

Abstract
Background
Catatonia is a debilitating disorder of movement and volition associated with schizophrenia and some other mental illnesses. People with catatonia are more likely to require hospitalisation and highly supervised care than those without the disorder. They also have an increased risk of secondary complications such as pneumonia, malnutrition and dehydration. The mainstay of treatment has been drug therapies and electroconvulsive therapy.
Objectives
To compare the effects of benzodiazepines with other drugs, placebo or electroconvulsive therapy for catatonia in people with schizophrenia or other similar serious mental illnesses (SMIs).
Search methods
We updated our previous search (28 February 2007) by searching the Cochrane Schizophrenia Group's Study‐Based Register of Trials (9 November 2016; 6 February 2019). This register is compiled by systematic searches of major resources (including CENTRAL, MEDLINE, Embase, AMED, BIOSIS, CINAHL, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings, with no language, date, document type, or publication status limitations for inclusion of records into the register. We also manually searched reference lists from studies selected by the search.
Selection criteria
All controlled clinical trials that randomised people who have schizophrenia or other similar SMI and experiencing catatonia to receive benzodiazepines or another relevant treatment. We included studies that met our inclusion criteria and reported usable data. We excluded those not meeting our inclusion criteria or those not reporting usable data. We contacted authors when we required further information; and if we received no response, we put those studies aside as 'awaiting assessment'.
Data collection and analysis
Review authors extracted data independently. For dichotomous data we calculated relative risks (RR) and their 95% confidence intervals (CI) on an intention‐to‐treat basis using a fixed‐effect model. We completed a 'Risk of bias' assessment for the included study and generated a 'Summary of findings' table using GRADE.
Main results
The searches found 130 citations, from which we could identify 22 possibly relevant studies. From these, we could only include one study. This study had a relatively small sample size of 17 participants who received lorazepam or oxazepam and were drug free for one week before the trial started. The only usable data reported by this study were clinically important change in symptoms of catatonia measured as 50% improvement on the Visual Analogue Scale (VAS). There was no difference in the numbers of participants showing a clinically important change in their catatonic symptoms (RR 0.95, 95% CI 0.42 to 2.16; participants = 17; studies = 1; very low quality evidence).
No data were reported for other important outcomes of hospital stay, clinically important change in satisfaction with care, global state, adverse effects or general functioning
We did find a few studies meeting our inclusion criteria but they reported no usable data. We had to exclude these. Although poorly reported, these studies do illustrate that relevant studies have been undertaken — they are not impossible to design and conduct.
Authors' conclusions
Analysis of the results from this review, which was a head‐to‐head comparison of two benzodiazepine monotherapies, does not show a clear difference in effect. No data were available for benzodiazepines compared to placebo or standard care. The lack of usable data and very low quality of data available makes it impossible to draw firm conclusions and further studies with a high‐quality methodology and reporting are required in order to determine more definitively the outcomes associated with benzodiazepine use in the clinical management of catatonia in persons with schizophrenia and other SMI.
Plain language summary
Benzodiazepines for catatonia in people with schizophrenia or similar serious mental illness
Review question Are the group of drugs called benzodiazepines an effective and tolerable treatment for catatonia in people with schizophrenia or other serious mental illnesses?
Background Catatonia is a debilitating condition that is characterised by diminished, excessive or peculiar movement and activity as well as diminished engagement with the social and physical environment. It can occur when a person has a number of different psychiatric conditions, including schizophrenia (an enduring mental illness whose hallmark is an altered perception of reality); and less frequently with medical conditions. Some of the other serious mental disorders that are associated with catatonia include bipolar disorder (an illness in which there are extremes of disturbed mood) and depression (another mood disorder characterized by low mood). Benzodiazepines are widely used in the treatment of catatonia, but there is no good quality evidence from randomised controlled trials concerning their effectiveness.
Searching for evidence The Information Specialist from Cochrane Schizophrenia ran electronic searches of the group's specialised register (the most recent in February 2019) for trials that randomised people with catatonia occurring in conjunction with schizophrenia or other similar serious mental illnesses to receive either benzodiazepines or any of the following: other drugs, placebo, or electroconvulsive therapy. One hundred and thirty records were found and checked by the review authors.
Evidence found One trial was found in the search which met the review requirements and provided limited, very low quality usable data for one outcome only. This trial compared two benzodiazepines (lorazepam vs oxazepam) and found no clear difference between these two treatments for improvement in the symptoms of catatonia for people who have catatonia and schizophrenia or similar serious mental illness.
Conclusions There is insufficient high quality evidence available to answer the review question. More high quality research is needed.
Summary of findings
Summary of findings for the main comparison. Lorazepam compared to Oxazepam for catatonia in people with schizophrenia or other SMI.
| Lorazepam compared to oxazepam for catatonia in people with schizophrenia and/or SMI | ||||||
| Patient or population: catatonia in people with schizophrenia or SMI Setting: hospital Intervention: lorazepam Comparison: oxazepam | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with oxazepam | Risk with lorazepam | |||||
| Catatonia: clinically important change in catatonia symptoms (improved at least 50% on the VAS) | Study population | RR 0.95 (0.42 to 2.16) | 17 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 3 | ||
| 600 per 1000 | 570 per 1000 (252 to 1000) | |||||
| Service use: duration of stay in hospital | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| Satisfaction with care: clinically important change in satisfaction by informal care‐givers | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| Satisfaction with care: clinically important change in satisfaction by recipients of care | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| Global state: clinically important change in global state (as defined by individual studies) | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| Adverse effects: incidence of clinically important adverse effect | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| General functioning: clinically important change in specific aspects of functioning, such as life skills ‒ as defined by each of the studies | ‐ | ‐ | ‐ | ‐ | ‐ | not reported |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Serious: downgraded by 1 level due to unclear study design whether any of the following factors were considered; randomisation, allocation concealment, blinding of participants and personnel and blinding of outcome assessment. The study did imply this was a "double‐blind crossover study" but no further description or details provided discussing study design. Study did not report adverse events. Data from 4 participants were excluded in the final analysis.
2 Serious: downgraded by 1 level. Intervention was only delivered once and outcomes measured. Reporting outcomes were measured for one day only after intervention. No long‐term points for outcome measurements. Fixed doses of both interventions used in participants.
3 Serious: downgraded by 1 level due to effect size from 1 small study with a sample size of 17 participants. Wide ranging CI interval around effect estimate.
Background
Description of the condition
Catatonia is a syndrome characterised by motor abnormalities such as purposeless activity, immobility and posturing, together with disturbances of consciousness and volition. Because of its debilitating nature and high risk for complications, it is usually treated in hospital. In 1874 observers described seventeen typical signs, although many more have since been added (Kahlbaum 1973). Catatonia has been described in the context of schizophrenia (a chronic mental illness presenting with features such as hallucinations, delusions, disorganized speech and disorganized behaviour), organic illnesses and mood disorders (APA 2000; WHO 1992). Regardless of the cause, when catatonia is not treated patients are at an increased risk for a number of negative outcomes such as extreme negativism, mutism and echolalia. People with catatonia may develop pneumonia and thromboembolic complications (Regestein 1977). Malnutrition and dehydration are also common (Penland 2006).
Description of the intervention
A number of different treatments have been advocated for catatonia. Attention to underlying causes is important although, in some instances, antipsychotics (used in the treatment of schizophrenia) may worsen catatonia (Penland 2006). Benzodiazepines are medications that act on the central nervous system; they are also referred to as anxiolytics and have sedative properties. Benzodiazepines, especially lorazepam, have been thought to be of some benefit to persons with catatonia (Rosebush 1990). Together with electroconvulsive therapy, benzodiazepines are perhaps the most widely reported treatment for the condition.
How the intervention might work
Neuronal pathways mediated by the neurotransmitter gamma‐aminobutyric acid (GABA) are believed to play a central role in the integration of emotional and cognitive functions. Catatonic symptoms are postulated to arise from dysregulation in these pathways (Ellul 2015). Benzodiazepines potentiate GABAergic activity and are believed to counteract the GABAergic dysregulation underlying catatonic symptoms.
Why it is important to do this review
Benzodiazepines are a common treatment strategy for catatonia; and much of the evidence base for their use in this capacity consists of case reports and small studies. This systematic review attempted to identify and summarise any relevant evidence from trials and thus provide clinicians with a stronger basis for making decisions about their use in the management of catatonia.
Objectives
To compare the effects of benzodiazepines with other drugs, placebo or electroconvulsive therapy for catatonia in people with schizophrenia or other similar serious mental illnesses.
Methods
Criteria for considering studies for this review
Types of studies
We included studies that were described as randomised controlled trials or as 'double blind', in which randomisation is implied, in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those that allocate intervention by alternate days of the week. Where people were given additional treatments as well as a benzodiazepine we only included studies and analysed the data if the adjunct treatment was evenly distributed between groups and it was only the benzodiazepine that was randomised.
Types of participants
Adults, however defined, with catatonia and schizophrenia or other serious mental illnesses, including schizophreniform disorder, schizoaffective disorder,and bipolar disorder, by any means of diagnosis.
Types of interventions
1. Benzodiazepines of any type, dose and means of administration; compared with 2. Any other class of pharmacological agent at any dose; 3. Placebo; 4. Electroconvulsive therapy.
Types of outcome measures
We would have divided all outcomes into short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks). Short‐term studies would also be distinguished according to whether they were single injection, one day or longer studies.
Primary outcomes
1. Catatonia
1.1 Clinically important change in catatonia symptoms (as defined by each study)
2. Hospital and service outcomes
2.1 Duration of stay in hospital
3. Satisfaction with care
3.1 Clinically important change in satisfaction (recipient or informal care‐giver)
4. Adverse effect
4.1 Adverse effects ‒ incidence of clinically important adverse effect
Secondary outcomes
1. Catatonia
1.1 Any change in symptoms ‒ as defined by individual studies (binary or continuous measures)
2. Global state
2.1 Clinically important change in global state (e.g. clinical response as defined by the individual studies ‒ e.g. global impression rated as 'much improved', or more than 50% improvement on a rating scale) 2.2 Relapse ‒ as defined by each study 2.3 Any change in global state 2.4 Average endpoint or change score global state scale 2.5 Use of other medications
3. Satisfaction with care
3.1 Clinically important change in satisfaction ‒ professional carers.
4. Adverse effects
4.1 General adverse effects
4.1.1 At least one adverse effect 4.1.2 Clinically important adverse effects 4.1.3 Average endpoint/change scores adverse‐effect scales
4.2 Specific adverse effects ‒ clinically important ‒ as defined by each of the studies
4.2.1 Respiratory depression 4.2.2 Hypotension 4.2.3 Syncope 4.2.4 Ataxia 4.2.5 Hepatitic (e.g. abnormal transaminase, abnormal liver function) 4.2.6 Haematology (e.g. haemogram, leukopenia, agranulocytosis/neutropenia) 4.2.7 Various other 4.2.8 Average endpoint or change score on specific adverse effect scale 4.2.9 Death
5. Leaving the study early
5.1 Due to any reason 5.2 Due to specific reason
6. General functioning
6.1 Overall
6.1.1 Clinically important change in general functioning ‒ as defined by each of the studies, including working ability 6.1.2 Any change in general functioning ‒ as defined by each of the studies, including working ability 6.1.3 Average endpoint or change score on general functioning scale
6.2 Specific
6.2.1 Clinically important change in specific aspects of functioning, such as life skills ‒ as defined by each of the studies 6.2.2 Any change in specific aspects of functioning, such as life skills ‒ as defined by each of the studies 6.2.3 Average endpoint or change score on specific aspects of functioning scale, such as life skills ‒ as defined by each of the studies
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008); and used GRADE profiler to import data from Review Manager 5 (RevMan 5) to create a 'Summary of findings' tables (GRADEpro; Review Manager). This table provides outcome‐specific information concerning the overall quality of evidence from the included study, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table:
Catatonia: clinically important change in symptoms
Service use: duration of stay in hospital
Satisfaction with care: clinically important change in satisfaction by informal care‐givers
Satisfaction with care: clinically important change in satisfaction by recipients of care
Global state: clinically important change in global state (as defined by individual studies)
Adverse effects: incidence of clinically important adverse effect
General functioning: clinically important change in specific aspects of functioning, such as life skills ‒ as defined by each of the studies
Search methods for identification of studies
Electronic searches
On 9 November 2016 and 6 February 2019, the information specialist searched Cochrane Schizophrenia's study‐based register of trials using the following search strategy:
(*Catatonia* in Health Care Condition) AND (*Benzodiazepine* in Intervention) of STUDY
In such study‐based registers, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017).
This register is compiled by systematic searches of major resources (CENTRAL, MEDLINE, Embase, AMED, BIOSIS, CINAHL, ClinicalTrials.Gov, ISRCTN, PsycINFO, PubMed, WHO ICTRP) and their monthly updates, ProQuest Dissertations and Theses A&I and its quarterly update, Chinese databases (CBM, CNKI, and Wanfang) and their annual updates, handsearches, grey literature, and conference proceedings (see Group's website). There are no language, date, document type, or publication status limitations for inclusion of records in the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We would have inspected references of all included studies for further relevant studies.
2. Personal contact
We would have contacted the first author of each included study for information regarding unpublished trials. We would have noted the outcome of this contact in the 'Characteristics of included studies' or 'Characteristics of studies awaiting classification' tables.
Data collection and analysis
Selection of studies
Review authors RG, GW and HZ independently inspected citations from the searches and identified relevant abstracts; we independently re‐inspected all abstracts. If disputes arose, we acquired the full report for more detailed scrutiny. We then independently inspected full reports of the abstracts or reports meeting the review criteria. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study concerned for clarification.
Data extraction and management
1. Extraction
Review author HZ and JO‐O (see Acknowledgements) independently extracted data from the included study. We presented data from graphs and figures only if there was consistency in extraction between HZ and JO‐O. If studies had been multi‐centre, then where possible we would have extracted data relevant to each. We would have discussed any disagreement and documented our decisions. If necessary, we would have attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification.
2. Management
2.1 Forms
We extracted data onto a standard, pre‐designed, simple form.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
a) the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000); and b) the measuring instrument had not been written or modified by one of the trialists for that particular trial. c) the instrument was a global assessment of an area of functioning and not sub‐scores which are not, in themselves, validated or shown to be reliable.
Ideally the measuring instrument should either be i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; in 'Description of studies' we would have noted if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data: change data can remove a component of between‐person variability from the analysis; however, calculation of change needs two assessments (baseline and endpoint) that can be difficult to obtain in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. If necessary, we would have combined endpoint and change data in the analysis, as we aimed to use mean differences (MDs) rather than standardised mean differences (SMDs) throughout (Higgins 2011a).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to relevant continuous data before inclusion.
For endpoint data from studies including fewer than 200 participants:
a) when a scale starts from the finite number zero, we subtracted the lowest possible value from the mean, and divided this by the standard deviation. If this value is lower than one, it strongly suggests that the data are skewed and we excluded these data. If this ratio is higher than one but less than two, there is suggestion that the data are skewed: we entered these data and, where possible, tested whether their inclusion or exclusion would change the results substantially. Finally, if the ratio is larger than two we would have included these data, because it is less likely that they are skewed (Altman 1996; Higgins 2011a).
b) if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210 (Kay 1986)), we would have modified the calculation described above to take the scale starting point into account. In these cases skewed data are present if 2 SD > (S − S min), where S is the mean score and 'S min' is the minimum score.
Please note: we would have entered all relevant data from studies of more than 200 participants in the analysis irrespective of the above rules, because skewed data pose less of a problem in large studies. We also would have entered all relevant change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether or not data are skewed.
2.5 Common measurement
To facilitate comparison between trials, where relevant we would have converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we would have made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS) (Overall 1962), or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a, Leucht 2005b). If data based on these thresholds were not available, we would have used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we would have entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for benzodiazepines. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'not un‐improved') we would have reported data where the left of the line indicates an unfavourable outcome and noted this in the relevant graphs.
Assessment of risk of bias in included studies
Review authors RG and GW would have worked independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011b). This set of criteria is based on evidence of associations between potential overestimation of effect and the level of risk of bias of the article that may be due to aspects of sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting, or the way in which these 'domains' are reported.
If the raters disagreed, we would have made the final rating by consensus. Where inadequate details of randomisation and other characteristics of trials were provided, we would have attempted to contact authors of the studies in order to obtain further information. We would have reported non‐concurrence in quality assessment, but if disputes arose regarding the category to which a trial was to be allocated, we would have resolved this by discussion.
We would have noted the level of risk of bias in both the text of the review, Figure 1, Figure 2, and the 'Summary of findings' table/s.
1.
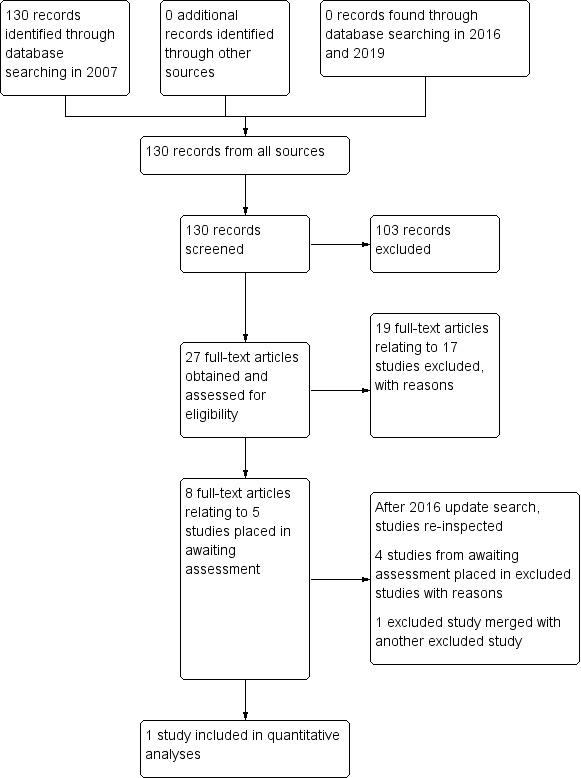
Study flow diagram for searches up to February 2019
2.
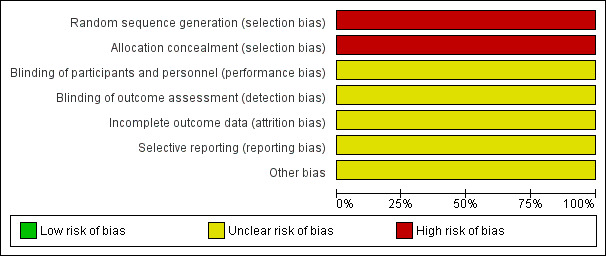
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Measures of treatment effect
1. Binary data
For binary outcomes we would have calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI), as it has been shown that RR is more intuitive than odds ratios (Boissel 1999); and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). Although the number needed to treat for an additional beneficial outcome (NNTB) and the number needed to treat for an additional harmful outcome (NNTH), with their CIs, are intuitively attractive to clinicians, they are problematic to calculate and interpret in meta‐analyses (Hutton 2009). For binary data presented in the 'Summary of findings' table/s we, where possible, would have calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we would have estimated MD between groups. We preferred not to calculate effect size measures (SMD). However if scales of very considerable similarity were used, we would have presumed there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a unit‐of‐analysis error whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated (Divine 1992). This causes type I errors (Bland 1997; Gulliford 1999).
Should clustering have been incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
Should clustering not have been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. We would have contacted first authors of studies to obtain intra‐class correlation coefficients for their clustered data and adjusted for this by using accepted methods (Gulliford 1999).
We sought statistical advice and have been advised that the binary data from cluster trials presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC): thus design effect = 1 + (m − 1) * ICC (Donner 2002). If the ICC had not been reported we would have assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed and intra‐class correlation coefficients and relevant data documented in the report taken into account, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. This occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, participants can differ significantly from their initial state at entry to the second phase, despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both carry‐over and unstable conditions are very likely in severe mental illness, we would have only used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant we would have presented the additional treatment arms in comparisons. If data were binary we simply would have added these and combined within the two‐by‐two table. If data were continuous we would have combined data following the formula for combining data in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). Where additional treatment arms were not relevant, we would not have reproduced these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for we would not reproduce these data or use them within analyses. If, however, more than 50% of those in one arm of a study had been lost, but the total loss had been less than 50%, we would have addressed this within the 'Summary of findings' table/s by down‐rating quality. Finally, we would have also downgraded quality within the 'Summary of findings' table/s should the loss have been 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome is between 0% and 50% and where these data are not clearly described, we would have presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis (ITT)). Those leaving the study early would have all been assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. For these outcomes the rate of those who stayed in the study — in that particular arm of the trial — would have been used for those who did not. We would have undertaken a sensitivity analysis testing how prone the primary outcomes would have been to change when data only from people who completed the study to that point were when compared to the intention‐to‐treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
We would have used data where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported.
3.2 Standard deviations
If standard deviations (SDs) had not been reported, we would have tried to obtain the missing values from the authors. If these were not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs were available for group means, and either P value or t value were available for differences in mean, we would have been able to calculate SDs according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). When only the SE is reported, SDs are calculated by the formula SD = SE * √(n). The Cochrane Handbook for Systematic Reviews of Interventions presents detailed formulae for estimating SDs from P, t or F values, CIs, ranges or other statistics (Deeks 2011). If these formulae did not apply, we would have calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. Nevertheless, we would have examined the validity of the imputations in a sensitivity analysis that excludes imputed values.
3.3 Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or were lost to follow‐up. Some trials just present the results of study completers; others use the method of last observation carried forward (LOCF); while more recently, methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences between groups in their reasons for doing so is often the core problem in randomised schizophrenia trials. We therefore did not exclude studies based on the statistical approach used. However, by preference we would have used the more sophisticated approaches, i.e. we would have preferred to use MMRM or multiple‐imputation to LOCF, and we would have only presented completer analyses if some kind of ITT data were not available at all. Moreover, we would have addressed this issue in the item 'Incomplete outcome data' of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We would have considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We would have simply inspected all studies for participants who are clearly outliers or situations that we had not predicted would arise and, where found, discussed such situations or participant groups.
2. Methodological heterogeneity
We would have considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We would have simply inspected all studies for clearly outlying methods which we had not predicted would arise and discussed any such methodological outliers.
3. Statistical heterogeneity
3.1 Visual inspection
We would have inspected graphs visually to investigate the possibility of statistical heterogeneity.
3.2 Employing the I² statistic
We would have investigated heterogeneity between studies by considering the I² statistic alongside the Chi² P value. The I² statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I² depends on the magnitude and direction of effects as well as the strength of evidence for heterogeneity (e.g. P value from Chi² test, or a confidence interval for I²). We would have interpreted an I² estimate greater than or equal to 50% and accompanied by a statistically significant Chi² statistic as evidence of substantial heterogeneity (Deeks 2011). When substantial levels of heterogeneity had been found in the primary outcome, we would have explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). We are aware that funnel plots may be useful in investigating reporting biases, but are of limited power to detect small‐study effects. We would not have used funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In other cases, where funnel plots were possible, we would have sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies, even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model: it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We would have chosen a fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We did not anticipate any subgroup analyses.
2. Investigation of heterogeneity
We would have reported if inconsistency was high. Firstly, we would have investigated whether data had been entered correctly. Secondly, if data were correct, we would have inspected the graph visually and removed outlying studies successively to see if homogeneity could be restored. For this review we decided that should this occur with data contributing to the summary finding of no more than 10% of the total weighting, we would have presented data. If not, we would not have pooled these data and discussed any issues. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
In cases where unanticipated clinical or methodological heterogeneity was obvious, we simply would have stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
1. Implication of randomisation
We would have included trials in a sensitivity analysis if they had been described in some way that implied randomisation. For primary outcomes, if the inclusion of these trials did not result in a substantive difference, they would have remained in the analyses. If their inclusion did result in statistically significant differences, we would not have added the data from these lower‐quality studies to the results of the higher‐quality trials, but presented these data within a subcategory.
2. Assumptions for lost binary data
Where assumptions would have to be made regarding people lost to follow‐up (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption compared with completer data only. If there had been a substantial difference, we would have reported results and discussed them, but continued to employ our assumption.
Where assumptions would have to be made regarding missing SD data (see Dealing with missing data), we would have compared the findings of primary outcomes when we used our assumption compared with complete data only. We would have undertaken a sensitivity analysis to test how prone results were to change when completer data only were compared to the imputed data using the above assumption. If there had been a substantial difference, we would have reported results and discussed them, but continued to employ our assumption.
3. Risk of bias
We would have analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the 'Risk of bias' domains (implied as randomised with no further details available, allocation concealment, blinding and outcome reporting) for the meta‐analysis of the primary outcome. If the exclusion of trials at high risk of bias had not altered the direction of effect or the precision of the effect estimates substantially, then we would have included relevant data from these trials.
4. Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials.
If we had noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but would have presented them separately.
5. Fixed‐effect and random‐effects
We synthesised data using a fixed‐effect model; however, we also would have synthesised data for the primary outcome using a random‐effects model to evaluate whether this would have altered the significance of the results.
Results
Description of studies
For further descriptions of the included and excluded studies please see Characteristics of included studies and Characteristics of excluded studies.
Results of the search
1. 2007 searching
We found 130 citations using the search strategies. Of the 130 citations, we obtained 30 full‐text articles related to 22 studies for detailed inspection. We excluded 17 of these studies and we need further information from five studies.
2. 2016 and 2019 searching
We found no new records in these searches; however we were able extract data from one of the studies awaiting assessment and have now included it in the review (Schmider 1999). We have now excluded the other four studies (Merlis 1962; Patra 1998; Ungvari 1999; Wetzel 1997). One previously excluded study, Smith 1961, was another citation of Smith 1960 and we have added it as a reference to Smith 1960. This review now has one included study and 20 are excluded. Please also see Figure 1.
Included studies
We could only include one study (Schmider 1999 ).
1. Length of study
The study duration was three days. Baseline observations were conducted on the first day of the trial; on day two participants received the intervention; and on day three, patients were crossed over.
2. Participants
Twenty‐one participants participated in this trial. They presented with significant psychomotor retardation, mutism and on the Bech‐Rafaelsen Melancholia Scale (BPRS) were rated 3 in each of the criteria for psychomotor retardation. Participants had a range of diagnoses based on the DSM‐III‐R criteria: severe major depressive episode; major depressive episode with psychotic features; bipolar disorder and concurrently depressed; schizoaffective disorder and concurrently depressed; schizophrenic disorder; and schizophreniform disorder. The mean age of the participants was 50.8 years (range 21 to 77 years).
3. Setting
The trial took place in a hospital setting.
4. Study size
Twenty‐one participants were originally enlisted in the trial, four participants were excluded from the final analysis. Two required medication during baseline evaluation and two were later diagnosed with Parkinson’s disease. The final analysis included 17 participants: 13 women and four men.
5. Interventions
The trial was a direct comparison between two benzodiazepines (lorazepam and oxazepam).
5.1 Lorazepam group
Seven participants received lorazepam in the first arm of the trial. They were given 2 mg lorazepam sublingually as a single dose (on day 2) then crossed over to receive 60 mg oxazepam sublingually as a single dose (on day 3).
5.2 Oxazepam group
Ten patients received oxazepam in the first arm of the trial. They were given 60 mg oxazepam sublingually as a single dose (on day 2) then crossed over to receive 2 mg lorazepam sublingually as a single dose (on day 3).
6. Outcomes
The study only measured one outcome which was to evaluate the degree of catatonic‐like symptoms with the Visual Analogue Scale at baseline day 1, day 2 and day 3 of the trial after intervention exposure. Due to the cross‐over trial design we could only extract data from the graph for day 2 where patients either received lorazepam or oxazepam before the cross‐over took place.
6.1 Rating scales used by the included study
The study used a Visual Analogue Scale (VAS) to measure the degree of catatonic‐like symptoms. The VAS has been used in other studies to measure symptom control, for example dyspnoea, asthma, pain and total hip arthroplasty.
The VAS, which measures 100 mm in length, was used through the 3‐day trial and was administered by resident psychiatrists trained in the use of this scale. After the administration of the intervention, VAS was used every two hours on seven occasions between 08:00 and 20:00 hours during baseline (day 1), days 2 and 3. The interpretation was that the higher the value (closer to 100 mm) that the patient scored on the VAS scale, the more the patient was experiencing increased levels of catatonic symptoms. Lower VAS scores on the scale, i.e. closer to the 10‐mm end, indicated the patient was experiencing fewer catatonic‐like symptoms.
Excluded studies
We excluded 20 studies. Six were not randomised; eight included participants with schizophrenia or other severe mental illnesses but these participants did not also have, or it was not clear that they had, catatonia; two trials did not use benzodiazepines as a treatment intervention; one trial did not collect pre‐cross‐over data for the intervention of interest; in two of the trials there was no usable data for our outcomes (see Types of outcome measures); and in one trial no independent data were presented for the participants who had catatonia.
Only 10 of these were randomised. Many of both the randomised and the non‐randomised excluded studies did not focus specifically on treatment outcomes in people with catatonia. Of the 10 randomised studies, only three included more than one person with catatonia (Fischer 1974; Kunigiri 2002; Ungvari 1997); and of these, none employed benzodiazepines as a therapeutic intervention.
1. Studies awaiting assessment
There are no studies awaiting assessment.
2. Ongoing studies
We did not identify any ongoing studies.
Risk of bias in included studies
Please also see Characteristics of included studies and Figure 2; Figure 3
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
There is no discussion within the Methods section of this study detailing whether participants were randomised to intervention arms but it is stated that the study was of a "double blind" design, implying randomisation. Methods for allocation concealment were not reported. We rated this study at high risk for selection bias.
Blinding
In the Methods section it states that this is a "double‐blind crossover study"; however no further information is provided as to how the participants were blinded to treatment, and there are no details concerning blinding of the personnel administering the VAS, i.e. if they were blinded to which intervention each participant received. We rated this unclear risk for performance and detection bias.
Incomplete outcome data
We rated this study at unclear risk for attrition bias. Data from four participants (19% of the total number of participants) were not used but the study clearly reported the reasons for this.
Selective reporting
There was only one measured outcome for this study: the degree of catatonic symptoms after each intervention was received, reported before and after cross‐over. However, it is to be noted that any adverse reactions to interventions were not reported during the trial. We rated the study to be at unclear risk for reporting bias.
Other potential sources of bias
The period of time between cross‐over from one drug to another could lead to a carry‐over effect from the previous drug which could contribute towards improved additive effect. The authors do mention the cross‐over time interval was "relatively short" due to ethical reasons preventing a longer unmediated period between the different medications. However, we only used data from the first arm of the trial and as no other potential sources of bias are apparent, we rated risk of bias to to be low for this domain.
Effects of interventions
See: Table 1
1. Lorazepam versus oxzepam
1.1 Catatonia: clinically important change in symptoms (improved at least 50% on VAS)
There was no clear difference between treatment groups (RR 0.95, 95% CI 0.42 to 2.16; participants = 17; studies = 1; very low quality evidence, Analysis 1.1).
1.1. Analysis.
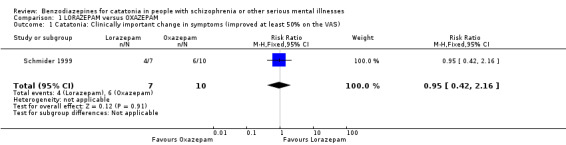
Comparison 1 LORAZEPAM versus OXAZEPAM, Outcome 1 Catatonia: Clinically important change in symptoms (improved at least 50% on the VAS).
2. Catatonia: any change in symptoms ‒ average total score (VAS, endpoint, high = poor)
There was no clear difference between treatment groups (MD 1.18, 95% CI −1.99 to 4.35; participants = 17; studies = 1, Analysis 1.2).
1.2. Analysis.
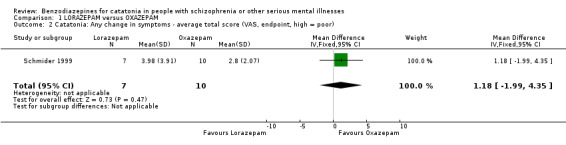
Comparison 1 LORAZEPAM versus OXAZEPAM, Outcome 2 Catatonia: Any change in symptoms ‐ average total score (VAS, endpoint, high = poor).
3. Missing outcomes
No data were available for any of our other outcomes listed in our protocol (see Types of outcome measures).
Discussion
Summary of main results
We found some small relevant trials, but we were only able to extract data for analysis from one study. This study was a direct comparison between two benzodiazepines and results showed no clear difference between the two treatments. The quality of the evidence is very low due to the low number of participants involved and risk of bias within this trial. Currently, to our knowledge, there is no high‐quality evidence available regarding the effects of benzodiazepines for catatonia in people with schizophrenia or similar SMI.
Overall completeness and applicability of evidence
The completeness of evidence available in this review is poor, only one small study was available. The applicability of the study is also poor. The dose of lorazepam (2 mg) used in the study was low compared to doses reported in literature for the management of catatonic symptoms (where doses used vary between 8 mg and 24 mg daily) (Dhossche 2016). The dose of oxazepam used in this study was based on the manufacturer's advice and in clinical practice is used rarely, lorazepam being preferred treatment of choice (Bush 1997). It is also important to note here that only a single dose of benzodiazepine was used in this trial. In clinical practice it is generally accepted that a person presenting with catatonic symptoms would continue to receive benzodiazepines until successful remission of symptoms (Gover 2011).
Quality of the evidence
The quality of evidence available for use in this review is 'very low' due to small sample size and high risks of bias in the methods. The need for well designed methodologically and clinically relevant RCTs with comprehensive reporting of short‐ and long‐term outcomes in relation to catatonic symptoms is required.
Potential biases in the review process
1. The search
It is feasible that we have failed to identify relevant studies by using a search that was biased. Every effort is made by Cochrane Schizophrenia to ensure that searches are as comprehensive as possible, but there is evidence that studies from low‐ and middle‐income countries are not well represented in some of the databases we employ, and it is from these countries that relevant trials may be more likely to come. We, however, do not think that we would have missed any large study as we think such a trial would have been widely reported.
Agreements and disagreements with other studies or reviews
Due to one study with a small data set being included in this review, we are unable to fully comment on agreement or disagreement with other studies or reviews. There still is insufficient trial‐derived evidence about the usefulness of benzodiazepines in the treatment of people with catatonia. At the time of writing this review, clinicians, patients and carers can base decisions about using benzodiazepines to treat people with catatonia only on anecdotal cases or small studies with some deficiencies in methodology or reporting of findings, or both. Such cases and studies have tended to support the use of benzodiazepines in the treatment of people with catatonia (Rosebush 1990).
Catatonia is a syndrome with a variety of disparate illnesses and range of symptoms responsive to benzodiazepines, although this response is poorly understood; it is dramatic and evident to see in patients. Due to the heterogeneous aetiology of catatonia it is difficult to predict how the anti‐catatonic effects of benzodiazepines are exerted; whether it is through the dopaminergic, GABA or cholinergic systems, it is unlikely that a single mode of action or mechanism will be found. Benzodiazepines remain the first choice of treatment and are regarded as safe, easy and effective to use with remission rates reported as high as 70% to 80% (Rosebush 2010). The most important indicator for efficacy in treatment is the dose used (Fink 2006). In the study included in this review the dose of lorazepam used was 2 mg as a single dose compared to more commonly used doses varying between 8 mg and 24 mg per day, which have been tolerated without any major adverse effects such as sedation (Dhossche 2016). Despite the low dose of lorazepam used, this was still associated with improvements in catatonic symptoms (4 out of 7 patients improved 50% on average for catatonic symptoms on day 2). Similarly, in the oxazepam intervention arm 6 out of 10 patients improved by 50% in catatonic symptoms but no statistical difference could be found (RR 0.95, 95% CI 0.42 to 2.16). It is also important to note that in clinical practice single dose administration of catatonic symptoms is very rarely used. Most authors suggest titrating doses according to response which can be seen usually within three to seven days of treatment (Daniels 2009). However, in the study included the total duration of trial was three days which could have been an inadequate time to see a clinically noticeable effect. Although lorazepam has generally been accepted as first line treatment for catatonia, successful use of diazepam (Hung 2006) and clonazepam (Lee 2000) has been reported in the literature. Analysis of results from this single study does not clearly demonstrate the effectiveness of benzodiazepines for treating catatonia (MD 1.18, 95% CI −1.99 to 4.35) which could be due to the small sample size and low doses used. From the data generated from this one study it is very difficult to determine and report whether one benzodiazapine is superior to another.
Authors' conclusions
Implications for practice.
1. For people with catatonia and carers
We are unable to give any clear statements on the benefits or risks associated with the use of benzodiazepines in the treatment of people with catatonia. As far as we can see, treatment of this distressing and serious condition will continue to be based on anecdotal or case report evidence other than that derived from high‐grade trials. Perhaps carers and recipients of care could highlight the need for good studies and their willingness to take part.
2. For clinicians
We are unable to give any clear statements on the benefits or risks associated with the use of benzodiazepines in the treatment of persons with catatonia. Clinicians will have to continue to make judgements on how to treat people that are probably more based on consensus than evidence from clinical trials. Again, should clinicians show a willingness to take part in clinically relevant evaluative studies, this would make this type of work much more possible.
3. For managers/policy makers
Policy will have to be based on non‐trial, low‐quality evidence and consensus until better data are available.
Implications for research.
1. General
Trialists undertaking research in this area should ensure that both methods and data reporting are of the highest quality. Some specific issues include the need for descriptions of both randomisation and the process and testing of blinding. Reporting outcomes using only graphs, summary statistics or P values should be avoided. Instead, authors should present means, standard deviations, confidence intervals, measures of association between intervention and outcome, e.g. relative risks and odds ratios, as well as the raw numbers, if possible. Finally, trialists should report on all findings related to the method they describe. Where cross‐over designs are used, findings from each cross‐over phase should be reported separately.
2. Specific
The use of specific treatments for people with catatonia is not supported by any evidence from randomised trials. The excluded studies and those awaiting assessment demonstrate that such studies are not impossible (Fischer 1974;Kunigiri 2002;Merlis 1962; Patra 1998; Ungvari 1997;Ungvari 1999; Wetzel 1997). We suggest that there is an urgent need to test the effects of benzodiazepines for this condition as, anecdotally, these drugs would appear to be a promising pharmacological approach (Rosebush 1990; Schmider 1999). We suggest a design for such a study in Table 2.
1. Suggested design of study.
| Methods | Participants | Interventions | Outcomes | Notes |
| Allocation: randomised, block, well described. Blinding: double, well described and tested. Duration: 12 to 24 weeks. | Diagnosis: catatonia. N = 300.* Age: adults. Sex: both. History: clearly reported. | 1. Lorazepam 4 mg/day. N = 150. 2. Placebo. N = 150. | General: relapse, general impression of clinician (CGI), carer/other (CGI), compliance with treatment, healthy days, time in hospital, satisfaction with care.
Mental state: CGI.
Catatonic symptoms: BFCRS Quality of life: CGI. Family burden: CGI. Social functioning: return to everyday living for 80% of time.* Adverse events: any adverse event recorded. Economic outcomes. |
* powered to be able to identify a difference of ˜20% between groups for primary outcome with adequate degree of certainty. |
CGI: Clinical Global Impression scale; BFCRS: Bush‐Francis Catatonia Rating Scale
What's new
| Date | Event | Description |
|---|---|---|
| 22 May 2019 | New search has been performed | Review updated with results from new searches. A study that was 'awaiting assessment' is now incuded in the review. |
| 22 May 2019 | New citation required but conclusions have not changed | New data do not change overall conclusions of the review. |
History
Protocol first published: Issue 2, 2007 Review first published: Issue 4, 2008
| Date | Event | Description |
|---|---|---|
| 6 February 2019 | Amended | Search updated and no new study or reference was found. |
| 9 November 2016 | Amended | Search updated and no new study or reference was found. |
| 31 January 2013 | Amended | Contact details updated. |
| 29 July 2008 | Amended | Converted to new review format. |
| 22 June 2008 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The Cochrane Schizophrenia Group Editorial Base at The University Of Nottingham, Nottingham, UK, produces and maintains standard text for use in the Methods section of their reviews. We have used this text as the basis of what appears here and adapted it as required.
We wish to thank the editorial base of the Cochrane Schizophrenia Group and the Department of Community Health and Psychiatry, University of the West Indies (Mona) for their support. Thanks also to Peter Strate, Calvin Ng, Andrea Cipriani and Stefan Leucht who provided help with translating information from some papers.
We would like to thank Javier Ortiz‐Orendain for his editorial input and help with extracting data.
We thank Sarah Abdel Sayed for peer reviewing this review.
Appendices
Appendix 1. Previous searches
Search in 2007
1. The Cochrane Schizophrenia Group Trials Register (March 2007)
We searched using the phrase:
[((catatoni*) in title, abstract and index fields in REFERENCE) OR ((benzodia* OR alprazo* OR chlordiaz* OR cloraze* OR estazo* OR medazepam* OR midazol* OR triazolam* OR clobazam* OR loprazol*) in interventions field in STUDY]
Appendix 2. Previous version of methods
Plain language summary
Benzodiazepines for extreme movement problems (catatonia) in people with schizophrenia and other serious mental illnesses. Some people who have schizophrenia or other serious mental illnesses develop catatonia, which consists of extreme lack of movement or constant repetitive movement over which they seem to have very little control. Whilst in a catatonic state these people are unable to interact with their environment and may go on to acquire secondary problems such as pneumonia, blood clotting problems (thrombosis), malnutrition or dehydration. Current treatments for this are either drugs, which are given by injection, or electric shock treatment (electroconvulsive therapy). The aim of this review is to look at how effective benzodiazepines are compared to placebo or other drug treatments in treating this problem. However, while some clinical trials that seemed relevant were identified, no usable data could be extracted from them. There is no good trial‐derived data on this subject. However, there are five trials on which more information needs to be collected. In the longer term, to make sure people with catatonia receive the most effective treatment, this is an area that would benefit from good research and well planned and reported trials. Also, since the condition is rare, there should be good communication between those involved in researching it.
(Plain language summary prepared for this review by Janey Antoniou of RETHINK, UK www.rethink.org)
Types of studies
Randomised controlled trials. Where a trial was described as 'double‐blind', but it was only implied that the study was randomised, these trials were included in a sensitivity analysis. If there was no substantive difference within primary outcomes (see types of outcome measures) when these 'implied randomisation' studies were added, then they were included in the final analysis. If there was a substantive difference, only clearly randomised trials were used and the results of the sensitivity analysis were described in the text. Quasi‐randomised studies, such as those allocating by using alternate days of the week, were excluded. For studies with cross‐over designs, only data from the first cross‐over phase were analysed.
Types of participants
We included all people with schizophrenia, other psychoses or affective disorders who have catatonia, however defined, as a principal feature of their clinical presentation.
Types of interventions
1. Benzodiazepines of any type, dose and means of administration.
2. Any other class of pharmacological agent at any dose or placebo.
3. Electroconvulsive therapy.
Types of outcome measures
*Primary outcomes: These primary outcomes will help focus the discussion of the review. Sensitivity analyses will also be restricted to these areas. All outcomes were grouped according to time ‐ short term (up to 12 weeks), medium term (13 to 26 weeks) and long term (over 26 weeks). Short‐term studies were also distinguished according to whether they were single injection, one day or longer studies.
Primary outcomes
1. Clinical response: Clinically significant reduction in severity of catatonic symptoms (as defined by each study).
2. Hospital and service outcomes: Duration of stay in hospital.
3. Satisfaction with care: Informal care givers.
Secondary outcomes
1. Death.
2. Clinical response 2.1 Any reduction in severity of symptoms. 2.2 Any increase in severity of symptoms. 2.3 Degree of change in severity of symptoms. 2.4 Clinically significant improvement in self care.3. Leaving the study early.4. Adverse effects. 4.1 Incidence of adverse effects (general and specific). 4.2 Measured acceptability of treatment. 5. Hospital and service outcomes. 5.1 Changes in hospital status (e.g. level of observation).6. Satisfaction with care. 6.1 Recipients of care. 6.2 Professional carers.
Data collection and analysis
[For definitions of terms used in this, and other sections, please refer to The Cochrane Library Glossary]
Selection of studies
We (RCG, GW) independently inspected citations identified from the search. We identified potentially relevant reports and ordered full papers for reassessment. If doubts remained we acquired the full article for further inspection. We obtained full reports and independently reassessed these for inclusion.This process was repeated for the full papers. If it was impossible to resolve disagreements these studies were added to those awaiting assessment and the authors of the papers contacted for clarification.
Data extraction and management
1. Extraction
We (RCG, GW) independently extracted data from the selected trials. Again, where disagreement occurred attempts were made to resolve this by discussion, where doubt still remained we acquired further information from authors. Data were extracted onto standard, simple forms.
2. Management
2.1 Continuous to binary
Where possible, efforts were made to convert outcome measures to binary data. This can be done by identifying cut off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a, Leucht 2005b). It was recognised that for many people, especially those with chronic or severe illness, a less rigorous definition of important improvement (e.g. 25% on the BPRS) would be equally valid. If individual patient data had been available, the 50% cut‐off would have been used for the definition in the case of non‐chronically ill people and 25% for those with chronic illness. If data based on these thresholds were not available, we would have used the primary cut‐off presented by the original authors.
2.2 Normal distribution For continuous data we calculated the weighted mean difference (WMD) between groups and its 95% confidence interval (CI) using a fixed effects model. Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data we applied the following standards to continuous final value endpoint data before inclusion: (a) standard deviations and means were reported in the paper or were obtainable from the authors; (b) when a scale started from zero, the standard deviation, when multiplied by two, should be less than the mean (otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996); in cases with data that are greater than the mean they were entered into 'Other data' table as skewed data. If a scale starts from a positive value (such as PANSS, which can have values from 30 to 210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score.
For change data (mean change from baseline on a rating scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not, unless individual patient data are available. After consulting the ALLSTAT electronic statistics mailing list, we presented change data in RevMan graphs to summarise available information. In doing this, we assumed either that data were not skewed or that the analysis could cope with the unknown degree of skew.
2.3 Final endpoint value versus change data Where both final endpoint data and change data were available for the same outcome category, only final endpoint data were presented. We acknowledge that by doing this much of the published change data may be excluded, but argue that endpoint data is more clinically relevant and that if change data were to be presented along with endpoint data, it would be given undeserved equal prominence. Authors of studies reporting only change data are being contacted for endpoint figures. Again, where loss to follow up is greater than 20%, we will not use data because we (RCG, GW) consider that, for short‐term studies such as those likely to be included in this review, this degree of loss would be indicative of poor study quality.
2.4 Scale‐derived data A wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and many are not valid, and are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore, continuous data from rating scales were included only if the measuring instrument had been described in a peer‐reviewed journal. Scales which had been rated by therapists, rather than an independent rater were reported as 'prone to bias'.
Assessment of risk of bias in included studies
Again working independently, RCG and GW assessed risk of bias using the tool described in the Cochrane Collaboration Handbook (Higgins 2005). This tool encourages consideration of how the sequence was generated, how allocation was concealed, the integrity of blinding at outcome, the completeness of outcome data, selective reporting and other biases. We would not have included studies where sequence generation was at high risk of bias or where allocation was clearly not concealed.
Measures of treatment effect
1. Binary data For binary outcomes we would have calculated the relative risk (RR) and its 95% confidence interval (CI) based on the fixed‐effect model. Relative Risk is more intuitive (Boissel 1999) than odds ratios and odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. Should the overall results have been significant we would have calculated the number needed to treat (NNT) and the number‐needed‐to‐harm (NNH).
2. Continuous data
For continuous outcomes we would have estimated a fixed‐effect weighted mean difference (WMD) between groups. We would not have calculated effect size measures.
Unit of analysis issues
1. Cluster trials Studies increasingly employ 'cluster randomisation' (e.g. randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type 1 errors (Bland 1997, Gulliford 1999).
Should clustering not have been accounted for in primary studies, we would have presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intraclass correlation co‐efficients of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Should clustering have been incorporated into the analysis of primary studies, we would have also presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect. We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation co‐efficient (ICC) [Design effect=1+(m‐1)*ICC] (Donner 2002). If the ICC had not been reported it would have been assumed to be 0.1 (Ukoumunne 1999). Should cluster studies have been appropriately analysed taking into account intraclass correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carryover effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, we only used data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Should a study have involved more than two treatment arms, if relevant, the additional treatment arms would have been presented in comparisons. Where the additional treatment arms were not relevant, these data were not reproduced.
Dealing with missing data
1. Unreported data
We would have contacted the primary author of each included study for any unreported data (e.g. standard deviations, details of dropouts, details of interventions received by control group).
2. Overall loss of credibility
At some degree of loss of follow up, data must lose credibility. We are forced to make a judgment where this is for the very short‐term trials likely to be included in this review. Should more than 20% of data have been unaccounted for we would not have reproduced these data or used them within analyses.
3. Binary
In the case where attrition for a binary outcome was between 0 and 20% and outcomes of these people had been described, we would have included these data as reported. Where these data were not clearly described, we would have assumed the worst primary outcome, and rates of adverse effects similar to those who did continue to have their data recorded.
4. Continuous
In the case where attrition for a continuous outcome had been between 0 and 20% and completer‐only data were reported, we would have reproduced the findings.
Assessment of heterogeneity
1. Clinical heterogeneity
Firstly, consideration of all the included studies within any comparison would have been undertaken to judge clinical heterogeneity.
2. Statistical
2.1 Visual inspection
Then visual inspection of graphs would have been used to investigate the possibility of statistical heterogeneity.
2.2 Employing the I‐squared statistic
Visual inspection would have been supplemented using, primarily, the I‐squared statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Should the I‐squared estimate have been greater than or equal to 50%, this would have been interpreted as indicating the presence of high levels of heterogeneity (Higgins 2003). If inconsistency had been high, data would not have been summated, but would have been presented separately and reasons for heterogeneity investigated.
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook (Higgins 2005). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We would not have used funnel plots for outcomes where there were ten or fewer studies, or where all studies were of similar sizes. In other cases, should funnel plots have been possible, we would have sought statistical advice in their interpretation.
Data synthesis
Should it have been possible we would have employed a fixed‐effect model for analyses. We understand that there is no closed argument for preference for use of fixed or random‐effect models. The random‐effect method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us, however, random‐effect does put added weight onto the smaller of the studies ‐ those trials that are most vulnerable to bias. For this reason we favour using fixed‐effect models and would have employed only random‐effect when investigating heterogeneity.
Subgroup analysis and investigation of heterogeneity
If data had been clearly heterogeneous we would have checked that data were correctly extracted and entered and that we had made no unit‐of‐analysis errors. If the high levels of heterogeneity had remained we would not have undertake a meta‐analysis at this point for if there is considerable variation in results, and particularly if there is inconsistency in the direction of effect, it may be misleading to quote an average value for the intervention effect. We would have wanted to explore heterogeneity. We pre‐specify no characteristics of studies that may be associated with heterogeneity except quality of trial method. If no clear association could have been shown by sorting studies by quality of methods a random‐effect meta‐analysis would have been preformed. Should other characteristics of the studies have been highlighted by the investigation of heterogeneity, perhaps some plausible but unpredicted clinical heterogeneity, these post‐hoc reasons would have been discussed, data analysed and presented. However, should the heterogeneity have been substantially unaffected by use of random‐effect meta‐analysis and no other reasons for heterogeneity be clear, the final data would have been presented without a meta‐analysis.
Sensitivity analysis
Should data have been permitting, sensitivity analyses would have been undertaken in order to see if sub‐grouping data resulted in important changes in the results. The following sub‐groupings were pre‐specified:
1. Published (in a journal/chapter) versus unpublished trials. 2. High quality (well‐described sequence generation and concealment of allocation) studies versus others. 3. Use of specific benzodiazepines versus use of all other benzodiazepines.
Data and analyses
Comparison 1. LORAZEPAM versus OXAZEPAM.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Catatonia: Clinically important change in symptoms (improved at least 50% on the VAS) | 1 | 17 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.42, 2.16] |
| 2 Catatonia: Any change in symptoms ‐ average total score (VAS, endpoint, high = poor) | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | 1.18 [‐1.99, 4.35] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Schmider 1999.
| Methods | Allocation: unclear (implied randomisation). Blindness: double. Duration: 3 days. Design: cross‐over. Setting: hospital, Germany. Consent: informed consent obtained from relative and patient. Loss: described. | |
| Participants | Diagnosis: schizoaffective disorder, schizophrenia, or schizophreniform disorder, major depressive disorder with or without psychotic features, bipolar disorder (all DSM‐III‐R) with a BPRS rating of 3 (criteria 1 to 3) for psychomotor retardation. N: 21* Age: over 18, average 50.8 years. Gender: Male 4, Female 13 (for participants included in analyses presented). History: psychomotor retardation and mutism. Exclusions: neurological conditions or other medical condition associated with psychomotor retardation. | |
| Interventions | 1. Lorazepam: lorazepam dose 2 mg for one day (on day 2 of study then switched to oxazepam 60 mg for one day on day 3 of study)**. N = 7.
2. Oxazepam: oxazepam dose 60 mg for one day (on day 2 of study then switched to lorazepam 2 mg for one day on day 3 of study)**. N = 10. (All participants were drug free for 1 week before the trial started). |
|
| Outcomes | Catatonia: clinically important change (> 50% change) and endpoint score (VAS)*** | |
| Notes | * data from 4 participants were excluded by the trial. ** data only extracted from VAS before cross‐over (day 2 data) *** data extracted from graphs |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomised not reported in text, but trial was a double‐blind design; (see Types of studies). |
| Allocation concealment (selection bias) | High risk | Not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐blind". No other information reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of assessors not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | "Four patients were excluded from analysis, 2 of whom required medication during baseline evaluation and 2 of whom were later diagnosed with Parkinson’s disease". This population accounted for 19% of the study. |
| Selective reporting (reporting bias) | Unclear risk | Only 1 outcome measured and this was reported; however the study did not seek to report on adverse effect/events. |
| Other bias | Unclear risk | Funding: not stated. We did not detect any other potential bias. |
BPRS: British Psychiatric Rating Scale
DSM‐III‐R: Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised
N = number
VAS: Visual Analogue Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arioni 1971 | Allocation: randomised Participants: people with schizophrenia but not catatonia |
| Barbee 1992 | Allocation: randomised Participants: people with schizophrenia but not catatonia |
| Fischer 1974 | Allocation: randomised Participants: people with schizophrenia including catatonic subtype Interventions: clozapine and chlorpromazine, not benzodiazepines |
| Hekimian 1967 | Allocation: not randomised |
| Holden 1968 | Allocation: randomised Participants: people with schizophrenia of various subtypes, only one participant with catatonia |
| Hu 2004 | Allocation: randomised Participants: people with psychosis, none with catatonia |
| Kunigiri 2002 | Allocation: randomised Participants: people with catatonia and schizophrenia or similar SMI and non‐responsiveness to lorazepam Interventions: electroconvulsive therapy and placebo versus electroconvulsive therapy and risperidone, not benzodiazepines |
| Kurland 1966 | Allocation: randomised Participants: did not include people with catatonia as a result of having schizophrenia or similar SMI |
| Merlis 1962 | Allocation: unclear Participants: people with catatonia and schizophrenia, psychosis with mental deficiency, psychosis with psychopathic personality Interventions: chlorpromazine, diazepam, chlordiazepoxide and placebo Outcomes: all data were unusable. Mental state: BPRS, MMS (no independent data specific for people with catatonic symptoms) |
| Patra 1998 | Allocation: unclear Participants: patients presenting with catatonic symptoms diagnosed using BFCRS (Bush Francis Catatonia Rating Scale) criteria Interventions: oral lorazepam and parenteral lorazepam Outcomes: no usable data presented in abstract evaluating catatonic symptoms |
| Smith 1960 | Allocation: not randomised |
| Ungvari 1997 | Allocation: randomised Participants: people with catatonia and schizophrenia or similar SMI Interventions: citalopram and placebo, not benzodiazepines |
| Ungvari 1999 | Allocation: randomised Participants people with schizophrenia and catatonic subtype (DSM‐IV) Intervention: lorazepam and placebo Outcomes: unable to use data; no data available before cross‐over point |
| Vasquez‐Gomez 2001 | Allocation: randomised Participants: people with agitation, no mention of catatonia specifically |
| Weckowicz 1960 | Allocation: not randomised |
| Wetzel 1997 | Allocation: randomised Participants: patients with history of catatonic syndrome with stupor and mutism Interventions: lorazepam and placebo Outcomes: unable to use any data. Global state: CGI. Catatonic symptoms: BFCRS (no means or SDs; no independent data specific for first cross‐over arm) |
| Wolkowitz 1988 | Allocation: not randomised |
| Wolkowitz 1992 | Allocation: not randomised |
| Wolkowitz 1993 | Allocation: randomised Participants: people with schizophrenia, no mention of catatonia |
| Wyant 1990 | Allocation: randomised Participants: people with schizophrenia of the paranoid and undifferentiated types, none with catatonia |
Differences between protocol and review
The protocol has been extensively re‐written for the new (version 5) of RevMan (2008) but Methods contains no substantive changes from the original, other than form and layout.
Contributions of authors
Hadar Zaman ‒ selection of trials, data extraction, data analysis, write‐up of review.
Roger Gibson ‒ protocol development, selection of trials, data extraction, data analysis, write‐up of review.
Geoffery Walcott ‒ protocol development, selection of trials, data extraction, write‐up of review.
Sources of support
Internal sources
Department of Community Health and Psychiatry, University of the West Indies (Mona), Jamaica.
External sources
No sources of support supplied
Declarations of interest
Hadar Zaman ‒ none.
Roger Gibson ‒ none.
Geoffery Walcott ‒ none.
Co‐lead author with Roger Carl Gibson
Co‐lead author with Hadar Zaman
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Schmider 1999 {published data only}
- Schmider J, Standhart H, Deuschle M, Drancoli J, Heuser I. A double blind comparison of lorazepam and oxazepam in psychogenic catatonia. 10th European College of Neuropsychopharmacology Congress; 1997 Sep 13‐17; Vienna, Austria. 1997. [CSzG: Ref2662]
- Schmider J, Standhart H, Deuschle M, Drancoli J, Heuser I. A double blind comparison of lorazepam and oxazepam in psychomotor retardation and mutism. Biological Psychiatry 1999;46(3):437‐41. [CSzG: Ref5244; MEDLINE: ; EMBASE 1999252184] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arioni 1971 {published data only}
- Arioni FBM, Gaston A, Pancheri P. Clinical experimentation with the new drug LM‐2717 [Sperimentazione clinica di un nuovo farmaco LM 2717]. Rivista di Psichiatria 1971;6(6):409‐94. [PsycINFO 1973‐23370‐001] [Google Scholar]
Barbee 1992 {published data only}
- Barbee JG, Mancuso DM, Freed CR, Todorov AA. Alprazolam as a neuroleptic adjunct in the emergency treatment of schizophrenia. American Journal of Psychiatry 1992;149:506‐10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fischer 1974 {published data only}
- Fischer Cornelssen K, Ferner U, Steiner H. Multifocal psychopharmaceutic testing ("Multihospital trial") [Multifokale Psychopharmakaprufung ("Multihospital Trial")]. Arzneimittel‐Forschung 1974;24(10):1706‐24. [MEDLINE: ] [PubMed] [Google Scholar]
Hekimian 1967 {published data only}
- Hekimian LJ, Friedhoff AJ. A controlled study of placebo, chlordiazepoxide and chlorpromazine with thirty male schizophrenic patients. Diseases of the Nervous System 1967;28(10):675‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Holden 1968 {published data only}
- Holden JM, Itil TM, Keskiner A, Fink M. Thioridazine and chlordiazepoxide, alone and combined, in the treatment of chronic schizophrenia. Comprehensive Psychiatry 1968;9(6):633‐43. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hu 2004 {published data only}
- 4. Investigation of administering BZD medicament in outpatients with psychosis. Heath Psychology 2004;12(1):43‐67. [116287] [Google Scholar]
Kunigiri 2002 {published data only}
- Girish K, Gill NS. Electroconvulsive therapy in lorazepam non‐responsive catatonia. Indian Journal of Psychiatry 2003;45:21‐5. [PMC free article] [PubMed] [Google Scholar]
- Kunigiri G, Gill NS, B GN, Naimmagadda J. Electroconvulsive therapy in lorazepam non‐responsive catatonia. 12th World Congress of Psychiatry; 2002 Aug 24‐29; Yokohama, Japan. 2002. [MEDLINE: ]
Kurland 1966 {published data only}
- Kurland AA, Bethon GD, Michaux MH, Agallianos DD. Chlorpromazine‐chlordiazepoxide and chlorpromazine‐imipramine treatment: side effects and clinical laboratory findings. Journal of New Drugs 1966;6:80‐95. [MEDLINE: ] [PubMed] [Google Scholar]
Merlis 1962 {published data only}
- Merlis S, Turner WJ, Krumholz W. A double‐blind comparison of diazepam, chlordiazepoxide and chlorpromazine in psychotic patients. Journal of Neuropsychiatry 1962;3(Suppl 1):S133‐8. [PsycINFO 1977‐03995‐001] [PubMed] [Google Scholar]
Patra 1998 {published data only}
- Patra A, Sarkar A, Kumar R. Outcome of lorazepam in catatonia oral vs parenteral administration. Indian Journal of Psychiatry 1998;40(Suppl):91. [Google Scholar]
Smith 1960 {published data only}
- Smith ME. A clinical study of chlorpromazine and chlordiazepoxide. Connecticut State Medical Journal 1961;25:153‐7. [Google Scholar]
- Smith ME. A comparative controlled study with chlordiazepoxide. American Journal of Psychiatry 1960;117:362‐3. [MEDLINE: ] [Google Scholar]
Ungvari 1997 {published data only}
- Ungvari GS, Chow LY, Chiu HFK, Lau B. Different treatment response of distinct schizophrenia subtypes: the group of systematic catatonias. 6th World Congress of Biological Psychiatry; 1997 Jun 22‐27; Nice, France. 1997.
Ungvari 1999 {published data only}
- Pang AHT, Ungvari G, Ng FS, Chiu H. Treatment response of systematic catatonia (leonhard) to lorazepam. 21st Collegium Internationale neuro‐Psychopharmacologicum Congress; 1998 Jul 12‐16; Glasgow, UK. 1998.
- Ungvari G, Pang A, Ng F, Chiu H, Wong C. Treatment response studies in systematic catatonia (leonhard). i. lorazepam challenge. 8th Congress of the Association of European Psychiatrists; 1996 Jul 7‐12; London, UK. 1996.
- Ungvari GS, Chiu HF, Chow LY, Lau BS, Tang WK. Lorazepam for chronic catatonia: a randomized, double blind, placebo controlled cross over study. Psychopharmacology 1999;142(4):393‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Vasquez‐Gomez 2001 {published data only}
- Vasquez‐Gomez F. The use of midazolam for control of acutely agitated outpatients in psychiatry emergency. 7th World Congress of Biological Psychiatry; 2001 Jul 1‐6; Berlin, Germany. 2001; Vol. 2, issue Suppl 1. [7th World Congress of Biological Psychiatry [CD‐ROM]: Conifer, Excerpta Medica Medical Communications BV, 2001 P034‐02]
Weckowicz 1960 {published data only}
- Weckowicz TE, Ward T. Clinical trial of RO 5‐0690 and chlorpromazine on disturbed chronic schizophrenic patients. Diseases of the Nervous System 1960;21:527‐8. [MEDLINE: ] [PubMed] [Google Scholar]
Wetzel 1997 {published data only}
- Wetzel H, Klawe CJ, Muller MJ, Wiesner J, Benkert O. Lorazepam for stupor and mutism: a double‐blind placebo‐controlled cross‐over study. 10th European College of Neuropsychopharmacology Congress; 1997 Sep 13‐17; Vienna, Austria. 1997. [MEDLINE: ]
Wolkowitz 1988 {published data only}
- Wolkowitz OM, Breier A, Doran A, Kelsoe J, Lucas P, Paul SM, et al. Alprazolam augmentation of the antipsychotic effects of fluphenazine in schizophrenic patients ‐ preliminary results. Archives of General Psychiatry 1988;45(7):664‐71. [MEDLINE: ; PsycINFO 76‐02568] [DOI] [PubMed] [Google Scholar]
- WolkowitzOM, Pickar D, Doran AR, Breier A, Tarell J, Paul SM. Combination alprazolam‐neuroleptic treatment of the positive and negative symptoms of schizophrenia. American Journal of Psychiatry 1986;143(1):85‐7. [DOI] [PubMed] [Google Scholar]
Wolkowitz 1992 {published data only}
- Wolkowitz OM, Turetsky N, Reus VI, Hargreaves WA. Benzodiazepine augmentation of neuroleptics in treatment resistant schizophrenia. Psychopharmacology Bulletin 1992;28(3):291‐5. [MEDLINE: ; PsycINFO 80‐18793] [PubMed] [Google Scholar]
Wolkowitz 1993 {published data only}
- Wolkowitz OM, Harris D, Turetsky NG, Reus VI, Johnson R, Gustafson M, et al. Alprazolam‐neuroleptic treatment of schizophrenia. 146th Annual Meeting of the American Psychiatric Association; 1993 May 22‐27; San Francisco, California, USA. 1993:119.
Wyant 1990 {published data only}
- Wyant M, Diamond BI, O'Neal E, Sloan A, Borison RL. The use of midazolam in acutely agitated psychiatric patients. Psychopharmacology Bulletin 1990;26(1):126‐9. [MEDLINE: ; PsycINFO 79‐28483] [PubMed] [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV TR). 4th Edition. Washington: American Psychiatric Association, 2000. [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315(7108):600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Aperçu sur la problématique des indices d'efficacité thérapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Bush 1997
- Bush G, Petrides G, Francis A. Catatonia and other motor syndromes in a chronically hospitalized psychiatric population. Schizophrenia research 1997;27(1):83‐92. [DOI] [PubMed] [Google Scholar]
Daniels 2009
- Daniels, J. Catatonia: clinical aspects and neurobiological correlates. Journal of Neuropsychiatry and Clinical Neurosciences 2009;21(4):371‐80. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG, editor(s). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Dhossche 2016
- Dhossche DM, Wachtel LE, Goetz M, Sienaert P. Catatonia in psychiatric illnesses. In: Fatemi SH, Clayton PJ editor(s). The Medical Basis of Psychiatry. 4th Edition. New York: Springer, 2016:517‐35. [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21(19):2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JP, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Ellul 2015
- Ellul P, Choucha W. Neurobiological approach of catatonia and treatment perspectives. Frontiers in Psychiatry / Frontiers Research Foundation 2015;6:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fink 2006
- Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Progress in Neuro‐psychopharmacology & Biological Psychiatry 2006;6(30):1182‐3. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Gover 2011
- Grover S, Aggarwal M. Long‐term maintenance lorazepam for catatonia: a case report. General Hospital Psychiatry 2011;33(1):82. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149(9):876‐83. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]. www.cochrane.org/resources/handbook/hbook.htm (accessed 04 February 2006).
Higgins 2011a
- Higgins JP, Green S, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JAC, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hung 2006
- Hung YY, Huang TL. Lorazepam and diazepam rapidly relieve catatonic features in major depression. Clinical Neuropharmacology 2006;29(3):144‐7. [DOI] [PubMed] [Google Scholar]
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [PUBMED: 19438480] [DOI] [PubMed] [Google Scholar]
Kahlbaum 1973
- Kahlbaum KL. Catatonia [Die Katatonie oder das spannungirresein]. Catatonia. Baltimore: Johns Hopkins University Press, 1973. [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Lee 2000
- Lee JW, Schwartz DL, Hallmayer J. Catatonia in a psychiatric intensive care facility: incidence and response to benzodiazepines. Annals of Clinical Psychiatry 2000;12(2):88‐96. [DOI] [PubMed] [Google Scholar]
Leon 2006
- Leon AC, Mallinckrodt CH, Chuang‐Stein C, Archibald DG, Archer GE, Chartier K. Attrition in randomized controlled clinical trials: methodological issues in psychopharmacology. Biological Psychiatry 2006;59(11):1001‐5. [PUBMED: 16905632] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale Scores. British Journal of Psychiatry 2005;187:366‐71. [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Penland 2006
- Penland HR, Weder N, Tampi RR. The catatonic dilemma expanded. Annals of General Psychiatry 2006;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Regestein 1977
- Regestein QR, Alpert JS, Reich P. Sudden catatonic stupor with disastrous outcome. Journal of the American Medical Association 1977;238:618‐20. [PubMed] [Google Scholar]
Rosebush 1990
- Rosebush PI, Hildebrand AM, Furlong BG, Mazurek MF. Catatonic syndrome in a general psychiatric inpatient population: frequency, clinical presentation, and response to lorazepam. Journal of Clinical Psychiatry 1990;51:357‐62. [PubMed] [Google Scholar]
Rosebush 2010
- Rosebush PI, Mazurek, MF. Catatonia and its treatment. Schizophrenia Bulletin 2009;36(2):239‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S, editor(s), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Shokraneh 2017
- Shokraneh F, Adams CE. Study‐based registers of randomized controlled trials: Starting a systematic review with data extraction or meta‐analysis. BioImpacts 2017;7(4):209‐17. [DOI: 10.15171/bi.2017.25] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 1961
- Smith ME. A clinical study of chlorpromazine and chlordiazepoxide. Connecticut State Medical Journal 1961;25:153‐7. [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Intervention. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. ICD‐10: international statistical classification of diseases and related health problems. Geneva: World Health Association, 1992. [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]