

Abstract
PURPOSE
The STK11 gene encodes a serine/threonine protein kinase that regulates cell polarity and functions as a tumor suppressor. Patients with non–small-cell lung cancer (NSCLC) and STK11 mutations often have other co-mutations. We evaluated the impact of KRAS and TP53 co-mutations on outcomes after first-line systemic therapy for patients with metastatic or recurrent NSCLC that harbors STK11 mutations.
METHODS
We conducted a retrospective review of patients with metastatic NSCLC and STK11 mutations treated at the University of Pennsylvania. STK11 mutations were identified through next-generation sequencing (NGS) in tissue or plasma. Cox proportional hazard models were used to determine the relationship between STK11 co-mutations and survival outcomes. The Kaplan-Meier method was used to estimate overall survival (OS) and progression-free survival (PFS).
RESULTS
From February 2013 to December 2016, samples from 1,385 patients with NSCLC were analyzed by NGS; of these, 77 patients (6%) harbored an STK11 mutation (n = 56, tissue; n = 21, plasma). Of the 62 patients included, 18 had an STK11 mutation alone, 19 had STK11/KRAS, 18 had STK11/TP53, and seven had STK11/KRAS/TP53. Patients with STK11/KRAS co-mutations had a worse median PFS (2.4 months) compared with STK11 alone (5.1 months; log-rank P = .048), STK11/TP53 (4.3 months; log-rank P = .043), and STK11/KRAS/TP53 (13 months; log-rank P = .03). Patients with STK11/KRAS co-mutation experienced shorter median OS (7.1 months) compared with STK11 alone (16.1 months; log-rank P < .001), STK11/TP53 (28.3 months; log-rank P < .001), and STK11/KRAS/TP53 (22 months; log-rank P = .025).
CONCLUSION
Among patients with advanced NSCLC and STK11 mutations treated with first-line systemic therapy, co-mutation with KRAS was associated with significantly worse PFS and OS. By contrast, co-mutation of STK11 with TP53 conferred a better prognosis.
INTRODUCTION
Lung cancer is the leading cause of cancer-related mortality in the United States, and non–small-cell lung cancer (NSCLC) represents 80% to 85% of all lung cancer.1 In nonsquamous NSCLC, it is routine practice to test for genetic abnormalities using comprehensive next-generation sequencing (NGS). The majority of the mutations found during routine testing are not actionable currently, but their presence likely has predictive and prognostic relevance.
CONTEXT
Key Objective
How do KRAS and TP53 co-mutations affect outcomes after first-line systemic therapy in patients with non–small-cell lung cancer and STK11 mutations?
Knowledge Generated
Among patients with metastatic NSCLC and tumor-associated STK11 mutations, co-mutation with TP53 conferred better progression-free survival (PFS) and overall survival (OS) after first-line therapy compared with patients who had a KRAS co-mutation.TP53 mutation in the presence of an STK11/KRAS co-mutation also conferred better PFS and OS compared with patients who had only the STK11/KRAS co-mutation.
Relevance
STK11/KRAS co-mutation has been associated with worse PFS after chemotherapy, but co-mutation with TP53 may modulate outcomes after first-line chemotherapy in this group and among patients with STK11 mutations without KRAS mutations.
STK11, also known as liver kinase B1 (LKB1), is a tumor suppressor and a negative regulator of mammalian target for rapamycin signaling. Loss-of-function mutations in germline STK11 are associated with Peutz-Jeghers hereditary cancer syndrome. STK11 mutations are estimated to be present in 8% to 39% of all NSCLC, with increased prevalence in smokers and patients with KRAS mutations.2,3 Animal studies suggest that STK11 mutations are critical in lung cancer differentiation, tumorigenesis, and metastasis.4,5 Mutations in STK11 have emerged as a potential prognostic and predictive marker in NSCLC. Somatic mutations in STK11 have been hypothesized as primarily oncogenic through loss of function, although gain-of-function alterations through mutations in exons 1 to 2 (STK11ex1-2) have also been described.6 In a report by Pécuchet et al,6 STK11ex1-2 mutations conferred significantly worse progression-free survival (PFS) and overall survival (OS) compared with mutations in exons 3 through 9 (STK11ex3-9) among patients undergoing curative intent surgery for NSCLC, which suggests that STK11 mutations may be a more heterogeneous group than previously thought.6,7
Co-mutation status may be another source of heterogeneity among patients with STK11 mutations. KRAS is frequently co-mutated with STK11, but the predictive and prognostic significance of this co-mutation is uncertain. In KRAS-mutant mice, co-mutation with STK11 was associated with resistance to anticancer therapy, whereas co-mutation with TP53 was not.8 In humans, STK11 mutation alone does not appear to be predictive of response to chemotherapy, whereas KRAS/STK11 co-mutation has been associated with worse PFS after chemotherapy.3,9-11 KRAS/STK11 co-mutation also is associated with inferior PFS and OS after immunotherapy compared with KRAS alone (PFS hazard ratio [HR], 1.98; P < .001) or KRAS/TP53 co-mutation (PFS HR, 1.77; P = .0072; OS duration, 6.4 v 16.1 v 16 months for KRAS/STK11 v KRAS v KRAS/TP53, respectively).12
Co-mutation with TP53 and STK11 is less common than KRAS/STK11 mutation but still may represent a distinct molecular subtype of NSCLC.11 TP53 is a DNA binding transcription factor that regulates multiple genes involved in DNA repair, metabolism, cell cycle arrest, apoptosis, and senescence.13,14 Gene expression studies have shown that, although KRAS, STK11, and KRAS/STK11 groups share a KRAS-mutant gene signature, the TP53 mutant group does not.11 NSCLC cell lines that harbor KRAS/TP53 also have a different drug sensitivity profile compared with KRAS/STK11 or KRAS/TP53/STK11 cell lines.10 In addition, TP53 is a known regulator of STK11 and has four potential binding sites in the STK11 promoter.13,15 These findings highlight the differential and context-dependent effects of a TP53 mutation and its potential interactions with STK11 and KRAS.
In this study, we evaluated patients with STK11-mutant NSCLC and the effect of concurrent mutations in KRAS and TP53 on treatment outcomes after first-line systemic for metastatic/recurrent disease.
METHODS
Patient Population
This was a retrospective study among patients with NSCLC diagnosed and treated at the University of Pennsylvania Abramson Cancer Center between February, 2013—when NGS testing, including for STK11, was first performed routinely on all patients with stage IV disease—and December, 2016. Eligible patients for this study had histologically confirmed stage IV NSCLC and had NGS performed on tissue or plasma as part of routine clinical testing. Patients who received treatment outside the institution or had another concurrent malignancy were excluded.
Mutational Analysis
Plasma was analyzed by Guardant Health (Redwood City, CA) as described previously.16 Solid tumor sequencing was performed at the Center for Personalized Diagnostics clinical laboratory at the University of Pennsylvania (Data Supplement). One KRAS amplification; one KRAS variant of unknown significance (VUS), Q61H; and one TP53 VUS (A161S) were not considered mutations in the respective genes. STK11 mutations were categorized as disease associated on the basis of the designation in the NGS report (ie, disease associated v VUS). Mutations were categorized using OncoPrinter by cBioPortal.17,18
Clinical Data
The following information was collected from the electronic medical record: age at diagnosis, sex, race/ethnicity, smoking status, Eastern Cooperative Oncology Group performance status at diagnosis, stage at diagnosis (TNM, according to American Joint Committee on Cancer, 7th edition, guidelines), histology, method of diagnosis, date of diagnosis, treatment (first-, second-, and third-line therapies, and chemotherapy v immunotherapy), and outcomes (date of progression, death, or last follow-up). All information was collected with approval from the institutional review board; informed consent was waived because of the retrospective, nontherapeutic nature of the study.
Statistical Analysis
Descriptive statistics, including mean, median, and proportions, were used to summarize patient demographics and tumor characteristics. PFS was calculated from the start of treatment of metastatic or recurrent disease to date of death or progression. The date of progression was based on radiologic progression, treatment change, or clinical deterioration that led to discontinuation of therapy, as documented in the electronic medical record. OS was calculated from the start of systemic treatment of metastatic or recurrent disease to the date of death or last follow-up. Patient data were censored at the last follow-up visit or on September 1, 2017, if still alive.
χ2 and Kruskal-Wallis analyses were used to assess differences in baseline characteristics between the mutation groups for categoric and continuous variables, respectively. Cox proportional hazard models were used to determine the relationship of STK11 co-mutations to survival. Kaplan-Meier method was used to estimate OS and PFS, and comparisons between groups were made using the log-rank test. The multivariable Cox regression models were selected by stepwise forward selection, and P < .2 was used for initial inclusion. Candidate models were refined using the likelihood ratio test for individual variables. Given the small sample size, a model with fewer covariables was selected if additional variables did not significantly change the model. The effect of co-mutation status on PFS and OS was investigated by looking at four mutation groups (STK11 alone, STK11/KRAS, STK11/TP53, and STK11/KRAS/TP53) separately as well as at individual mutation effects and interactions in a Cox regression model. HRs from the Cox model were reported.
RESULTS
Baseline Characteristics
During a 42-month period, 1,385 unique patients had sequencing of a lung neoplasm in either tissue (n = 1,526 samples) or plasma (n = 245 samples). A total of 77 patients (6%) harbored an STK11 mutation (n = 56, tissue; n = 21, plasma). Fifteen patients were excluded (Fig 1). The majority (51 of 62, or 82%) of patients received platinum doublet–based therapy as the first-line regimen (Table 1). Nine patients had a driver mutation and received targeted therapy at some point during treatment (Data Supplement). Five patients received immunotherapy as first-line systemic therapy (Data Supplement). Among the 62 included patients, 44 had tissue NGS, and 18 had plasma NGS testing (Fig 1). The baseline characteristics were well balanced among these co-mutation groups, except that patients in the STK11 alone or STK11/KRAS/TP53 group were slightly older (P = .015; Table 1). There was no statistically significant difference in the proportion of KRAS or TP53 alterations detected by tissue versus plasma testing (Pearson’s χ2 test; P = .34 and P = .51, respectively).
FIG 1.

Flowchart of the study cohort. NGS, next-generation sequencing; NSCLC, non–small-cell lung cancer.
TABLE 1.
Baseline Characteristics of Patients With STK11 Mutations by Co-Mutation Status

Mutation Characteristics
A total of 46 (74%) of 62 STK11 mutants were confirmed as disease associated (DA-STK11), as defined by the sequencing report. STK11ex1-2 mutations were found in 22 patients, and 40 patients had STK11ex3-9 mutations (Data Supplement). The most common STK11 mutation was p.L282Afs*3, which resulted in a frameshift mutation in exon 6 (Fig 2).
FIG 2.
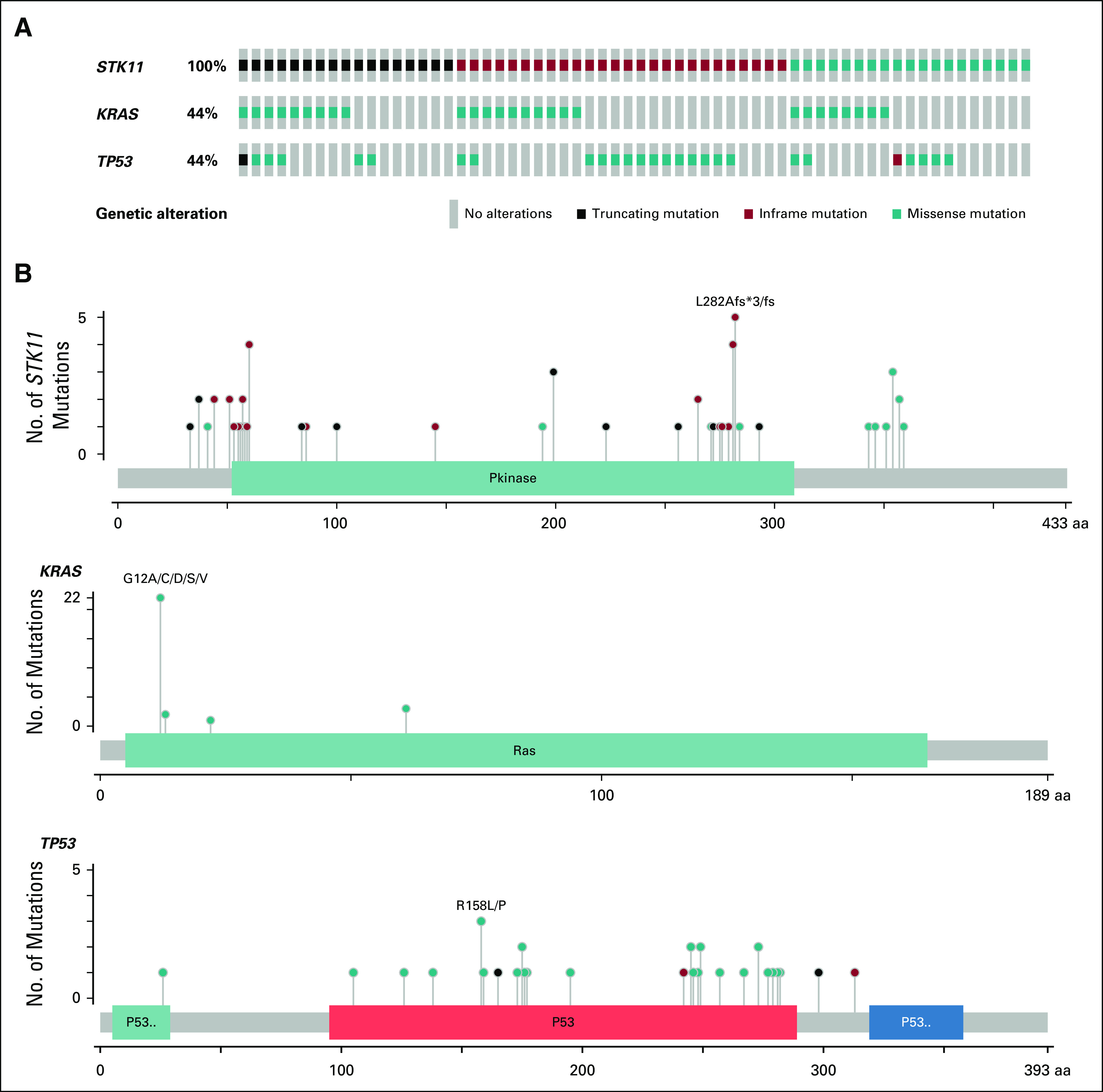
Distribution of STK11, KRAS and TP53 mutations. (A) Columns represent individual patients with mutation type specified by color; missense mutations in STK11 were found in six patients, but specific point mutations were not identified. Five missense mutations were in splice sites, and one was a deletion in exon 5. (B) Lollipop plots mapping specific mutation location (x-axis) and frequency (y-axis) for STK11, KRAS and TP53. aa, amino acids.
There was no correlation between the position of the STK11 mutation and co-mutation status. Among the 22 STK11ex1-2 mutations identified, 17 had additional co-mutations (n = 8, KRAS; n = 6, TP53; n = 3, KRAS/TP53). There was also no correlation between STK11ex1-2 or STK11ex3-9 and the presence of a KRAS or TP53 mutation (χ2 P = .34 and P = .637, respectively). KRAS alterations occurred at codon positions 12, 13, 22, and 61, and each was considered disease associated by pathology report using publicly available databases. The most frequent mutation seen in TP53 was a missense alteration of R158L or P (Fig 2).
Prognostic Relevance of Co-Mutation Status Among Patients With STK11 Mutations
Patients with STK11/KRAS co-mutations had a worse median PFS (2.4 months) compared with STK11 alone (5.1 months; log-rank P = .048), STK11/TP53 (4.3 months; log-rank P = .043), and STK11/KRAS/TP53 (13 months; log-rank P = .03; Table 2; Fig 3A). In an unadjusted, univariable Cox proportional hazards model of PFS, male sex was the only factor independently associated with an increased risk of progression (HR, 1.82; 95% CI, 1.04 to 3.17; P = .035; Tables 3 and 4). This effect persisted in the multivariable model after the analysis was controlled for DA-STK11 mutations, KRAS mutations, and TP53 mutations (HR, 2.08; 95% CI, 1.15 to 3.78; P = .016). In the multivariable model, the interactions between DA-STK11 and KRAS or DA-STK11 and TP53 were not statistically significant. However, when the interaction between DA-STK11 and KRAS was included in the model, there was an increased risk of progression among patients with KRAS/DA-STK11 mutations compared with the KRAS/non–DA-STK11 group (HR, 2.03; 95% CI, 1.05 to 3.92; P = .035; Tables 3 and 4). There was no change in risk of progression among patients with TP53/DA-STK11 compared with TP53/non–DA-STK11.
TABLE 2.
Median PFS and OS for STK11 Co-Mutation Groups
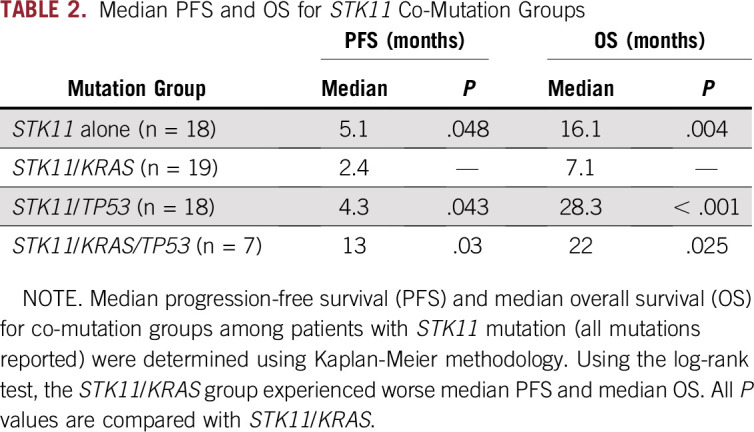
FIG 3.
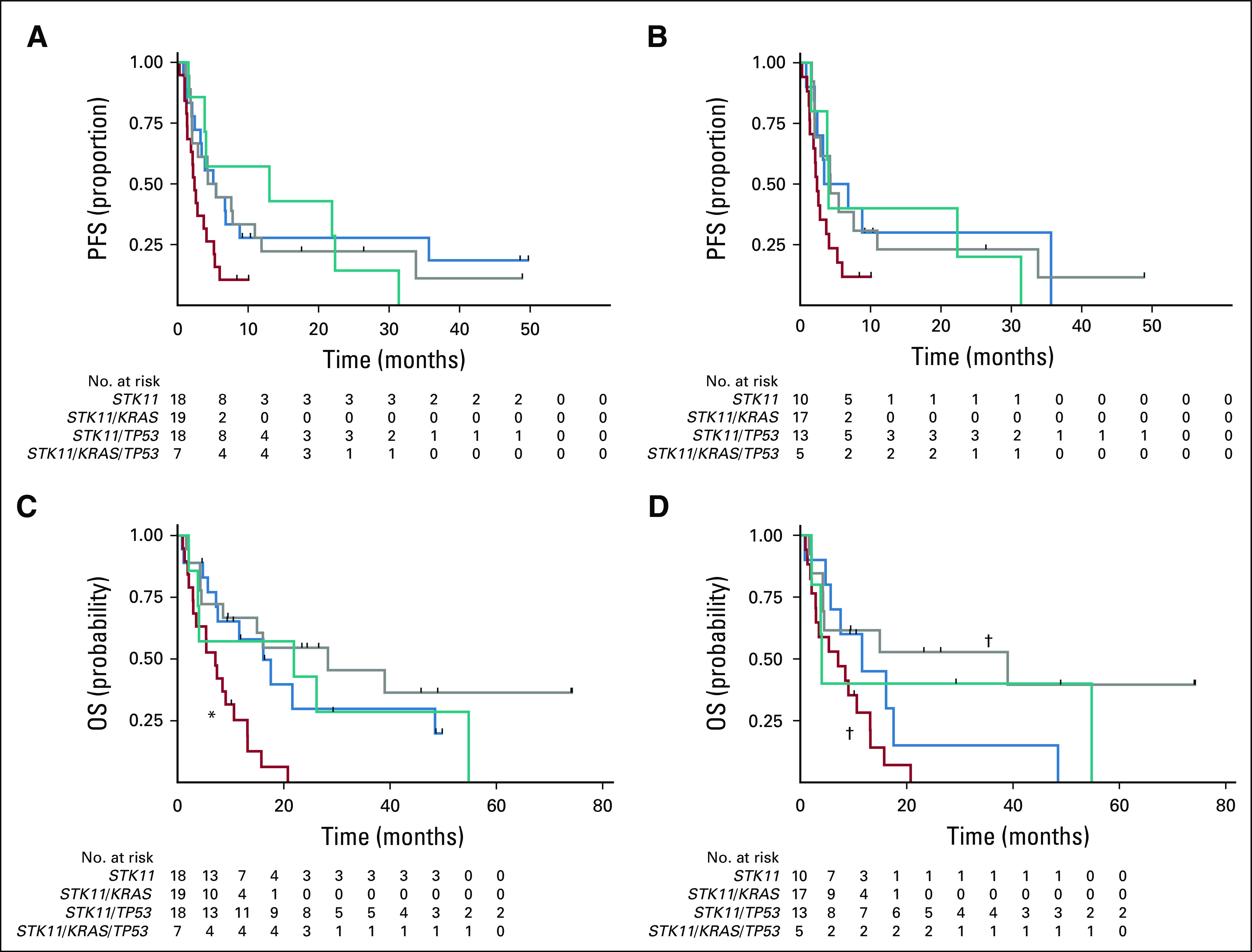
Progression-free survival (PFS) and overall survival (OS) by STK11 co-mutation status. Kaplan-Meier curves of (A) PFS and (C) OS of patients with stage IV or recurrent disease and tumors with STK11 mutation. (*) STK11/KRAS versus STK11/KRAS/TP53, log-rank P = .03. Kaplan-Meier curves of (B) PFS and (D) OS of patients with stage IV or recurrent disease and tumors with disease-associated STK11 mutation. (†) STK11/KRAS versus STK11/TP53, log-rank P = .01.
TABLE 3.
Univariable and Multivariable Models of PFS

TABLE 4.
Univariable and Multivariable Models of OS

Patients with STK11/KRAS mutations experienced shorter median OS (7.1 months) compared with STK11 alone (16.1 months; log-rank P < .001), STK11/TP53 (28.3 months; log-rank P < .001), and STK11/KRAS/TP53 (22 months; log-rank P = .025; Table 2; Fig 3C). Male sex conferred an increased risk of death in the univariable and multivariable models of OS (Tables 3 and 4). KRAS mutations were associated with an increased risk of death in the univariable model (HR, 2.46; 95% CI, 1.4 to 4.5; P = .003) but not in the multivariable model (Tables 3 and 4). TP53 mutation was associated with a decreased risk of death in the univariable analysis (HR, 0.48; 95% CI, 0.25 to 0.91; P = .025) but not in the multivariable analysis. As was the case with PFS, the interaction between KRAS and DA-STK11 was not significant on its own for OS, but patients who had a KRAS mutation and a DA-STK11 mutation had an increased risk of death compared with patients who had a KRAS mutation and a non–DA-STK11 mutation (HR, 2.18; 95% CI, 1.08 to 4.4; P = .031). Interestingly, we did not find that STK11ex1-2 mutations were associated with an increased risk of progression or death compared with STK11ex3-9 mutations, in contrast to the recent report from Pécuchet et al6 (Appendix Fig A1). Median PFS and OS did not significantly differ according to the source of NGS (tissue v plasma; Tables 3 and 4; Data Supplement). Other co-factors considered in the analysis but found not to be independent predictors in the univariable model or significant contributors to the multivariable model were smoking status, performance status at diagnosis, age at diagnosis, stage at presentation, and race/ethnicity (Tables 3 and 4). Analyses that excluded patients who received immunotherapy or targeted therapy still showed superior outcomes for patients with STK11/TP53 co-mutations, even in the presence of a KRAS mutation (Data Supplement).
Relevance of Disease-Associated Variants of STK11
As part of an exploratory subset analysis, we repeated the PFS and OS Kaplan-Meier analyses using only the STK11 mutations characterized as disease associated (DA-STK11). When only DA-STK11 mutations were included, there was no difference in median PFS between co-mutation groups of interest (Fig 3B). There remained a significant difference in median OS when STK11/KRAS and STK11/TP53 were compared (7.1 months v 39 months; log-rank P = .01; Fig 3D).
DISCUSSION
In the era of precision medicine, it has become increasingly important to understand the full implications of an ever-increasing quantity of tumor genetic information obtained as part of routine sequencing of NSCLCs. This study includes one of the largest cohorts of patients with STK11 mutations (n = 62), and to our knowledge it is the only study to specifically evaluate STK11 co-mutations with TP53 and KRAS and their relationship to response to first-line systemic therapy in patients with metastatic or recurrent disease. The results show that STK11/KRAS co-mutation is associated with a worse median PFS and OS after front-line chemotherapy compared with patients who had STK11 mutation alone, whereas patients who had the STK11/TP53 co-mutation had improved outcomes.
We found that, in the context of an STK11 mutation, TP53 mutation is associated with better outcomes even in the presence of mutant KRAS. The STK11/TP53 and STK11/KRAS/TP53 co-mutation groups had superior PFS and OS compared with the STK11/KRAS group. Of interest, when examined outside the context of STK11, TP53 mutations reportedly have a deleterious effect on OS and response to platinum-based therapy, especially in early-stage disease.19-22 TP53 has not previously been found to be predictive or prognostic in the presence of a KRAS mutation.9,10
Mutations in the TP53 binding sites of the STK11 promoter have been associated with decreased STK11 expression in endometrial cancer.15 In NSCLC, 82% of TP53 mutations are in the DNA binding region; therefore, a mutation in TP53 in NSCLC could lead to decreased expression of a deleterious STK11 protein, such as a gain-of-function STK11 mutation in exons 1 to 2.6,23 However, we observed a survival benefit for TP53 co-mutation with STK11 that was independent of the location of the STK11 mutation. The significance of STK11 mutation location and potential interactions with co-mutations are still poorly understood and should be explored in a larger study.
There was no difference in outcomes when patients with STK11ex1-2 and STK11ex3-9 mutations were compared. The proportion of patients with STK11 mutations in exons 1 and 2 in our cohort was similar to that of the cohort described by Pécuchet et al6 (35% v 25%), who came to a different conclusion. However, the study populations differed slightly. Although some patients in our study had early-stage progression (37.1%), patients with early-stage nonprogressive disease and patients who never received any systemic therapy were formally excluded (Fig 1). Therefore, if STK11ex1-2 mutations do confer a higher risk of recurrence after early-stage disease, we would not have been able to identify this risk, given the study design.
In the context of an STK11 mutation, we found that KRAS mutation in the absence of TP53 co-mutation conferred a significantly worse PFS and OS after first-line systemic therapy for metastatic or recurrent disease. Facchinetti et al3 also found that STK11/KRAS co-mutated tumors had a higher metastatic burden and a trend toward worse OS. This deleterious interaction between STK11 and KRAS may be explained by previous data showing that STK11 mutations enhance KRAS mutation–associated gene expression.11 In theory, this interaction would lead to augmentation of downstream KRAS signaling driving tumorigenesis. This is also supported by the observation that an acceleration of KRAS-induced tumorigenesis and metastasis has been found in STK11-null mice as well as in humans who lack STK11 expression.4,15
In a separate study, Arbour et al9 found that KRAS/STK11 co-mutations were associated with shorter OS in univariable analysis but not in multivariable analysis. In their cohort, STK11 co-mutation status with KEAP1 or NFE2L2 could have contributed to a shorter OS.9 KEAP1/NFE2L2 co-mutation occurred in 63% (60 of 95) of STK11 mutations in their cohort and was highly correlated with the KRAS/STK11 subgroup in another study.9,10 They did not report a correlation between KEAP1/NFE2L2 and TP53 mutations. The tumor and plasma NGS panels reported in our study did not include KEAP1 or NFE2L2 mutations, so we were unable to assess the effect of these mutations on outcome.
Detection of KRAS and TP53 mutations via plasma or tissue raises the possibility that the detected mutations may be due to clonal hematopoiesis (CH) in the blood. In another series, five of 33 TP53 mutations detected by plasma NGS were found in peripheral-blood cells but not in the tumor.24 The same series reported that most JAK2, some TP53, and rare KRAS mutations detected in cell-free DNA are from CH and not from the tumor. In our cohort, there was no significant difference in the proportion of TP53 or KRAS mutations detected in tumor versus plasma (Table 1). According to the series by Hu et al,24 it is possible that approximately one of the seven TP53 mutations detected by plasma testing was from CH; even if true, this small proportion is unlikely to change our results. In addition, CH is associated with worse outcome after therapy, and we report better outcomes with a TP53 mutation.25 Therefore, this possible misclassification would bias our result toward the null and imply that the observed association may be stronger than reported.26
Our study has additional limitations that must be addressed. First, this analysis is based on a relatively small cohort of patients, and the results must be validated in a larger study. Second, given the retrospective nature of this study, we used a real-world measurement of PFS defined as time from the start of treatment until radiologic progression, clinical deterioration, death, or change of therapy. This has been shown to be an appropriate surrogate for Response Evaluation Criteria in Solid Tumors (RECIST)–based PFS used in clinical trials.27
Many commercially available assays do not disclose how they determine whether an alteration is disease associated; thus, there may be variation among vendors in how they categorize mutations. In addition, studies that look to characterize the prognostic or predictive significance of mutations in a specific gene have used different definitions of mutation (eg, nonsynonymous, pathogenic). We initially used all nonsynonymous mutations in STK11 but then performed a subset analysis using only disease-associated STK11 mutations (ie, mutations classified as disease associated or pathogenic on the molecular report). With the disease-associated categorization of STK11 mutations, there was no longer a difference in median PFS between co-mutation groups. Importantly, there was still a significant difference in median OS between STK11/KRAS and STK11/TP53. This analysis is limited by the small sample size—only five patients were in the DA-STK11/KRAS/TP53 co-mutation group—and so should be considered exploratory. Future work must be done to standardize how classification of molecular alterations and identification of mutations that influence response to therapy and prognosis.
In summary, this study shows that the co-mutation status of STK11-mutated NSCLC contributes to the heterogeneity of this molecular subgroup. The study also highlights the need for a more complete understanding of the biologic interplay that multiple, seemingly unrelated mutations have on prognosis and response to therapy.
Appendix
Fig A1.
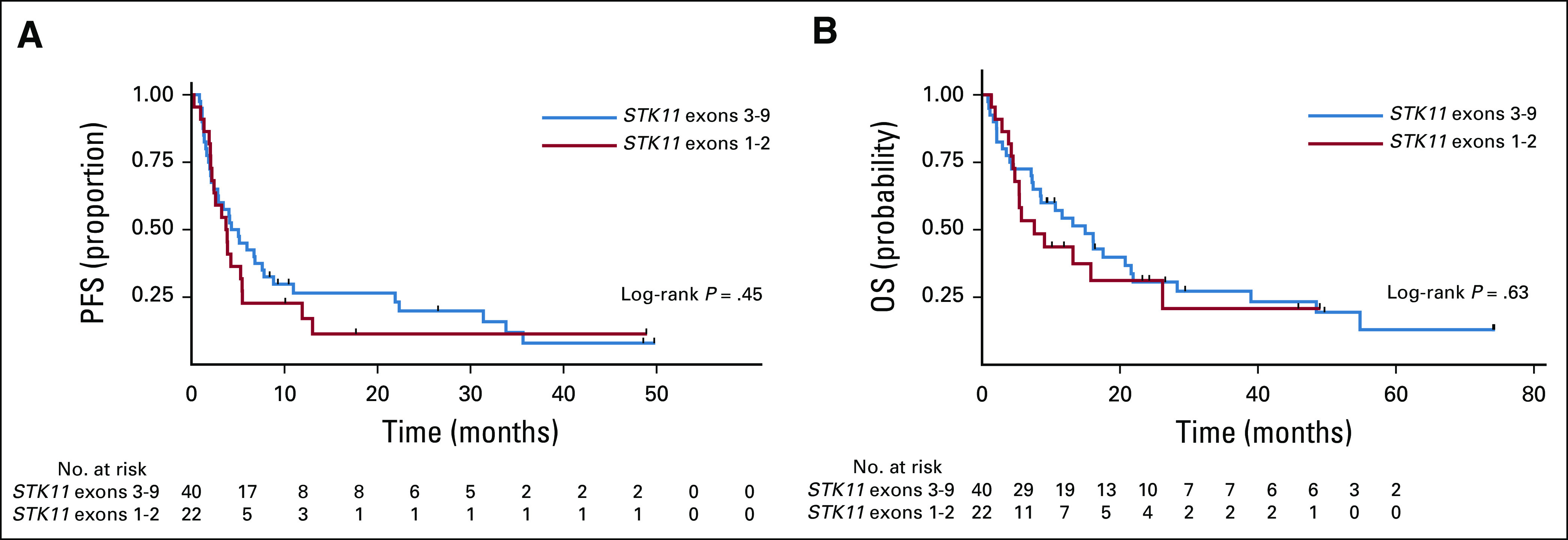
Kaplan-Meier curves of (A) progression-free survival (PFS) and (B) overall survival (OS) of patients with stage IV or recurrent disease and tumors with STK11 mutations stratified by STK11 mutation location.
Footnotes
Presented in part at the Annual Meeting of the American Association for Cancer Research, Chicago, IL, April 14-18, 2018.
AUTHOR CONTRIBUTIONS
Conception and design: Erin Bange, Melina E. Marmarelis, Corey J. Langer, Charu Aggarwal
Collection and assembly of data: Erin Bange, Melina E. Marmarelis, Jeffrey C. Thompson, Evan W. Alley, Erica Carpenter, Charu Aggarwal
Provision of study material or patients: Joshua M. Bauml, Christine Ciunci, Evan W. Alley
Data analysis and interpretation: Erin Bange, Melina E. Marmarelis, Wei-Ting Hwang, Yu-Xiao Yang, Jeffrey C. Thompson, Jason Rosenbaum, Joshua M. Bauml, Christine Ciunci, Roger B. Cohen, Corey J. Langer, Erica Carpenter, Charu Aggarwal
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Melina E. Marmarelis
Stock and Other Ownership Interests: Gilead Sciences, Portola Pharmaceuticals, Merck, Bluebird Bio
Consulting or Advisory Role: Boehringer Ingelheim
Other Relationship: Novartis
Research Funding: Lilly (Inst)
Travel, Accommodations, Expenses: Trizell
Wei-Ting Hwang
Research Funding: Janssen (I)
Jeffrey C. Thompson
Consulting or Advisory Role: OncoCyte
Jason Rosenbaum
Consulting or Advisory Role: Genentech, Roche, AbbVie, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Agilent, Bristol-Myers Squibb, Takeda, AbbVie, Genentech, Roche
Joshua M. Bauml
Consulting or Advisory Role: Bristol-Myers Squibb, Merck, AstraZeneca, Genentech, Celgene, Boehringer Ingelheim, Guardant Health, Takeda
Research Funding: Merck (Inst), Carevive Systems (Inst), Novartis (Inst), Incyte (Inst), Bayer (Inst), Janssen (Inst), AstraZeneca (Inst), Takeda (Inst)
Christine Ciunci
Research Funding: Celgene (Inst), Merck (Inst)
Roger B. Cohen
Consulting or Advisory Role: Heat Biologics, Takeda, Alkermes, Kyn Therapeutics, Innate Pharma, Cantargia AB, Genocea Biosciences
Research Funding: Heat Biologics (Inst), Merck (Inst), Celldex (Inst), Innate Pharma (Inst), Kyn Therapeutics (Inst), Xencor (Inst), Genocea Biosciences (Inst)
Travel, Accommodations, Expenses: Heat Biologics, Takeda, Innate Pharma, Kyn Therapeutics, Alkermes
Corey J. Langer
Honoraria: Bristol-Myers Squibb, Genentech, Roche, Lilly, ImClone
Consulting or Advisory Role: Genentech, Roche, Lilly, ImClone, Merck, Abbott Biotherapeutics, Bayer, Onyx, Clarient, Clovis Oncology, Celgene, Cancer Support Community, Bristol-Myers Squibb, ARIAD, Takeda, AstraZeneca, Pfizer, Novocure
Research Funding: Merck (Inst), Advantagene (Inst), Clovis Oncology (Inst), Celegene (Inst), Inovio Pharmaceuticals (Inst), ARIAD (Inst), GlaxoSmithKline (Inst), Genentech (Inst), Roche (Inst), Stem CentRx (Inst), Lilly (Inst)
Other Relationship: Lilly, Amgen, Peregrine Pharmaceuticals, Synta
Erica Carpenter
Honoraria: AstraZeneca
Research Funding: Merck, Janssen, Becton Dickinson
Patents, Royalties, Other Intellectual Property: Invention disclosure entitled, “Methods and Compositions for Treating Neuroblastoma”; invention disclosure entitled, “Methods and Compositions for Identifying, Diagnosing and Treating Neuroblastoma”
Travel, Accommodations, Expenses: AstraZeneca, Foundation Medicine
Charu Aggarwal
Consulting or Advisory Role: Genentech, Bristol-Myers Squibb, Lilly, Celgene, MedImmune
Research Funding: Genentech (Inst), Roche (Inst), Incyte (Inst), Macrogenics (Inst), Merck Sharp & Dohme (Inst), AstraZeneca (Inst), MedImmune (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Cheng TYD, Cramb SM, Baade PD, et al. The international epidemiology of lung cancer: Latest trends, disparities, and tumor characteristics. J Thorac Oncol. 2016;11:1653–1671. doi: 10.1016/j.jtho.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsumoto S, Iwakawa R, Takahashi K, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911–5918. doi: 10.1038/sj.onc.1210418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Facchinetti F, Bluthgen MV, Tergemina-Clain G, et al. LKB1/STK11 mutations in non–small-cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer. 2017;112:62–68. doi: 10.1016/j.lungcan.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Gill RK, Yang SH, Meerzaman D, et al. Frequent homozygous deletion of the LKB1/STK11 gene in non–small-cell lung cancer. Oncogene. 2011;30:3784–3791. doi: 10.1038/onc.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 6.Pécuchet N, Laurent-Puig P, Mansuet-Lupo A, et al. Different prognostic impact of STK11 mutations in non-squamous non–small-cell lung cancer. Oncotarget. 2017;8:23831–23840. doi: 10.18632/oncotarget.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahmani R, Just PA, Delay A, et al. A novel LKB1 isoform enhances AMPK metabolic activity and displays oncogenic properties. Oncogene. 2015;34:2337–2346. doi: 10.1038/onc.2014.182. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Cheng K, Walton Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arbour KC, Jordan E, Kim HR, et al. Effects of co-occurring genomic alterations on outcomes in patients with KRAS-mutant non–small-cell lung cancer. Clin Cancer Res. 2018;24:334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schabath MB, Welsh EA, Fulp WJ, et al. Differential association of STK11 and TP53 with KRAS mutation–associated gene expression, proliferation, and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35:3209–3216. doi: 10.1038/onc.2015.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karuman P, Gozani O, Odze RD, et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell. 2001;7:1307–1319. doi: 10.1016/s1097-2765(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 14.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Co NN, Iglesias D, Celestino J, et al. Loss of LKB1 in high-grade endometrial carcinoma: LKB1 is a novel transcriptional target of p53. Cancer. 2014;120:3457–3468. doi: 10.1002/cncr.28854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res. 2016;22:5772–5782. doi: 10.1158/1078-0432.CCR-16-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deben C, Deschoolmeester V, Lardon F, et al. TP53 and MDM2 genetic alterations in non–small-cell lung cancer: Evaluating their prognostic and predictive value. Crit Rev Oncol Hematol. 2016;99:63–73. doi: 10.1016/j.critrevonc.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Ahrendt SA, Hu Y, Buta M, et al. p53 mutations and survival in stage I non–small-cell lung cancer: Results of a prospective study. J Natl Cancer Inst. 2003;95:961–970. doi: 10.1093/jnci/95.13.961. [DOI] [PubMed] [Google Scholar]
- 21.Mitsudomi T, Hamajima N, Ogawa M, et al. Prognostic significance of p53 alterations in patients with non–small-cell lung cancer: A meta-analysis. Clin Cancer Res. 2000;6:4055–4063. [PubMed] [Google Scholar]
- 22.Kandioler D, Stamatis G, Eberhardt W, et al. Growing clinical evidence for the interaction of the p53 genotype and response to induction chemotherapy in advanced non–small-cell lung cancer. J Thorac Cardiovasc Surg. 2008;135:1036–1041. doi: 10.1016/j.jtcvs.2007.10.072. [DOI] [PubMed] [Google Scholar]
- 23.Shajani-Yi Z, de Abreu FB, Peterson JD, et al. Frequency of somatic TP53 mutations in combination with known pathogenic mutations in colon adenocarcinoma, non–small-cell lung carcinoma, and gliomas as identified by next-generation sequencing. Neoplasia. 2018;20:256–262. doi: 10.1016/j.neo.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Y, Ulrich BC, Supplee J, et al. False positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437–4443. doi: 10.1158/1078-0432.CCR-18-0143. [DOI] [PubMed] [Google Scholar]
- 25.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374–382e4. doi: 10.1016/j.stem.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauml J, Levy B. Clonal hematopoiesis: A new layer in the liquid biopsy story in lung cancer. Clin Cancer Res. 2018;24:4352–4354. doi: 10.1158/1078-0432.CCR-18-0969. [DOI] [PubMed] [Google Scholar]
- 27.Bartlett C, Mardekian J, Cotter M, et al. Concordance of real-world progression-free survival (PFS) on endocrine therapy as first line treatment for metastatic breast cancer using electronic health record with proper quality control versus conventional PFS from a phase 3 trial. Cancer Res. 2018;78 [Google Scholar]