

SUMMARY
Abnormal subthalamic nucleus (STN) activity is linked to impaired movement in Parkinson’s disease (PD). The autonomous firing of STN neurons, which contributes to their tonic excitation of the extrastriatal basal ganglia and shapes their integration of synaptic input, is downregulated in PD models. Using electrophysiological, chemogenetic, genetic, and optical approaches, we find that chemogenetic activation of indirect pathway striatopallidal neurons downregulates intrinsic STN activity in normal mice but this effect is occluded in Parkinsonian mice. Loss of autonomous spiking in PD mice is prevented by STN N-methyl-D-aspartate receptor (NMDAR) knockdown and reversed by reactive oxygen species breakdown or KATP channel inhibition. Chemogenetic activation of hM3D(Gq) in STN neurons in Parkinsonian mice rescues their intrinsic activity, modifies their synaptic integration, and ameliorates motor dysfunction. Together these data argue that in PD mice increased indirect pathway activity leads to disinhibition of the STN, which triggers maladaptive NMDAR-dependent downregulation of autonomous firing.
Graphical Abstract
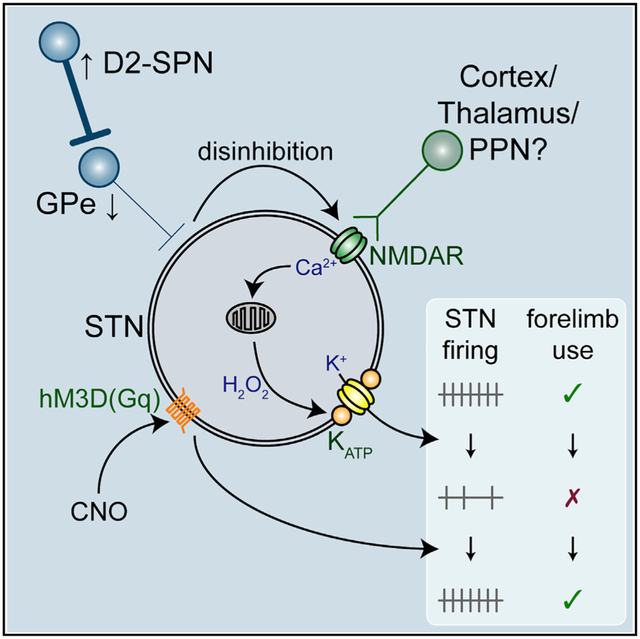
In Brief
McIver et al. describe the cellular and circuit mechanisms responsible for the loss of autonomous subthalamic nucleus (STN) spiking in dopamine-depleted mice and demonstrate that chemogenetic rescue of intrinsic STN activity reduces Parkinsonian motor dysfunction.
INTRODUCTION
The glutamatergic subthalamic nucleus (STN) is part of the basal ganglia, a group of subcortical brain nuclei critical for movement and a key site of pathology in Parkinson’s disease (PD) (Albin et al., 1989). The STN is a component of the so-called hyperdirect and indirect pathways, which contribute to the execution of action sequences through elevation of GABAergic basal ganglia output (Nambu et al., 2002; Kravitz et al., 2010; Tecuapetla et al., 2016; Markowitz et al., 2018). In PD, increases in the frequency and synchronization of STN activity have been linked to the expression of akinesia, bradykinesia, and rigidity (Zaidel et al., 2009; Shimamoto et al., 2013; Sharott et al., 2014). However, PD motor symptoms are ameliorated by both inhibition of STN activity through lesions (Bergman et al., 1990) or pharmacological/optogenetic approaches (Levy et al., 2001; Yoon et al., 2014) and elevation of STN activity through direct electrical stimulation (Benabid et al., 2009; Wichmann et al., 2018). In addition, therapeutic dopamine (DA) receptor activation and STN deep brain stimulation decorrelate but do not necessarily diminish STN activity (Hashimoto et al., 2003; Eusebio et al., 2012). Together these data argue that an abnormal pattern rather than frequency of STN activity is particularly detrimental to movement in PD.
In toxin models of PD, abnormal STN activity takes days to weeks to manifest, suggesting that plasticity triggered by loss of DA contributes to its emergence (Vila et al., 2000; Mallet et al., 2008b). Indeed, recent studies showed that the strength of external globus pallidus (GPe) inhibition relative to cortical excitation of the STN quadruples through proliferation and removal of GPe and cortical synapses, respectively (Chu et al., 2015; Mathai et al., 2015; Chu et al., 2017). In parallel, the intrinsic autonomous firing of STN neurons is strongly downregulated (Zhu et al., 2002a; Wilson et al., 2006). The available evidence suggests that increased inhibition of the GPe by D2-striatal projection neurons (D2-SPNs) leads to disinhibition of the STN and excessive activation of STN N-methyl-D-aspartate receptors (NMDARs), triggering synaptic plasticity (Chu et al., 2015, 2017). Although the cause of autonomous firing downregulation is unknown, it may be triggered by a similar mechanism because NMDAR activation can produce oxidant stress and opening of KATP channels, which hyperpolarize STN neurons and inhibit their spiking (Atherton et al., 2016). Autonomous firing is thought to underlie the resting, tonic discharge of STN neurons in vivo and thus support their function as a “driving force” of extrastriatal basal ganglia activity (Kitai and Kita, 1987; Bevan and Wilson, 1999; Nambu et al., 2000; Do and Bean, 2003). Autonomous spiking also influences the synaptic patterning of STN activity because the responses to inputs are related to the frequency and timing of the postsynaptic firing cycle (Bevan et al., 2002b; Wilson, 2013). Thus, the loss of autonomous firing together with plastic alterations in the strength of synaptic input may contribute to the emergence of abnormal STN activity in PD. Using the unilateral 6-hydroxydopamine (6-OHDA)-lesion model of PD, we therefore addressed the underlying mechanisms and functional consequences of downregulated autonomous STN activity.
RESULTS
Data are reported as median and interquartile ranges. Unpaired data are represented as boxplots, with the median, interquartile range, and 10%–90% range denoted. Paired data are represented as tilted line-segment plots. Non-parametric, two-tailed statistical tests were applied: Mann-Whitney U (MWU) test for unpaired data, Wilcoxon signed rank (WSR) test for paired comparisons, and Fisher’s exact test for contingency analyses. p values were corrected for multiple comparisons (Holm, 1979) (Tables S1-S8).
Autonomous STN Activity Is Downregulated in DA-Depleted Mice
Previous studies have demonstrated that autonomous STN activity is downregulated in toxin models of PD (Zhu et al., 2002a; Wilson et al., 2006; Chu et al., 2017). Furthermore, STN neurons identified as receiving input from the primary motor cortex also exhibited decreased activity (Chu et al.,2017). We first confirmed the result of Chu et al. (2017) using an identical approach, but a more extensive sample. Thus, the autonomous activities of STN neurons that were excited by optogenetic stimulation of motor cortical axon terminals were compared in ex vivo brain slices from mice that had received unilateral injection of 6-OHDA or vehicle into the medial forebrain bundle (MFB). The cell-attached, current-clamp configuration of the patch clamp technique was used to record autonomous activity without perturbation. In concordance with earlier reports, the frequency, regularity, and incidence of autonomous STN firing was reduced in slices from 6-OHDA-injected DA-depleted mice (Figures 1 and S1A; Table S1). Thus, loss of DAergic neuromodulation leads to the downregulation of autonomous activity in the motor territory of the STN.
Figure 1. The Autonomous Activity of STN Neurons Was Downregulated in 6-OHDA-Injected, DA-Depleted Mice.

(A and B) ChR2(H134R)-eYFP expression (green) and tyrosine hydroxylase (TH) immunoreactivity (red) ipsilateral (A) and contralateral (B) to injections of an AAV vector expressing ChR2(H134R)-eYFP in the primary motor cortex (M1) and 6-OHDA in the MFB.
(C) ChR2(H134R)-eYFP (green) expressing cortico-STN axon terminals in the vicinity of NeuN-immunoreactive (red) neurons (asterisks) in the ipsilateral STN.
(D and E) The frequency, regularity, and incidence of autonomous activity in STN neurons that receive motor cortical input was reduced in slices from DA-depleted mice (D, examples; D insets, response to optogenetic stimulation (blue) of motor cortical input; E, population data).
*p < 0.05.
Elevation of D2-SPN Transmission Is Sufficient to Downregulate Autonomous STN Activity
In PD and its experimental models, loss of striatal DAergic innervation disinhibits D2-SPNs, which increases inhibition of the GPe, which in turn disinhibits the STN (Mallet et al., 2006; Lemos et al., 2016; Sharott et al., 2017; Parker et al., 2018; Ryan et al., 2018). This change in circuit activity triggers alterations in the strength of synaptic transmission in the STN (Chu et al., 2015, 2017). We therefore hypothesized that the downregulation of autonomous STN activity is similarly triggered by Parkinsonian circuit activity. However, SN DA neurons also directly and positively modulate STN firing (Zhu et al., 2002a, 2002b; Loucif et al., 2008; Ramanathan et al., 2008) arguing that reduced STN activity in PD models may result from loss of local DA signaling. To determine whether an increase in D2-SPN activity is sufficient to downregulate autonomous firing in the STN, D2-SPNs were chemogenetically stimulated in DA-intact adora2A-rM3D(Gs)-mCherry mice that express rM3D(Gs)-mCherry in D2-SPNs (Farrell et al., 2013). Consistent with chemogenetic activation of D2-SPNs in these mice (1) subcutaneous (s.c.) administration of 1 mg/kg clozapine-N-oxide (CNO) reduces both GPe activity in vivo and movement in the open field (Farrell et al., 2013; Chu et al., 2017; Bouabid and Zhou, 2018) and (2) application of 10 μM of CNO elevates the frequency of GABAergic synaptic currents in GPe neurons ex vivo (Chu et al., 2017). We found that the frequency, regularity, and incidence of autonomous STN activity in ex vivo brain slices from CNO-treated adora2A-rM3D(Gs)-mCherry mice (1 mg/kg s.c. every 12 h for 60 h) was reduced relative to activity in slices from vehicle-treated adora2A-rM3D(Gs)-mCherry mice (Figures 2A-2D; Table S2). In regular C57BL/6 mice that lacked designer receptor exclusively activated by designer drug (DREADD) expression, the same schedule of CNO administration had no effect (Figures S1B and S1C; Tables S1 and S2), consistent with a recent report showing no non-specific effect of CNO on basal ganglia circuit activity (Bouabid and Zhou, 2018). Together these data demonstrate that elevation of D2-SPN transmission is sufficient to downregulate autonomous STN activity.
Figure 2. Chemogenetic Aactivation of D2-SPNs Was Sufficient to Downregulate Autonomous STN Activity in DA-Intact Mice but This Effect Was Occluded in DA-Depleted Mice.

(A) Schedule of s.c. CNO or vehicle injection and ex vivo electrophysiological recording in adora2a-rM3D(Gs)-mCherry mice.
(B) Expression of rM3D(Gs)-mCherry (red) in a subset of NeuN-immunoreactive striatal neurons (green) in an adora2a-rM3D(Gs)-mCherry mouse (arrows, expressing; asterisks, non-expressing).
(C and D) Autonomous STN activity was downregulated in slices from CNO-treated mice (C, examples; D, population data).
(E and F) In MFB vehicle-injected adora2a-rM3D(Gs)-mCherry mice, autonomous STN activity was downregulated in slices from CNO- compared to vehicle-treated mice. However, in MFB 6-OHDA-injected adora2a-rM3D(Gs)-mCherry mice, autonomous STN activity was downregulated in slices from both CNO- and vehicle-treated mice, arguing that the effect of chemogenetic stimulation of D2-SPNs was occluded by DA depletion (E, examples; F, population data).
*p < 0.05. ns, not significant.
If elevated striato-pallidal transmission and subsequent disinhibition of the STN underlie the downregulation of autonomous STN activity in 6-OHDA-treated mice, a corollary is that the effect of chemogenetic activation of D2-SPNs should be occluded by DA depletion. To test this prediction, adora2A-rM3D(Gs)-mCherry mice were injected unilaterally in the MFB with either 6-OHDA or vehicle. Approximately 2–3 weeks later, mice received s.c. injections of either vehicle or CNO (as above) and the autonomous activity of STN neurons from the hemisphere ipsilateral to MFB injection was analyzed ex vivo. Consistent with this prediction, the effect of D2-SPN chemogenetic stimulation on autonomous STN activity was occluded by DA depletion, as evinced by the similarity of data from MFB 6-OHDA-injected mice that received s.c. vehicle or CNO treatment (Figures 2E and 2F; Table S2).
NMDARs Are Necessary for the Downregulation of Autonomous STN Activity in PD Mice
In 6-OHDA-injected mice, disinhibition of the STN leads to NMDAR-dependent alterations in the strength of synaptic transmission (Chu et al., 2015, 2017). Furthermore, pre-incubation of brain slices from DA-intact mice with 25 μM of NMDA persistently downregulates autonomous STN activity (Atherton et al., 2016). Therefore, we hypothesized that the loss of autonomous STN spiking in 6-OHDA-injected mice is due to increased activation of STN NMDARs in vivo. To test this hypothesis, STN NMDARs were knocked down by stereotaxic injection of an adeno-associated virus (AAV) carrying a cre recombinase expression construct into the STN of mice in which GRIN1, the gene encoding the obligatory subunit of the NMDAR, was floxed (Tsien et al., 1996; Chu et al., 2015, 2017) (Figures 3A-3D; Table S3). As a control, another set of these mice received injection of an AAV expressing only eGFP (Figures 3A-3D; Table S3). Viral injections were performed at the same time as 6-OHDA or vehicle injection in the MFB. Consistent with our hypothesis, STN NMDAR knockdown (KD) prevented the downregulation of autonomous STN activity in 6-OHDA-injected mice (Figures 3C and 3D; Table S3). If activation of STN NMDARs in vivo is responsible for the downregulation of autonomous STN activity in 6-OHDA-injected mice, then a corollary is that the downregulatory effect of NMDAR activation ex vivo (Atherton et al., 2016) should be occluded in slices from 6-OHDA-injected but not vehicle-injected C57BL/6 mice. Consistent with this prediction, NMDAR activation ex vivo downregulated autonomous STN activity in slices from vehicle- but not 6-OHDA-injected mice relative to untreated slices (Figures 3E and 3F; Table S3).
Figure 3. Downregulation of Autonomous STN Activity in DA-Depleted Mice Was Dependent on STN NMDAR Activation In Vivo.

(A and B) Confocal micrographs of viral-mediated eGFP (A, green) or cre-eGFP (B, green) expression in NeuN-immunoreactive (red) STN neurons (arrowheads) in Grin1lox/lox mice.
(C and D) In eGFP-expressing, NMDAR-intact STN neurons autonomous activity was downregulated in slices from 6-OHDA-injected mice. However, in cre-eGFP-expressing, NMDAR KD STN neurons autonomous firing was normal in slices from 6-OHDA-injected mice (C, examples; D, population data).
(E and F) Activation of STN NMDARs ex vivo downregulated autonomous firing in slices from vehicle-injected, NMDAR-intact mice but its effects were occluded in slices from 6-OHDA-injected, NMDAR-intact mice. As expected, the autonomous firing of STN neurons in untreated slices from 6-OHDA-injected, NMDAR-intact mice was downregulated relative to that in untreated slices from vehicle-injected, NMDAR-intact mice (E, examples; F, population data).
*p < 0.05. ns, not significant.
See also Table S3.
STN Neurons Exhibit Elevated Oxidant Stress and Hydrogen Peroxide-Triggered KATP Channel Activation in PD Mice
NMDAR activation can stimulate opening of ATP-sensitive K+ (KATP) channels, which limit the firing of STN neurons (Shen and Johnson, 2010; Atherton et al., 2016). NMDAR activation can promote KATP channel opening through a fall in the ratio of ATP to ADP (Nichols, 2006; Wang and Michaelis, 2010) or the generation of reactive oxygen (ROS) and/or reactive nitrogen species (RNS), which act directly (Ichinari et al., 1996; Kawano et al., 2009; Lee et al., 2015; Atherton et al., 2016) or indirectly via second messenger pathways (Shen and Johnson, 2010; Zhang et al., 2014). To determine whether NMDAR-dependent downregulation of autonomous STN firing in 6-OHDA-injected mice is dependent on activation of KATP channels, we conducted a series of experiments. To test whether disinhibition of STN neurons in 6-OHDA-injected mice is associated with elevated oxidant stress, we virally expressed the mitochondrial persistent downregulation of autonomous spiking (Atherton et al., 2016) (Table S4).
To determine whether elevated production of ROS is linked to the downregulation of autonomous STN activity, membrane permeable catalase, which metabolizes hydrogen peroxide, was applied. Catalase application rapidly reversed the firing deficit in slices from 6-OHDA-injected mice (Figures 4C and 4D; Table S4). This treatment also modestly increased firing in slices from vehicle-injected mice, although the effects were smaller. To determine whether KATP channels are responsible for downregulated STN activity, we compared the effect of the KATP channel inhibitor glibenclamide (100 nM) on autonomous firing in slices from 6-OHDA- and vehicle-injected mice. Glibenclamide rescued autonomous activity in slices from 6-OHDA-injected mice. Although this treatment also modestly increased the frequency and regularity of firing in slices from vehicle-injected mice, the effects were smaller (Figures 4E and 4F; Table S4). The effects of catalase or KATP channel inhibition on spiking were similar, implying that they were mediated through a common pathway (Figure 4G; Table S4). Consistent with this inference, the effects of catalase were fully occluded by prior application of glibenclamide (Figures 4H and 4I; Table S4).
Figure 4. STN Neurons Exhibited Elevated Mitochondrial Oxidant Stress and Hydrogen Peroxide-Triggered KATP Channel Activation in PD Mice.

(A) The expression of mito-roGFP (green) in STN neurons (arrowheads) and their somatic morphology (grayscale) were imaged under simultaneous 2-photon laser scanning fluorescent and Dodt contrast microscopy, respectively.
(B) The relative oxidation of mitochondria in STN neurons was elevated in slices from 6-OHDA- versus vehicle-injected mice.
(C and D) Breakdown of hydrogen peroxide with catalase rescued autonomous STN activity in slices from 6-OHDA-injected mice but had relatively minimal effects on neurons from vehicle-injected mice (C, examples; D, population data, catalase-associated changes).
(E and F) Inhibition of KATP channels with glibenclamide rescued autonomous STN activity in slices from 6-OHDA-injected mice but had relatively minimal effects on neurons from vehicle-injected mice (E, examples; F, population data, glibenclamide-associated changes).
(G) Autonomous firing in slices from 6-OHDA-injected mice that had been treated with glibenclamide or catalase were not significantly different from autonomous firing in slices from DA-intact control mice (Figures 1C and 1D).
(H and I) The effect of catalase on autonomous STN firing in slices from 6-OHDA-injected mice was occluded by prior application of glibenclamide (H, population data; I, example).
(J and K) In slices from vehicle- or 6-OHDA-injected mice, autonomous STN activity first recorded in the cell-attached configuration was not altered by subsequent establishment of the whole-cell configuration with pipettes containing 2 mM of ATP (J, examples; K, population data).
*p < 0.05. ns, not significant.
See also Figures S2 and S3 and Table S4.
To determine whether ATP depletion was also a factor, the spiking rate in cell-attached recordings was compared to that after breaking into the cell with an internal solution containing 2 mM of ATP. Dialysis with ATP did not change spiking in neurons from lesioned mice, arguing that ATP depletion did not contribute (Figures 4J and 4K; Table S4). Finally, we tested whether the downregulation of autonomous STN firing produced by chemogenetic stimulation of D2-SPNs was also mediated through KATP channels. Consistent with this mechanism, glibenclamide rescued autonomous STN firing in slices from CNO-treated mice but had minimal effects in vehicle-treated mice (Figures S2A and S2B; Table S4). Together, the data argue that following the loss of DA, disinhibition of the STN leads to increased activation of STN NMDARs, which increases ROS-triggered KATP channel opening, resulting in the downregulation of autonomous firing.
It was recently reported that expression of small-conductance Ca2+-activated K+ (SK) channels, which are important regulators of autonomous STN activity (Hallworth et al., 2003), is upregulated in 6-OHDA-injected rodents (Mourre et al., 2017). To determine their role, the effect of 10 nM of apamin (an SK channel blocker) was examined. The effects of SK channel blockade were negligible in 6-OHDA- (and vehicle-) injected mice, arguing that SK channels are not responsible for the downregulation of autonomous activity in 6-OHDA-injected mice (Figure S3; Table S4).
Chemogenetic Restoration of Autonomous STN Activity Ameliorates Motor Dysfunction in PD Mice
To gain insight into the effect of autonomous firing loss on motor function in PD mice, we first determined whether intrinsic STN activity could be restored through direct chemogenetic activation (Roth, 2016) (Figures 5, 6, and 7; Tables S5, S6, and S7). An AAV vector carrying a synapsin promoter-driven hM3D(Gq)-mCherry construct was injected into the STN ipsilateral to MFB injection of 6-OHDA or vehicle (Figure 5A). Two to three weeks matrix-targeted thiol redox sensor mito-roGFP in STN neurons (Hanson et al., 2004; Guzman et al., 2009; Atherton et al., 2016). 2-photon laser scanning microscopic imaging of mito-roGFP confirmed that mitochondrial oxidant stress was increased in STN neurons in slices from 6-OHDA-injected mice (the dynamic range of the sensor was similar in slices from 6-OHDA- and vehicle-injected mice; Figures 4A and 4B; Table S4). Mitochondrial oxidant stress was also elevated in STN neurons in slices from DA-intact mice that were pre-treated with a concentration of exogenous NMDA that induces the later the effect of chemogenetically activating hM3D(Gq)-mCherry through administration of 10 μM of CNO was tested ex vivo. CNO application increased the frequency and regularity of autonomous activity in hM3D(Gq)-mCherry-expressing STN neurons in slices from both 6-OHDA- and vehicle-injected mice, presumably through the generation of a persistent inward current that could be measured under voltage clamp at −60 mV (Figures 5B-5E; Table S5). These effects occurred within a minute of CNO application and persisted for the remainder of the recording, which was usually a further 10–30 min. Administration of CNO to slices from DA-intact mice that did not express hM3D(Gq)-mCherry had no effect on autonomous activity, ruling out off-target effects of CNO (Figure S4; Table S5). In addition, no effect on the amplitude or short-term plasticity of electrically evoked postsynaptic currents in hM3D(Gq)-mCherry-expressing STN neurons was observed (Figures 5F and 5G; Table S5). Together these data demonstrate that chemogenetic activation of hM3D(Gq)-mCherry rapidly and persistently restores autonomous STN activity in 6-OHDA-injected mice with no effect on afferent synaptic transmission.
Figure 5. Chemogenetic Activation of hM3D(Gq)-mCherry in STN Neurons Augmented Their Intrinsic Activity.

(A) Confocal micrograph of hM3D(Gq)-mCherry expression (red) in NeuN-immunoreactive STN neurons.
(B and C) Chemogenetic activation of hM3D(Gq)-mCherry with 10 μM of CNO generated a persistent inward current in STN neurons that were voltage clamped at −60 mV in ex vivo brain slices prepared from 6-OHDA- (green) or vehicle- (black) injected mice (B, example from a 6-OHDA-injected mouse; C, population).
(D and E) Chemogenetic activation of hM3D(Gq)-mCherry increased the frequency and regularity of autonomous STN activity ex vivo (D, example from a 6-OHDA-injected mouse; E, population, black and green from vehicle- and 6-OHDA-injected mice, respectively).
(F and G) Pairs of inhibitory postsynaptic currents (iPSCs; upper black and red traces; holding voltage, −60 mV) and excitatory synaptic currents (EPSCs; lower black and red traces; holding voltage, −80 mV) electrically evoked in STN neurons with an interval of 100 ms were unaffected by chemogenetic activation (F, examples from a 6-OHDA-injected mouse; G, population data, black and green from vehicle- and 6-OHDA-injected mice, respectively).
(H–J) The patterning of STN activity by motor cortical inputs optogenetically stimulated at 20 Hz for 1 s (blue) in ex vivo brain slices from 6-OHDA-injected mice was reduced following chemogenetic rescue of intrinsic activity (H, example cell-attached recordings; I, population peristimulus time histogram [magenta line, pre-stimulus mean; cyan line, pre-stimulus mean + 3 SD]; J, population data; data were derived from 5 trials of stimulation per neuron). APs, action potentials.
*p < 0.05. ns, not significant.
See also Table S5.
Figure 6. Chemogenetic Activation of STN Neurons In Vivo Increased Immediate Early Gene Expression in the STN and Its Targets.

(A–C) The density of hM3D(Gq)-mCherry-expressing STN neurons that expressedc-fos was elevated in DA-intact and -depleted mice that were treated with CNO versus those treated with vehicle (A and B, examples; C, population data).
(D–F) The density of PV-immunoreactive SN neurons that expressed c-fos was elevated in DA-intact and -depleted mice that were treated with CNO versus those treated with vehicle (D and E, examples; F, population data).
*p < 0.05.
See also Table S6.
Figure 7. Chemogenetic Activation of the STN Improved Motor Function in Unilateral 6-OHDA-Injected Mice.

(A) In unilateral 6-OHDA-injected mice (green) contralateral forelimb use during vertical exploration was lower than in DA-intact, vehicle-injected mice (black).
(B and E) In vehicle-injected, DA-intact mice forelimb use was symmetric and not affected by chemogenetic activation (CNO) of hM3D(Gq)-mCherry expressed in the STN and zona incerta (ZI) (B, left panel, hM3D(Gq)-mCherry expression; B, right panel, population data, example highlighted).
(C and E) In 6-OHDA-injected, DA-depleted mice forelimb usage was asymmetric but ameliorated by chemogenetic activation of hM3D(Gq)-mCherry expressed in the STN and ZI (C, left panel) or the STN alone (C, middle panel; expression was restricted to the STN in a GABRR3-cre mouse through injection of an AAV carrying a cre-dependent construct; C, right panel, population data with examples highlighted).
(D and E) In 6-OHDA-injected, DA-depleted mice asymmetric forelimb use was moderately ameliorated by chemogenetic activation of hM3D(Gq)-mCherry expressed in the ZI (D, left panel, hM3D(Gq)-mCherry expression; D, right panel, population data, example highlighted).
(E) However, the chemogenetic rescue of forelimb usage in 6-OHDA-injected mice was significantly greater when hM3D(Gq)-mCherry expression involved the STN.
*p < 0.05. ns, not significant.
Given the voltage- and time-dependent properties of STN neuron ion channels and neurotransmitter receptors, the impact of synaptic input is exquisitely sensitive to the presence, frequency, and timing of postsynaptic autonomous activity (Bevan et al., 2002a; Wilson, 2013). Therefore, to determine whether chemogenetic restoration of autonomous firing affected synaptic integration in STN neurons, responses to optogenetic stimulation of their motor cortical inputs were compared before and after chemogenetic activation of hM3D(Gq)-mCherry in ex vivo brain slices from 6-OHDA-injected mice. ChR2(H134R)-eYFP was virally expressed in motor cortical projections to the STN through AAV injection in the motor cortex, as described previously (Chu et al., 2015, 2017). Two to three weeks later ex vivo brain slices were prepared and cortical inputs were stimulated using 1-ms duration blue light pulses delivered at 20 Hz for 1 s (Figures 5H and 5I). The number of “synaptically driven” action potentials was defined as the number of spikes that exceeded the mean + 3 SD of spikes in the second prior to stimulation (Figure 5J). Chemogenetic restoration of autonomous STN activity reduced the amount of synaptically driven activity both in absolute terms and as a fraction of the total number of intrinsically and extrinsically generated spikes (Figures 5H-5J). These data confirm that restoration of autonomous STN activity in PD mice profoundly alters the impact of synaptic inputs on STN activity.
Given that chemogenetic activation of STN neurons rescued their intrinsic activity and modified the impact of synaptic inputs that have been implicated in patterning Parkinsonian activity (Zaidel et al., 2009; Shimamoto et al., 2013; Sharott et al., 2014), we hypothesized that chemogenetic activation of STN neurons in vivo would ameliorate motor dysfunction in PD mice. To confirm chemogenetic activation of STN neurons in hM3D(Gq)-mCherry expressing vehicle- or 6-OHDA-injected mice in vivo, CNO (s.c., 1 mg/kg) or vehicle was administered and 90 min later, mice were perfused-fixed and then processed for immunohistochemical detection of (1) c-fos, a protein whose expression is linked to elevated neuronal activity (Morgan et al., 1987), and (2) parvalbumin, a Ca2+-binding protein that can be used to identify GABAergic neurons in the SN (Rajakumar et al., 1994). Consistent with the chemogenetic activation of the STN, the density of STN neurons that expressed c-fos was elevated in tissue from CNO-treated mice (Figures 6A-6C; Table S6). Furthermore, the density of PV-immunoreactive SNr neurons that expressed c-fos was increased in tissue from CNO-treated mice, consistent with the chemogenetic elevation of synaptic input from the STN (Figures 6D-6F; Table S6). The impact of chemogenetic activation of the STN on motor function was then assessed using the “cylinder test” (Schallert et al., 2000). When placed in a small-diameter cylinder, rodents explore their vertical environment by rearing upward and in so doing place their forelimbs on the side of the cylinder. In unilateral MFB vehicle-injected, DA-intact mice forelimb contacts with the side of the cylinder were symmetric, and chemogenetic stimulation of the STN (s.c., 1 mg/kg of CNO) had no consistent effect (Figures 7A and 7B; Table S7). However, in unilateral MFB 6-OHDA-injected, DA-depleted rodents, forelimb usage was asymmetric. Forelimb use contralateral to toxin injection was reduced relative to the ipsilateral forelimb and also to contralateral forelimb use in MFB vehicle-injected, DA-intact mice (Schallert et al., 2000) (Figures 7A-7D; Table S7). Within 30 min of chemogenetic stimulation (s.c., 1 mg/kg of CNO), unilateral 6-OHDA-injected mice with ipsilateral STN hM3D(Gq)-mCherry expression increased their usage of the contralateral Parkinsonian forelimb and decreased their usage of the ipsilateral forelimb relative to baseline (Figure 7C; Table S7; Video S1). Twenty-four hours following CNO injection, forelimb usage had returned to its asymmetric state (data not shown). In unilateral MFB 6-OHDA-injected, DA-depleted mice, with hM3D(Gq)-mCherry expressed ipsilaterally in regions adjacent to but not including the STN, chemogenetic stimulation modestly increased contralateral forelimb usage (Figure 7D; Table S7). However, the rescue of forelimb usage following STN chemogenetic stimulation was greater than that produced by stimulation of surrounding structures (Figure 7E; Table S7). Administration of CNO to MFB 6-OHDA-injected, DA-depleted mice without hM3D(Gq)-mCherry expression had no effect on forelimb usage (Figure S5; Table S7), ruling out a non-specific effect of CNO (Bouabid and Zhou, 2018). Together these data demonstrate that the chemogenetic rescue of autonomous STN activity in unilateral 6-OHDA-injected mice improved usage of the contralateral Parkinsonian forelimb during vertical exploratory behavior.
DISCUSSION
Autonomous firing is a fundamental property of STN neurons that supports their resting excitation of the extrastriatal basal ganglia (Kitai and Kita, 1987; Bevan and Wilson, 1999; Nambu et al., 2000; Do and Bean, 2003; Atherton et al., 2008) and influences the manner in which STN activity is patterned by synaptic input (Bevan et al., 2002a; Wilson, 2013). Here, we studied the cellular and circuit mechanisms that underlie the loss of autonomous STN firing in the 6-OHDA model of PD and the functional consequences of chemogenetically restoring this intrinsic activity.
In PD models, loss of inhibitory D2 receptor-dependent neuromodulation leads to an increase in the frequency and synchronization of D2-SPN output (Mallet et al., 2006; Lemos et al., 2016; Sharott et al., 2017; Parker et al., 2018; Ryan et al., 2018). These changes have been attributed to disinhibition of D2-SPN dendrites and synapses and remodeling of cortical and fast-spiking interneuron inputs (Day et al., 2006; Gittis et al., 2011; Fieblinger et al., 2014; Lemos et al., 2016). Thus, in response to cortical input, D2-SPNs generate abnormally strong, phasic suppression of GPe activity at approximately the same time that the STN receives direct cortical excitation (Walters et al., 2007; Zold et al., 2007; Mallet et al., 2008a). In DA-intact mice, we found that chemogenetic stimulation of D2-SPNs was sufficient to downregulate autonomous STN activity, suggesting that the loss of autonomous STN activity in PD models is triggered by elevated D2-SPN output. Consistent with this mechanism, prior 6-OHDA lesioning occluded the downregulatory effect of chemogenetically stimulating D2-SPNs. Although the loss of dopaminergic neuromodulation within the STN (Zhu et al., 2002b; Loucif et al., 2008; Ramanathan et al., 2008) cannot be completely excluded as a factor, the extent to which STN activity was downregulated in PD mice was reproduced solely by D2-SPN stimulation in DA-intact mice. Furthermore, DA receptor antagonist application has no effect on autonomous STN activity in ex vivo brain slices from DA-intact mice (Tofighy et al., 2003; Loucif et al., 2008).
The proposition that disinhibition of the STN leads to downregulation of autonomous spiking is consistent with our observation that KD of STN NMDARs completely prevented the effect of DA depletion on autonomous STN activity. Furthermore, the ability of NMDAR stimulation to suppress STN spiking ex vivo (Atherton et al., 2016) was occluded in slices from 6-OHDA-lesioned mice, suggesting that this mechanism had already been engaged in vivo. These data, together with recent studies of synaptic excitation (Chu et al., 2015) and inhibition (Chu etal., 2017), argue that both the level of autonomously generated STN activity and the balance of synaptic excitation and inhibition of STN neurons are regulated by the level of STN NMDAR activation in vivo.
How do NMDARs regulate autonomous STN activity? NMDAR activation can stimulate mitochondrial respiration through several mechanisms, including mitochondrial Ca2+ loading and/or reducing the cytosolic ATP to ADP ratio, leading to an increase in the production of ROS (Dugan et al., 1995; Stanika et al., 2010). Consistent with this mechanism, mitochondrial matrix oxidant stress was elevated in STN neurons from 6-OHDA-injected mice. This effect could also be mimicked in STN neurons from DA-intact mice by pre-incubation of slices in exogeneous NMDA. Elevated mitochondrial oxidant stress can impair ATP production and could therefore limit autonomous firing through an ATP-dependent mechanism (Wang and Michaelis, 2010). However, we found that dialysis of STN neurons with ATP did not rescue autonomous activity. Instead, breakdown of the ROS hydrogen peroxide rapidly reversed autonomous firing deficits in slices from DA-depleted mice. KATP channels are key metabolic sensors that limit cellular excitability in response to oxidative stress and/or a reduction in the ATP-to-ADP ratio (Nichols, 2006; Lee et al., 2015). In DA-depleted mice, inhibition of KATP channels restored autonomous STN activity and occluded the effect of hydrogen peroxide breakdown. Because hydrogen peroxide directly promotes KATP channel opening (Ichinari et al., 1996; Lee et al., 2015) and its breakdown rapidly reversed firing deficits, it appears to be directly responsible for increased KATP channel activation in STN neurons from PD mice, rather than, e.g., RNS-dependent modulation (Kawano et al., 2009; Shen and Johnson, 2010). Indeed, in DA-intact mice elevation of hydrogen peroxide through inhibition of glutathione peroxidase reduces the rate of autonomous STN activity through activation of KATP channels (Atherton et al., 2016). Interestingly, inhibition of KATP channels also restored autonomous firing following chemogenetic stimulation of D2-SPNs in DA-intact mice, pointing to the engagement of a common mechanism of downregulation in response to D2-SPN hyperactivity. Precisely why ROS levels and KATP channel activation remain persistently elevated in ex vivo brain slices is unclear, but possible mechanisms include (1) increased ROS production by mitochondria because of damage to electron transport chain proteins (Adam-Vizi, 2005; Wang and Michaelis, 2010); (2) compromised antioxidant defense (Wang and Michaelis, 2010); and (3) long-term modification of KATP channel gating and expression (Shen and Johnson, 2012).
NMDAR-dependent STN plasticity appears to be maladaptive in the 6-OHDA model of late-stage PD because its obstruction through STN NMDAR KD prevented motor dysfunction (Chu et al., 2017). Also consistent with the maladaptive nature of STN plasticity is our demonstration that the chemogenetic rescue of autonomous firing improves motor function in PD mice. It would be interesting to know when, and to what degree, cellular and synaptic STN plasticity are engaged during the progressive degeneration of DA neurons in PD. STN plasticity may become progressively extreme and track the emergence of motor symptoms as DA neurons degenerate. Alternatively, STN plasticity may be compensatory initially and engaged maximally prior to motor dysfunction. Increasingly, pathological upstream activity in conjunction with aberrant cellular and synaptic STN properties may then contribute to symptomatic onset. In this circumstance, chemogenetic restoration of autonomous firing could be therapeutic because it opposes the pathological patterning of STN activity by the hyper-direct and -indirect pathways (Mallet et al., 2008a; Tachibana et al., 2011). Consistent with this mechanism, we found that the impact of hyperdirect pathway cortical inputs ex vivo was largely vetoed by chemogenetically created intrinsic STN activity. Chemogenetic elevation of autonomous STN firing and output could also oppose Parkinsonian indirect pathway activity by modifying the impact of GPe inhibitory synaptic inputs on STN activity (Bevan et al., 2002a; Wilson, 2013) and counteracting the excessive inhibitory synaptic patterning of prototypic GPe neurons by D2-SPNs (Walters et al., 2007; Zold et al., 2007; Mallet et al., 2008a). Indeed, STN NMDAR KD, which also ameliorates motor dysfunction, largely disconnects the indirect pathway by downregulating GPe-STN transmission (Chu et al., 2015). Interruption of Parkinsonian hyperdirect and indirect pathway activity may ultimately explain why diverse STN manipulations—including lesioning (Bergman et al., 1990); optogenetic or pharmacological silencing (Levy et al., 2001; Yoon et al., 2014); high-frequency optogenetic stimulation of afferents (Gradinaru et al., 2009; Sanders and Jaeger, 2016); direct high-frequency electrical stimulation (Benabid et al., 2009; Wichmann et al., 2018); genetic manipulation (Zhuang et al., 2018); and chemogenetic elevation of autonomous activity—are therapeutic. Ultimately, simultaneous, large-scale measurement of neuronal activity throughout the cortico-basal ganglia-thalamo-cortical circuit will be necessary to determine why such radically different manipulations of the STN exhibit therapeutic efficacy in PD and its experimental models.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to the Lead Contact, Mark Bevan (m-bevan@northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experiments were performed using naive adult male C57BL/6 (RRID:IMSR_JAX:000664; age = 76, 70-92 days; n = 152), B6.129S4-Grin1tm2Stl/J (RRID:IMSR_JAX:005246; age = 89, 82-198 days; n = 9), B6.Cg-Tg(Adora2a-Chrm3*,-mCherry)AD6Blr/J (RRID:IMSR_JAX:017863; age = 125, 87-205 days; n = 18), and Tg(Gabrr3-cre)KC112Gsat (RRID:MGI:5528400; age = 68 days; n = 2) mice (Table S8) according to Northwestern University Feinberg School of Medicine IACUC and NIH guidelines for the care and use of animals. Mice were housed singly or with up to 4 other littermates per cage under a 14-hour light, 10-hour dark cycle with food and water ad libitum, and were inspected daily by Northwestern University Feinberg School of Medicine animal care technicians, veterinarians, and laboratory staff. Where possible littermates were assigned evenly to different treatment groups.
METHOD DETAILS
Surgery
6-OHDA, vehicle, and viral vectors were injected stereotaxically (Neurostar, Tubingen, Germany) under (1%–2%) isoflurane anesthesia (Smiths Medical ASD, Inc., Dublin, OH, USA). 6-OHDA (3-4 mg/ml) or vehicle was injected unilaterally into the medial forebrain bundle (MFB; from Bregma: AP −0.7 mm, ML +1.2 mm, DV +4.7 mm; 1.5 μl) to lesion midbrain DA neurons or control for injection, respectively. Pargyline (50 mg/kg) and desipramine (25 mg/kg) were used to increase the potency and specificity of 6-OHDA, respectively. In C57BL/6 mice AAV expressing hSyn-ChR2(H134R)-eYFP (AAV9.hSyn.hChR2(H134R)-eYFP.WPRE.hGH; 1 × 1013 GC/ml; University of Pennsylvania Viral Vector Core) was injected at 3 sites in primary motor cortex (AP +0.6 mm, +1.2 mm, and +1.8 mm, ML +1.5 mm, DV +1.0 mm; 300 nL per site), or AAV expressing CMV-mito-roGFP (AAV9-CMV-mito-roGFP-SV40; 2.5 × 1012 GC/ml; Virovek Inc., Hayward, CA), or hSyn-HA-hM3D(Gq)-IRES-mCitrine (AAV2/5-hSyn-HA-hM3D(Gq)-IRES-mCitrine; 2 × 1012 GC/ml) or hSyn-hM3D(Gq)-mCherry (AAV8-hSyn-hM3D(Gq)-mCherry; 0.5-3 × 1012 GC/ml; UNC Vector Core, Chapel Hill, NC or Addgene, Cambridge, MA) was injected in the STN (AP −2.06 mm, ML +1.4 mm, DV +4.45 mm; 300-500 nl), ipsilateral to the MFB injection. In Grin1lox/lox mice AAV expressing eGFP (AAV9-hSyn-eGFP-WPRE-bGH; 2 × 1012 GC/ml; University of Pennsylvania Viral Vector Core) or cre-eGFP (AAV9-hSyn-HI-eGFP-Cre-WPRE-SV40; 2 × 1012 GC/ml; University of Pennsylvania Viral Vector Core) was injected in the STN (AP −2.06 mm, ML +1.4 mm, DV +4.45 mm; 150-300 nl) ipsilateral to the MFB injection. Gabrr3-cre mice, in which there is cre-recombinase expression in STN neurons and minimal expression in adjacent structures, were injected with AAV expressing cre-dependent hSyn-DIO-hM3D(Gq)-mCherry (AAV8-hSyn-DIO-hM3D(Gq)-mCherry; 2–3 × 1012 GC/ml; UNC Vector Core, Chapel Hill, NC) in the STN. Finally, a subset of C57BL/6 mice were injected with AAV8-hSyn-hM3D(Gq)-mCherry dorsal to the STN (AP −2.06, ML +1.4, DV +3.75) to test the behavioral effects of hM3D(Gq) activation of structures adjacent to the STN. Electrophysiological, imaging and behavioral measurements were made 20, 16-22 days (n = 173) after surgery (Table S8).
Electrophysiology
Animals were first anesthetized through IP injection of ketamine/xylazine (87/13 mg/kg) and then transcardially perfused with ice-cold sucrose-based artificial cerebro-spinal fluid (sACSF: 230 mM sucrose, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 10 mM MgSO4, 10 mM glucose, and 26 mM NaHCO3), equilibrated with 95% O2 and 5% CO2. The brain was then removed and sectioned at 250 μm in the parasagittal plane at 30 μm/s in a chamber containing ice-cold sACSF using a vibratome (VT1200S; Leica Microsystems Inc., Buffalo Grove, IL, USA). Cut slices were transferred to ACSF (126 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM CaCl2, 2 mM MgSO4, 10 mM glucose, 26 mM NaHCO3, 1 mM sodium pyruvate, and 5 μM L-glutathione), equilibrated with 95% O2 and 5% CO2 at 35°C for 30 minutes and then held in ACSF at room temperature until recording.
In the recording chamber slices were perfused at a rate of 4-5 ml/min with synthetic interstitial fluid (SIF: 126 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.6 mM CaCl2, 1.5 mM MgSO4, 10 mM glucose and 26 mM NaHCO3), equilibrated with 95% O2 and 5% CO2 at 35°C. A microscope (Axioskop FS2 microscope (Carl Zeiss, Oberkochen, Germany) equipped with a LUMPlanFl/IR 60 × 0.9 NA objective (Olympus, Tokyo, Japan) or a BX51WI microscope (Olympus) equipped with a UIS2 LUMPFL 60 × 0.9 NA objective (Olympus)) employing infrared Dodt Gradient Contrast illumination (Luigs & Neumann, Ratingen, Germany) was used to visualize cell bodies for patch clamp recording. 590 nm LED illumination (Cairn Instruments, Faversham, Kent, UK) was used to identify neurons expressing hM3D(Gq)-mCherry and 890 nm 2-photon illumination (Mira 900F and G8 532 nm OPSL pump laser, Coherent Inc., Santa Clara, USA; as for mito-roGFP imaging, detailed below) was used to identify neurons expressing eGFP or cre-eGFP.
Recordings were obtained using computer-controlled manipulators (Luigs & Neumann) and a Multiclamp 700B amplifier, and a Digidata 1440A digitizer controlled by PClamp10 (Molecular Devices, Sunnyvale, CA, USA). Signals were low-pass filtered online at 10 kHz and sampled at 50 kHz. Cell and electrode capacitance, series resistance, and junction potential were compensated electronically. Recordings utilized borosilicate glass pipettes (Warner Instruments, Hamden, CT, USA) pulled using a P-97 Flaming/Brown Micropipette Puller (Sutter Instruments, Novato, CA, USA). Loose-seal cell-attached recordings were made with 3-5 MΩ impedance pipettes containing HEPES-buffered SIF solution (HBS: 140 mM NaCl, 23 mM glucose, 15 mM HEPES, 3 mM KCl, 1.5 mM MgCl2, 1.6 mM CaCl2; pH 7.2 with NaOH; 300–310 mOsm/L). Cell-attached recordings were excluded if the membrane became disrupted. Whole-cell voltage clamp recordings were made using 4-5 MΩ impedance pipettes filled with a K-gluconate-based internal solution (140 mM K-gluconate, 3.8 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 0.1 mM Na4-EGTA, 0.4 mM Na3GTP, and 2 mM Mg1.5ATP). Voltage clamp recordings were excluded if series resistance changed by more than 20% during the course of recording. Whole-cell current clamp recordings were made using 12-15 MΩ impedance pipettes filled with KCH3SO4-based internal solution (130 mM KCH3SO4, 3.8 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 5 mM phosphocreatine, 0.1 mM Na4-EGTA, 0.4 mM Na3GTP, and 2 mM Mg1.5ATP).
Optogenetic stimulation was delivered via the objective lens using a 470 nm light emitting diode (OptoLED; Cairn Research, Faversham, Kent, UK). Cortico-STN transmission was evoked by 1 ms duration optogenetic stimulation of cortico-STN axon-terminals. EPSCs and IPSCs were also evoked by electrical stimulation of the internal capsule bordering the rostro-ventral STN using a constant current isolator (A365R, World Precision Instruments, Sarasota, FL). The poles of stimulation were selected from a custom-built matrix of 10 stimulation electrodes (MX52CBWMB2, Frederick Haer, Bowdoin, ME).
Autonomous activity was typically recorded in the presence of 20 μM DNQX, 50 μM D-APV, 10 μM SR-95531, and 2 μM CGP55845 (Abcam, Cambridge, MA, USA) to antagonize AMPARs, NMDARs, GABAARs and GABABRs, respectively. DNQX and D-APV were excluded when the response of STN neurons to optogenetic cortico-STN stimulation or electrically evoked EPSCs were studied. SR-95531 and CGP55845 were excluded when electrically evoked IPSCs were studied. The effect of catalase (polyethylene glycolcatalase; 250 U/mL; Sigma-Aldrich), glibenclamide (100 nM; Sigma-Aldrich), apamin (10 nM; Sigma-Aldrich), or CNO (10-100 μM; Sigma-Aldrich) on firing was also studied.
2-Photon Imaging of mito-roGFP
Brain slices were prepared from mice expressing the mitochondrially-targeted redox-sensitive probe mito-roGFP in STN neurons, as for electrophysiology. mito-roGFP-expressing neurons were imaged at 890 nm with 76 MHz pulse repetition and ~250 fs pulse duration at the sample plane. Two-photon excitation was provided by a G8 OPSL pumped Mira 900 F laser (Coherent, Santa Clara, CA, USA). Sample power was regulated by a Pockels cell electro-optic modulator (model M350-50-02-BK, Con Optics, Danbury, CT, USA). Images were acquired using an Ultima 2 P system running PrairieView 5.3 (Bruker Nano Fluorescence Microscopy, Middleton, WI, USA) and a BX51WI microscope (Olympus, Tokyo, Japan) with a 60 × 0.9 NA objective (UIS2 LUMPFL; Olympus). Mito-roGFP fluorescence and simultaneous laser-scanned Dodt contrast images were acquired from each field of STN neurons in the presence of 20 μM DNQX, 50 μM APV, 10 μM SR-95531, and 2 μM CGP55845 under baseline conditions, following the addition of 2 mM dithiothrietol (DTT) to fully reduce the tissue, and following the addition of 200 μM aldrithiol (ALD) to fully oxidize the tissue (Sanchez-Padilla et al., 2014; Atherton et al., 2016). Average fluorescence intensities of mitochondria in STN neurons under baseline (F), reduced (FDTT), and oxidized (FALD) conditions were quantified using ImageJ (NIH, Bethesda, MD, USA). Mitochondrial measurements were restricted to the somata of STN neurons through registration of mito-roGFP image and Dodt contrast images (Figure 4A). Baseline mitochondrial oxidation in STN neurons was expressed relative to that under conditions of minimum and maximum oxidation from the Equation 1−[(F − FALD)/(FDTT − FALD)].
Behavioral Testing
To assess forelimb usage during vertical exploration, animals were placed in a 600 mL cylindrical glass beaker (9.5 cm diameter; 12 cm height) and imaged using an HD digital camcorder recording at 60 fps (VIXIA HF R40 Full HD Camcorder; Canon, Melville, NY, USA). Forelimb placements on the vertical walls of the beaker were counted manually during video review (Schallert et al., 2000). Placement of the left and right forepaw were considered simultaneous (“both”) if they occurred within 3 frames of each other (i.e., < 33 ms apart). Mice expressing hM3D(Gq)-mCherry were tested ~20-60 minutes following SC injection of vehicle or 1 mg/kg clozapine-n-oxide (CNO) or in the absence of an injection.
Histology
In order to study the expression of ChR2(H134)-eYFP, rM3D(Gs)-mCherry, cre-eGFP, eGFP, mito-roGFP, hM3D(Gq)-mCherry, c-fos, NeuN, PV, and TH, brain tissue was first fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, pH7.4. Tissue prepared for electrophysiology or mito-roGFP imaging was immersion-fixed. Mice used for behavioral analyses were perfused-fixed under deep anesthesia (IP: 87 mg/kg ketamine, 13 mg/kg xylazine). Tissue was held in fixative at 4°C for at least 12 hours before rinsing in phosphate buffered saline (PBS; 0.05 M; pH 7.4). Tissue exceeding 250 μm in thickness was re-sectioned at 70 μm using a vibratome (Leica VT1000S; Leica Microsystems Inc.). Immunohistochemical detection of c-fos, NeuN, PV, and TH was carried out in PBS containing 0.5% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) and 2% normal donkey serum (Jackson ImmunoResearch, West Grove, PA, USA). Tissue was incubated in primary antibodies for 48-72 hours at 4°C or overnight at RT (rabbit anti-c-fos: 1:500 dilution; cat# 2250S; Cell Signaling Technology, Danvers, MA; mouse anti-NeuN: 1:200 dilution; cat# MAB377; EMD Millipore, Darmstadt, Germany; guinea pig anti-PV: 1:1,000; cat# 195 004; Synaptic Systems, Gottingen, Germany; mouse anti-TH: 1:500 dilution; cat# MAB318; EMD Millipore), washed in PBS, and then incubated in their respective secondary antibodies (donkey anti-rabbit Alexa Fluor 488: dilution 1:250; cat # 711-545-152; Jackson ImmunoResearch; donkey anti-mouse Alexa Fluor 488: dilution 1:250; cat # 715-545-150; Jackson ImmunoResearch; donkey anti-mouse Alexa Fluor 594: dilution 1:250; cat # 715-585-150; Jackson ImmunoResearch; donkey anti-guinea pig Alexa Fluor 647: dilution 1:250; cat # 706-605-148; Jackson ImmunoResearch) for 90-120 minutes at room temperature before a final wash with PBS. All tissue was mounted on glass slides using ProLong Diamond Antifade Reagent (ThermoFisher Scientific, Waltham, MA, USA) and coverslipped. Sections were imaged using an Axioskop 2 microscope (Carl Zeiss) equipped with a Neurolucida system (MBF Bioscience, Williston, VT, USA) and/or a confocal laser scanning microscope (A1R; Nikon, Melville, USA).
The densities of c-fos-positive neurons were assessed using NIH ImageJ and quantified using the optical dissector method (West, 1999). Confocal images were collected using a 60X, 1.4NA oil immersion Plan Apo objective lens on a Nikon A1R confocal microscope (0.205 micron/pixel; Nikon, Melville, USA). Sample sites were chosen using a grid (frame size, 50 × 50 μm) superimposed randomly on each image stack. Stereological counting commenced and was terminated at an optical section 5 μm and 21 μm below the slice surface, respectively. Look-up and reference planes were separated by 2 μm. Dopaminergic innervation was assessed from striatal TH immunoreactivity (Table S8), as described previously (Chu et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Only data that were compromised by technical failure (e.g., failure of viral expression or immunohistochemical detection) were excluded. Each dataset, together with the associated number and type of observations, and the statistical tests that were applied are summarized in Tables S1-S8. In addition, paired data points or box-and-whisker plots displaying medians and inter-quartile and 10%–90% ranges calculated in Prism 7 (GraphPad Software, Inc., La Jolla, CA) are displayed in their respective figures to reflect the distribution and central tendency of the data. To minimize assumptions concerning the distribution of the data, exact, non-parametric (two-sided) statistics were used throughout: the Mann-Whitney U (MWU) test for unpaired data, the Wilcoxon signed rank (WSR) test for paired comparisons, and Fisher’s exact test for contingency analyses. To ensure our experiments were adequately powered sample sizes for MWU and WSR tests were estimated to achieve a minimum of 80% power. For the MWU test, a 50th percentile difference in median requires 10 samples. For the WSR test, if all pairs exhibit the same direction of change at least 6 observations are required. Exact p values were calculated using the fisher.test and the Wilcox.exact function (exactRankTests package) in R (http://www.r-project.org). Where datasets were subjected to multiple comparisons the p value was adjusted to maintain the family-wise error rate at 0.05 using the Holm-Bonferroni method (notated ph; Holm, 1979).
DATA AND CODE AVAILABILITY
The data associated with this paper have been deposited on the Open Science Framework: https://doi.org/10.17605/OSF.IO/7KWD2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-c-fos | Cell Signaling Technology | cat#2250S; RRID:AB_2247211 |
| mouse anti-NeuN | EMD Millipore | cat#MAB377; RRID:AB_2298772 |
| guinea pig anti-PV | Synaptic Systems | cat#195004; RRID:AB_2156476 |
| mouse anti-TH | EMD Millipore | cat# MAB318; RRID:AB_2201528 |
| donkey anti-rabbit Alexa Fluor 488 | Jackson ImmunoResearch | cat#711-545-152; RRID:AB_2313584 |
| donkey anti-mouse Alexa Fluor 488 | Jackson ImmunoResearch | cat#715-545-150; RRID:AB_2340846 |
| donkey anti-mouse Alexa Fluor 594 | Jackson ImmunoResearch | cat#715-585-150; RRID:AB_2340854 |
| donkey anti-guinea pig Alexa Fluor 647 | Jackson ImmunoResearch | cat#706-605-148; RRID:AB_2340476 |
| Bacterial and Virus Strains | ||
| AAV9.hSyn.hChR2(H134R)-eYFP.WPRE.hGH | U. Penn. Vector Core | Addgene viral prep # 26973-AAV9 |
| AAV9-CMV-mito-roGFP-SV40 | Virovek Inc.; Sanchez-Padilla et al., 2014 | N/A |
| AAV2/5-hSyn-HA-hM3D(Gq)-IRES-mCitrine | U.N.C. Vector Core | discontinued |
| AAV8-hSyn-hM3D(Gq)-mCherry | Addgene | Addgene viral prep # 50474-AAV8 |
| AAV9-hSyn-eGFP-WPRE-bGH | U. Penn. Vector Core | Addgene viral prep # 105539-AAV9 |
| AAV9.hSyn.HI.eGFP-Cre.WPRE.SV40 | U. Penn. Vector Core | Addgene viral prep # 105540-AAV9 |
| AAV8-hSyn-DIO-hM3D(Gq)-mCherry | Addgene; Krashes et al., 2011 | Addgene viral prep # 44361-AAV8 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Apamin | Sigma-Aldrich | Cat#A9459 |
| Catalase-polyethylene glycol | Sigma-Aldrich | Cat#C4963 |
| 6-Hydroxydopamine hydrobromide | Tocris Bioscience | Cat#2547 |
| CGP 55845 hydrochloride | Abcam Inc | Cat#ab120337 |
| Clozapine N-oxide | Tocris Bioscience | Cat#4936 |
| D-APV | Abcam Inc | Cat#ab120003 |
| Desipramine hydrochloride | Sigma-Aldrich | Cat#P3900 |
| DNQX disodium salt | Abcam Inc | Cat#ab120169 |
| Glibenclamide | Sigma-Aldrich | Cat#G0639 |
| L-Glutathione | Sigma-Aldrich | Cat#G4251 |
| N-Methyl D-aspartic acid | Tocris Bioscience | Cat#0114 |
| Pargyline hydrochloride | Sigma-Aldrich | Cat#P8013 |
| Sodium pyruvate | Sigma-Aldrich | Cat#P2256 |
| SR95531 (Gabazine) | Abcam Inc | Cat#ab120042 |
| Deposited Data | ||
| Raw and analyzed data | This paper | https://doi.org/10.17605/OSF.IO/7KWD2 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| B6.Cg-Tg(Adora2a-Chrm3*,-mCherry) AD6Blr/J mice | The Jackson Laboratory | RRID:IMSR_JAX:017863 |
| B6.129S4-Grin1tm2Stl/J mice | The Jackson Laboratory | RRID:IMSR_JAX:005246 |
| Tg(Gabrr3-cre)KC112Gsat mice | Mutant Mouse Resource & Research Centers | RRID:MGI:5528400 |
| Software and Algorithms | ||
| Clampfit (version 10.5) | Molecular Devices | N/A |
| Igor Pro (version 6.37) | Wavemetrics Inc | RRID: SCR_000325 |
| Neurolucida (version 11.03) | MBF Bioscience | RRID: SCR_001775 |
| Neurolucida Explorer (version 11.08) | MBF Bioscience | N/A |
| NIH ImageJ (version 1.47) | NIH | RRID: SCR_003070 |
| OriginPro (version 2015) | OriginLab Co. | RRID: SCR_014212 |
| pClamp (version 10.5) | Molecular Device | RRID: SCR_011323 |
| Prism (version 6.0h) | Graphpad software Inc | RRID: SCR_002798 |
| R (version 3.3.1) | http://www.R-project.org/ | RRID: SCR_001905 |
Highlights.
Autonomous subthalamic nucleus (STN) activity is downregulated after loss of dopamine
Elevated D2-striatal projection neuron transmission is sufficient for downregulation
Downregulation is dependent on activation of STN NMDA receptors and KATP channels
Chemogenetic restoration of autonomous spiking reduces Parkinsonian dysfunction in mice
ACKNOWLEDGMENTS
This study was funded by NIH NINDS grants 2R37 NS041280, P50 NS047085, 5T32 NS041234, and F31 NS090845. Confocal imaging work was performed at the Northwestern Center for Advanced Microscopy, supported by NCI CCSG grant P30 CA060553.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.076.
REFERENCES
- Adam-Vizi V (2005). Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal 7, 1140–1149. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci. 12, 366–375. [DOI] [PubMed] [Google Scholar]
- Atherton JF, Wokosin DL, Ramanathan S, and Bevan MD (2008). Autonomous initiation and propagation of action potentials in neurons of the subthalamic nucleus. J. Physiol 586, 5679–5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JF, McIver EL, Mullen MR, Wokosin DL, Surmeier DJ, and Bevan MD (2016). Early dysfunction and progressive degeneration of the subthalamic nucleus in mouse models of Huntington’s disease. eLife 5, e21616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabid AL, Chabardes S, Mitrofanis J, and Pollak P (2009). Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 8, 67–81. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, and DeLong MR (1990). Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249, 1436–1438. [DOI] [PubMed] [Google Scholar]
- Bevan MD, and Wilson CJ (1999). Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J. Neurosci 19, 7617–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Hallworth NE, Bolam JP, and Wilson CJ (2002a). Regulation of the timing and pattern of action potential generation in rat subthalamic neurons in vitro by GABA-A IPSPs. J. Neurophysiol 87, 1348–1362. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, and Wilson CJ (2002b). Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci. 25, 525–531. [DOI] [PubMed] [Google Scholar]
- Bouabid S, and Zhou FM (2018). Cyclic AMP-producing chemogenetic activation of indirect pathway striatal projection neurons and the downstream effects on the globus pallidus and subthalamic nucleus in freely moving mice. J. Neurochem 145, 436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Atherton JF, Wokosin D, Surmeier DJ, and Bevan MD (2015). Heterosynaptic regulation of external globus pallidus inputs to the subthalamic nucleus by the motor cortex. Neuron 85, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HY, Mciver EL, Kovaleski RF, Atherton JF, and Bevan MD (2017). Loss of Hyperdirect Pathway Cortico-Subthalamic Inputs Following Degeneration of Midbrain Dopamine Neurons. Neuron 95, 1306–1318.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, et al. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci 9, 251–259. [DOI] [PubMed] [Google Scholar]
- Do MT, and Bean BP (2003). Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 39, 109–120. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, and Choi DW (1995). Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J. Neurosci 15, 6377–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eusebio A, Cagnan H, and Brown P (2012). Does suppression of oscillatory synchronisation mediate some of the therapeutic effects of DBS in patients with Parkinson’s disease? Front. Integr. Nuerosci 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MS, Pei Y, Wan Y, Yadav PN, Daigle TL, Urban DJ, Lee HM, Sciaky N, Simmons A, Nonneman RJ, et al. (2013).AGαs DREADD mouse for selective modulation of cAMP production in striatopallidal neurons. Neuropsychopharmacology 38, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, and Surmeier DJ (2014). Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat. Commun 5, 5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Finkbeiner S, and Kreitzer AC (2011). Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron 71, 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, and Deisseroth K (2009). Optical deconstruction of parkinsonian neural circuitry. Science 324, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, and Surmeier DJ (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci 29, 11011–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallworth NE, Wilson CJ, and Bevan MD (2003). Apamin-sensitive small conductance calcium-activated potassium channels, through their selective coupling to voltage-gated calcium channels, are critical determinants of the precision, pace, and pattern of action potential generation in rat subthalamic nucleus neurons in vitro. J. Neurosci 23, 7525–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, and Remington SJ (2004). Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem 279, 13044–13053. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, and Vitek JL (2003). Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J. Neurosci 23, 1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S (1979).A simple sequentially rejective multiple test procedure. Scand. J. Stat 6, 65–70. [Google Scholar]
- Ichinari K, Kakei M, Matsuoka T, Nakashima H, and Tanaka H (1996). Direct activation of the ATP-sensitive potassium channel by oxygen free radicals in guinea-pig ventricular cells: its potentiation by MgADP. J. Mol. Cell. Cardiol 28, 1867–1877. [DOI] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Kimura M, Liang MY, Wu HE, Gemes G, McCallum JB, Kwok WM, Hogan QH, and Sarantopoulos CD (2009). Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol. Pain 5, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai ST, and Kita H (1987). Anatomy and physiology of the subthalamic nucleus: a driving force of the basal ganglia In The Basal Ganglia II, Carpenter MB and Jayaraman A, eds. (Plenum), pp. 357–373. [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, and Kreitzer AC (2010). Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Patel JC, O’Neill B, and Rice ME (2015). Inhibitory and excitatory neuromodulation by hydrogen peroxide: translating energetics to information. J. Physiol 593, 3431–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Friend DM, Kaplan AR, Shin JH, Rubinstein M, Kravitz AV, and Alvarez VA (2016). Enhanced GABA Transmission Drives Bradykinesia Following Loss of Dopamine D2 Receptor Signaling. Neuron 90, 824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Lang AE, Dostrovsky JO, Pahapill P, Romas J, Saint-Cyr J, Hutchison WD, and Lozano AM (2001). Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain 124, 2105–2118. [DOI] [PubMed] [Google Scholar]
- Loucif AJ, Woodhall GL, Sehirli US, and Stanford IM (2008). Depolarsation and suppression of burst firing activity in the mouse subthalamic nucleus by dopamine D1/D5 receptor activation of a cyclic-nucleotide gated non-specific cation conductance. Neuropharmacology 55, 94–105. [DOI] [PubMed] [Google Scholar]
- Mallet N, Ballion B, Le Moine C, and Gonon F (2006). Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J. Neurosci 26, 3875–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Márton LF, Bolam JP, Brown P, and Magill PJ (2008a). Parkinsonian beta oscillations in the external globus pallidus and their relationship with subthalamic nucleus activity. J. Neurosci 28, 14245–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, and Magill PJ (2008b). Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J. Neurosci 28, 4795–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz JE, Gillis WF, Beron CC, Neufeld SQ, Robertson K, Bhagat ND, Peterson RE, Peterson E, Hyun M, Linderman SW, et al. (2018). The Striatum Organizes 3D Behavior via Moment-to-Moment Action Selection. Cell 174, 44–58.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Ma Y, Paré JF, Villalba RM, Wichmann T, and Smith Y (2015). Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain 138, 946–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL, and Curran T (1987). Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237, 192–197. [DOI] [PubMed] [Google Scholar]
- Mourre C, Manrique C, Camon J, Aidi-Knani S, Deltheil T, Turle-Lorenzo N, Guiraudie-Capraz G, and Amalric M (2017). Changes in SK channel expression in the basal ganglia after partial nigrostriatal dopamine lesions in rats: Functional consequences. Neuropharmacology 113 (Pt A), 519–532. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, Hamada I, Kita H, Imanishi M, Akazawa T, Ikeuchi Y, and Hasegawa N (2000). Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. J. Neurophysiol 84, 289–300. [DOI] [PubMed] [Google Scholar]
- Nambu A, Tokuno H, and Takada M (2002). Functional significance of the cortico-subthalamo-pallidal ‘hyperdirect’ pathway. Neurosci. Res 43, 111–117. [DOI] [PubMed] [Google Scholar]
- Nichols CG (2006). KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Parker JG, Marshall JD, Ahanonu B, Wu YW, Kim TH, Grewe BF, Zhang Y, Li JZ, Ding JB, Ehlers MD, and Schnitzer MJ (2018). Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 557, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumar N, Elisevich K, and Flumerfelt BA (1994). Parvalbumin-containing GABAergic neurons in the basal ganglia output system of the rat. J. Comp. Neurol 350, 324–336. [DOI] [PubMed] [Google Scholar]
- Ramanathan S, Tkatch T, Atherton JF, Wilson CJ, and Bevan MD (2008). D2-like dopamine receptors modulate SKCa channel function in subthalamic nucleus neurons through inhibition of Cav2.2 channels. J. Neurophysiol 99, 442–459. [DOI] [PubMed] [Google Scholar]
- Roth BL (2016). DREADDs for Neuroscientists. Neuron 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, Bair-Marshall C, and Nelson AB (2018). Aberrant striatal activity in Parkinsonism and levodopa-induced dyskinesia. Cell Rep. 23, 3438–3446.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, Schieber S, Oertel W, Wokosin D, Schumacker PT, and Surmeier DJ (2014). Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci 17, 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders TH, and Jaeger D (2016). Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiol. Dis 95, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, and Bland ST (2000). CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39, 777–787. [DOI] [PubMed] [Google Scholar]
- Sharott A, Gulberti A, Zittel S, Tudor Jones AA, Fickel U, Münchau A, Köppen JA, Gerloff C, Westphal M, Buhmann C, et al. (2014). Activity parameters of subthalamic nucleus neurons selectively predict motor symptom severity in Parkinson’s disease. J. Neurosci 34, 6273–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharott A, Vinciati F, Nakamura KC, and Magill PJ (2017). A Population of Indirect Pathway Striatal Projection Neurons Is Selectively Entrained to Parkinsonian Beta Oscillations. J. Neurosci 37, 9977–9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, and Johnson SW (2010). Ca2+ influx through NMDA-gated channels activates ATP-sensitive K+ currents through a nitric oxide-cGMP pathway in subthalamic neurons. J. Neurosci 30, 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, and Johnson SW (2012). Chronic dopamine depletion augments the functional expression of K-ATP channels in the rat subthalamic nucleus. Neurosci. Lett 531, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto SA, Ryapolova-Webb ES, Ostrem JL, Galifianakis NB, Miller KJ, and Starr PA (2013). Subthalamic nucleus neurons are synchronized to primary motor cortex local field potentials in Parkinson’s disease. J. Neurosci 33, 7220–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Winters CA, Pivovarova NB, and Andrews SB (2010). Differential NMDA receptor-dependent calcium loading and mitochondrial dysfunction in CA1 vs. CA3 hippocampal neurons. Neurobiol. Dis 37, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana Y, Iwamuro H, Kita H, Takada M, and Nambu A (2011). Subthalamo-pallidal interactions underlying parkinsonian neuronal oscillations in the primate basal ganglia. Eur. J. Neurosci 34, 1470–1484. [DOI] [PubMed] [Google Scholar]
- Tecuapetla F, Jin X, Lima SQ, and Costa RM (2016). Complementary Contributions of Striatal Projection Pathways to Action Initiation and Execution. Cell 166, 703–715. [DOI] [PubMed] [Google Scholar]
- Tofighy A, Abbott A, Centonze D, Cooper AJ, Noor E, Pearce SM, Puntis M, Stanford IM, Wigmore MA, and Lacey MG (2003). Excitation by dopamine of rat subthalamic nucleus neurones in vitro-a direct action with unconventional pharmacology. Neuroscience 116, 157–166. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, and Tonegawa S (1996). The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Vila M, Périer C, Féger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, and Hirsch EC (2000). Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur. J. Neurosci 12, 337–344. [DOI] [PubMed] [Google Scholar]
- Walters JR, Hu D, Itoga CA, Parr-Brownlie LC, and Bergstrom DA (2007). Phase relationships support a role for coordinated activity in the indirect pathway in organizing slow oscillations in basal ganglia output after loss of dopamine. Neuroscience 144, 762–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Michaelis EK (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ (1999). Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 22, 51–61. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, and DeLong MR (2018). Basal ganglia, movement disorders and deep brain stimulation: advances made through non-human primate research J. Neural Transm (Vienna: ) 125, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ (2013). Active decorrelation in the basal ganglia. Neuroscience 250, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CL, Cash D, Galley K, Chapman H, Lacey MG, and Stanford IM (2006). Subthalamic nucleus neurones in slices from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mice show irregular, dopamine-reversible firing pattern changes, but without synchronous activity. Neuroscience 143, 565–572. [DOI] [PubMed] [Google Scholar]
- Yoon HH, Park JH, Kim YH, Min J, Hwang E, Lee CJ, Suh JK, Hwang O, and Jeon SR (2014). Optogenetic inactivation of the subthalamic nucleus improves forelimb akinesia in a rat model of Parkinson disease. Neurosurgery 74, 533–540, discussion 540-541. [DOI] [PubMed] [Google Scholar]
- Zaidel A, Arkadir D, Israel Z, and Bergman H (2009). Akineto-rigid vs. tremor syndromes in Parkinsonism. Curr. Opin. Neurol 22, 387–393. [DOI] [PubMed] [Google Scholar]
- Zhang DM, Chai Y, Erickson JR, Brown JH, Bers DM, and Lin YF (2014). Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J. Physiol 592, 971–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Bartol M, Shen K, and Johnson SW (2002a). Excitatory effects of dopamine on subthalamic nucleus neurons: in vitro study of rats pretreated with 6-hydroxydopamine and levodopa. Brain Res. 945, 31–40. [DOI] [PubMed] [Google Scholar]
- Zhu ZT, Shen KZ, and Johnson SW (2002b). Pharmacological identification of inward current evoked by dopamine in rat subthalamic neurons in vitro. Neuropharmacology 42, 772–781. [DOI] [PubMed] [Google Scholar]
- Zhuang QX, Li GY, Li B, Zhang CZ, Zhang XY, Xi K, Li HZ, Wang JJ, and Zhu JN (2018). Regularizing firing patterns of rat subthalamic neurons ameliorates parkinsonian motor deficits. J. Clin. Invest 128, 5413–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zold CL, Ballion B, Riquelme LA, Gonon F, and Murer MG (2007). Nigrostriatal lesion induces D2-modulated phase-locked activity in the basal ganglia of rats. Eur. J. Neurosci 25, 2131–2144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data associated with this paper have been deposited on the Open Science Framework: https://doi.org/10.17605/OSF.IO/7KWD2.