. We acknowledge the use of the Integrated Genomics Operation Core. funded by the Memorial Sloan Kettering Cancer Center Support Grant (NIHP30 CA008748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Disclosure of Potential Conflicts of Interest
J.J. Harding reports receiving a commercial research grant from Bristol-Myers Squib and is a consultant/advisory board member for Bristol-Myers Squibb, Eli Lilly, Eisai, and CytomX. M.A. Lowery is a consultant/advisory board member for Agios, Celgene, EMD Serono, and Roche. J.M. Schvartzman is an Associate Physician at Best Doctors. M. Roshal reports receiving a commercial research grant from Agios and is a consultant/advisory board member for Agios and Celgene. R.K. Do is a consultant/advisory board member for Bayer. M.S. Tallman reports receiving commercial research grants from Arog, Cellerant, Orsenix, ADCT Therapeutics, and AbbVie and is a consultant/advisory board member for Daiichi Sankyo, AbbVie, Nohla, Danbury Hospital, and American Society of Hematology. D.M. Hyman reports receiving commercial research grants from Loxo Oncology, Puma Biotechnology, and AstraZeneca and is a consultant/advisory board member for Atara Bio-therapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, and Genentech. E. Papaemmanuil reports receiving a commercial research grant from MDS Foundation, has been a speaker at conference symposia, and is a consultant/advisory board member for the Novartis Scientific Advisory Board. A. Tosolini is Director, Clinical Science at Celgene Corporation and has ownership interest (including stock, patents, etc.) in the same. M.G. Frattini is Executive Medical Director at Celgene Corporation and has ownership interest (including stock, patents, etc.) in the same. K.J. MacBeth is Senior Director at Celgene Corporation and has ownership interest (including stock, patents, etc.) in the same. G. Liu is Director at Agios Pharmaceuticals and has ownership interest (including stock, patents, etc.) in the same. S. Choe has ownership interest (including stock, patents, etc.) in Agios. B. Wu has ownership interest (including stock, patents, etc.) in Agios. Y.Y. Janjigian reports receiving commercial research grants from Boehringer Ingelheim, Bayer, Genentech/Roche. Bristol-Myers Squibb, Eli Lilly, and Merck and is a consultant/advisory board member for Merck Serono, Bristol-Myers Squibb, Eli Lilly, Pfizer, Bayer, Imugene, and Merck. I.K. Mellinghoff reports receiving commercial research support from GE Global Research and is a consultant/advisory board member for Agios, Puma Biotechnology, Merck, Deciphera, Roche, and Debiopharm Group. L.A. Diaz is a member of the Board of Directors of Jounce Therapeutics and Personal Genome Diagnostics, has ownership interest (including stock, patents, etc.) in PapGene, Personal Genome Diagnostics, Phoremost, Jounce Therapeutics, and Zydecom, and is a consultant/advisory board member for Merck, Personal Genome Diagnostics, Caris, Lyndra, and Genocea. R.L. Levine is on the Supervisory Board of Qiagen and the Scientific Advisory Board of Loxo, reports receiving commercial research grants from Celgene, Roche, and Prelude, has received honoraria from the speakers bureaus of Gilead and Lilly, has ownership interest (including stock, patents, etc.) in Qiagen and Loxo, and is a consultant/advisory board member for Novartis, Roche, Janssen, Celgene, and Incyte. G.K. Abou-Alfa is a consultant/advisory board member for 3DMedcare, Agios, Beigene, Bioline, BMS, Boston Scientific, Bridgebio, Carsgen, Celgene, Casi, Cipla, CytomX, Alignmed, Daiichi, Debio, Delcath, Eisai, Exelixis, Genoscience, Gilead, Halozyme, Hengrui, Incyte, Amgen, Invio, Jazz, Janssen, Kyowa Kirin, Lamonoco, Lilly, Loxo, Merck, Mina, Newlink Genetics, Antengene, Novella, Onxeo, PCI Biotech, Pfizer, Pharmacyte, Pharmacyclics, Pieris, QED, Redhill, Sanofi, Aptus, Servier, Silenseed, Sillajen, Sobi, Targovax, Tekmira, Twoxar, Vicus, Yakult, Yiviva, Aslan, Astellas, AstraZeneca, and Bayer. E. Stein reports receiving commercial research grants from Celgene Pharmaceuticals, Agios Pharmaceuticals, Syros Pharmaceuticals, and Bayer Pharmaceuticals, and is a consultant/advisory board member for Celgene Pharmaceuticals, Agios Pharmaceuticals, Daiichi Sankyo Pharmaceuticals, Astellas Pharmaceuticals, AbbVie Pharmaceuticals, Bayer Pharmaceuticals, Syros Pharmaceuticals, Novartis Pharmaceuticals, Pfizer Pharmaceuticals, and Seattle Genetics. A.M. Intlekofer is a consultant/advisory board member for Foundation Medicine.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
No potential conflicts of interest were disclosed by the other authors.
REFERENCES
- 1.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011;224:334–43. [DOI] [PubMed] [Google Scholar]
- 5.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 2017;18:2780–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 2013;27:836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483:474–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013;339:1621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018;34:186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018;378:2386–98. [DOI] [PubMed] [Google Scholar]
- 16.Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett 2018;9:300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017;130:732–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Intlekofer AM, Shih AH, Wang B, Nazir A, Rustenburg AS, Albanese SK, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018;559:125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med 2018;24:1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng DT, Cheng J, Mitchell TN, Syed A, Zehir A, Mensah NYT, et al. Detection of mutations in myeloid malignancies through paired-sample analysis of microdroplet-PCR deep sequencing data. J Mol Diagn 2014;16:504–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fathi AT, DiNardo CD, Kline I, Kenvin L, Gupta I, Attar EC, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol 2018;4:1106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan SM, Thomas D, Corces-Zimmerman MR., Xavy S, Rastogi S, Hong WJ, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 2015;21:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward PS, Lu C, Cross JR, Abdel-Wahab O, Levine RL, Schwartz GK, et al. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J Biol Chem 2013;288:3804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23: 703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salamanca-Cardona L, Shah H, Poot AJ, Correa FM, Di Gialleonardo V, Lui H, et al. In vivo imaging of glutamine metabolism to the onco-metabolite 2-hydroxyglutarate in IDH1/2 mutant tumors. Cell Metab 2017;26:830–41e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.