

Abstract
Recent studies reported the development of biocatalytic heme carbenoid Si-H insertions for the selective formation of carbon-silicon bonds, but many mechanistic questions remain unaddressed. To this end, a DFT mechanistic investigation was performed which reveals an FeII-based concerted hydride transfer mechanism with early transition state feature. The results from these computational analyses are consistent with experimental data of radical trapping, kinetic isotope effects, and structure-reactivity data using engineered variants of hemoproteins. Detailed geometric and electronic profiles along the heme catalyzed Si-H insertion pathways were provided to help understand the origin of experimental reactivity trends. Quantitative relationships between reaction barriers and some properties such as charge transfer from substrate to heme carbene and Si-H bond length change from reactant to transition state were found. Results suggest catalyst modifications to facilitate the charge transfer from the silane substrate to the carbene, which was determined to be a major electronic driving force of this reaction, should enable the development of improved biocatalysts for Si-H carbene insertion reactions.
Keywords: biocatalysis, silanes, carbenoids, heme proteins, density functional calculations
Graphical Abstract
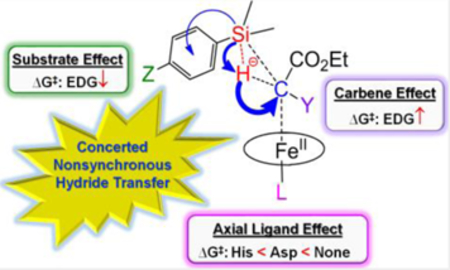
An FeII-based concerted hydride transfer mechanism with nonsynchronous and early transition state was found for biocatalytic heme carbenoid Si-H insertions, with key insights into effects of substrates, carbenes, and protein axial ligands to help understand experimental structure-reactivity trends and facilitate design of improved (bio)catalysts for challenging Si-H insertion reactions.
Introduction
Artificial hemoproteins have been proven useful for promoting a broad range of carbene-mediated transformations, including carbene insertion into X-H bonds (X = N, S, Si, C, B),[1] cyclopropanations,[2] aldehyde olefinations,[3] and sigmatropic rearrangements.[4] Among these reactions, the synthesis of organosilicon compounds find important applications in the area of material science[5] and medicinal chemistry.[6] While synthetic methodologies for the transition metal-catalyzed carbene insertion into Si-H bonds have been reported,[7] these protocols require organic solvents and are typically characterized by limited catalytic turnovers (<100). Recently, biocatalytic methods for accomplishing this type of transformations in aqueous solvents and under mild reaction conditions were reported using engineered variants of cytochrome c[1c] and myoglobin.[1h] These systems offer several attractive features for sustainable chemistry due to the accessibility and biocompatibility of these iron-based metalloprotein biocatalysts, along with their promising catalytic activity in these transformations (>1,500–8,000 catalytic turnovers).[1c, 1h]
Despite this progress, many mechanistic questions remain unanswered. For instance, what is the major electronic driving force for this reaction? What is the origin of the experimentally observed substrate and carbene reactivity trends? What kind of roles do conformation and spin state play on the transition state (TS)? What factors are critical to catalyst design? Computational work on Si-H insertion is scarce[8] and detailed analyses of geometric or electronic property changes along these metal-carbenoid Si-H insertion pathways have been missing.
To address these mechanistic questions, a quantum chemical investigation of heme-carbenoid Si-H insertions was performed here, using basically the same computational approach in recent accurate predictions of iron porphyrin carbenes’ (IPCs) experimental X-ray crystal structures, Mössbauer and NMR properties, and their experimental formation, cyclopropanation, and C-H insertion reactivity and selectivity results,[9] see details in Experimental section. Reproducing experimentally observed structure-activity trends and kinetic isotope effects (KIEs), our computational analyses provide key insights into the basic mechanism of heme-catalyzed Si-H carbene insertion and into the impact of structural changes at the level of the substrate, carbene, and heme catalyst on this reactivity.
Results and Discussion
Basic mechanism
Since the previously reported active biocatalysts for this reaction (i.e., cytochrome c and myoglobin) both contain a histidine-ligated heme cofactor,[1c, 1h] [Fe(Por)(His)(C(Me)CO2Et)] was first investigated to model the core part of these biocatalysts in the Si-H carbene insertion reaction 1 of Scheme 1. Por is a non-substituted porphyrin, His is modeled as 5-methylimidazole as done previously,[9b-d] and C(Me)CO2Et is the carbene group derived from the corresponding diazo ester reagent.[1c] This carbene structure was also confirmed in a recent X-ray crystallographic study.[8c] Since there is no prior computational study of effects of substrates, carbenes, and catalysts on heme carbenoid Si-H insertions, this work focuses on the Si-H insertion reactivities of the core part of the protein due to these effects.
Scheme 1.

Si-H insertion pathways in reactions 1–5.
Both a concerted mechanism as proposed for other metal carbenoid Si-H insertions[7b, c, 8b, 10] and a stepwise radical mechanism reminiscent of heme-enzyme catalyzed oxidation reactions[11] were investigated for reaction 1 to explore the basic mechanism, with details in Supporting Information (SI) and key results discussed here.
Comparisons of different mechanisms, conformations, and spin states.
We first studied the concerted transition state. Because electronic state and conformation may influence the mechanism, such effects were examined first to identify the most favorable conformations and spin states, see SI for details. Overall, the conformation effect was found to be small. For example, the Gibbs free energy differences for different conformations of TS(1) (the reaction number is in parenthesis) (see Figure S1) are within ~ 0.3 kcal/mol, indicating a weak steric interaction due to the long C…Si distance (~3.1 Å).
In contrast, the spin state effect on mechanism is much larger. Here, we focused on both singlet and quintet transition states because the iron-containing product is of quintet ground state,[12] which is different from the reactant’s singlet ground state for various IPCs.[13] These experimental ground states of reactants and products were reproduced in our calculations.[9] Regarding concerted TS(1), the FeII-based quintet transition state has Fe spin density of 3.885 e and carbene’s carbon spin density of only −0.165 e. It has significantly higher energy (ΔΔG‡ of 10.27 kcal/mol) than the FeII-based closed-shell singlet (Table S4). In contrast, the third TS spin state with S=1 is of the highest energy, by ΔΔG‡ of 12.81 kcal/mol (Table S4). These results support the singlet TS(1) as the most favorable spin state, and therefore there is no spin crossover from the singlet reactant. The results obtained for the other reactions considered here (Scheme 1) also indicated that the singlet state TS is the most favorable transition state (see SI for details).
Compared with such FeII-based concerted transition states, for the radical mechanism, the initial broken-symmetry setup of FeIII (S=1/2), carbene (S=−1/2), H (S=1/2) from the Si-H bond, and the remaining substrate (S=−1/2) for the overall singlet transition state was optimized the same as FeII-based concerted TS(1) described above. Similarly the initial broken-symmetry setup of FeIII (S=5/2), carbene (S=−1/2), H (S=1/2), and the remaining substrate (S=−1/2) for the overall quintet radical-based transition state was also optimized to the same quintet concerted TS(1) mentioned above. In contrast with these results at singlet and quintet levels, the triplet TS(1) has obvious radical feature with 1.092, 0.558, and 0.377 e spin densities for Fe, the carbene α-carbon atom, and Si, respectively. However, it has the highest Gibbs free energy of activation, with ΔΔG‡ of 12.81 kcal/mol larger than that of the lowest energy TS(1), see Table S4.
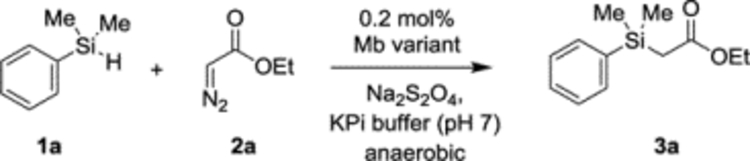
In previous work, an engineered active site variant of sperm whale myoglobin, Mb(H64V,V68A), was determined to catalyze a Si—H carbene insertion reaction between dimethyl(phenyl)silane (1a) and ethyl α-diazoacetate (2a) with up to 1,545 total turnovers.[1h] To probe experimentally the occurrence of a radical mechanism in this hemoprotein-catalyzed Si-H insertion process, the reaction was performed in the absence and in the presence of a large excess (100 mM) of the spin trap reagent 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). As shown in Scheme 2a, the addition of DMPO did not have a significant impact on the yield of the Mb(H64V,V68A)-catalyzed Si-H insertion reaction (52 ± 5% vs. 45 ± 7%), suggesting the absence of a radical intermediate. To gain further insights into the mechanism of this reaction, kinetic isotope effect (KIE) experiments were performed using a deuterated form of the dimethyl(phenyl)silane substrate (d-1a). Competition experiments with equimolar amount of 1a and d-1a yielded a positive KIE (kH/kD) corresponding to 1.19 ± 0.05 (Scheme 2b and Figure S4). A similar KIE value of 1.3 ± 0.2 was also obtained by comparing the rate of parallel reactions with the protiated vs. the deuterated silane substrate (Figure S5). These results indicate that the cleavage of the Si—H bond is part of the rate-limiting step of the reaction. Importantly, using the concerted TS(1), the calculated KIE at room temperature for reaction 1 and other reactions discussed later (2 and 3) using the same catalyst are 1.19, 1.26, and 1.31, respectively, thus showing an excellent agreement between the computational predictions and the experimental data obtained with the Mb variant. The KIE values obtained for the hemoprotein-catalyzed reaction fall in the same range of those reported for Rh- and Ir-carbenoid Si-H insertions (1.08–1.6).[7b, 10]
Scheme 2.

Radical trap (a) and kinetic isotope effect experiments (b) for Mb(H64V,V68A)-catalyzed Si-H carbene insertion reaction.
Taken together, the results above suggest that the heme carbene Si-H insertion favors an FeII-based concerted mechanism, rather than a stepwise radical mechanism. This FeII feature is consistent with experimental reaction studies where the starting heme proteins were reduced to the catalytically competent ferrous form,[1c, 2b, 2e] and experimental spectroscopic studies using UV/Vis and NMR spectroscopies,[13b, 14] X-ray crystallography,[13a, 14–15] and XANES (directly sensitive to Fe oxidation state)[15] which show that IPC has a dominant FeII feature (i.e. closed-shell singlet). The proposed concerted mechanism is reminiscent of that proposed for rhodium- and iridium-carbenoid Si-H insertions[7b, c, 8b, 10] and heme carbene cyclopropanations.[9d]
Major electronic driving force and hydride transfer feature.
Since charge and geometry changes along the reaction pathway have not been reported before for any metal-catalyzed carbenoid Si-H insertion, these parameters were examined here to gain further insights into the mechanism of this reaction.
A large negative charge transfer (QCT: −0.223 e) occurs from PhMe2SiH to carbene from reactants to TS(1), as shown by the data in Figure 1A and additional data in Figure S3 and Tables S7 and S8. As the spin densities of the atoms for the most favorable singlet TS(1) do not support an electron transfer mechanism (either coupled or uncoupled with proton transfer), the large negative charge transfer mentioned above suggests a hydride transfer feature. This is also the largest charge transfer in the system, indicating that it may represent a major electronic driving force for this reaction. The charge transfer direction shows the electrophilicity of the heme-carbene group, which is consistent with the experimentally observed lower and higher reactivity of electron-deficient and electron-rich silane substrates, respectively, in biocatalytic reactions.[1c] It is also interesting to note that while Si donates more charge (0.165 e, see data in blue in Figure 1A) to help the hydride formation for subsequent charge transfer, it also donates 0.040 e to the remaining part of substrate as a result of electronegativity difference[16] between C (2.55) and Si (1.90). Altogether, these effects result in a very large positive charge for the Si atom at the TS, corresponding to ~1.6 e (Table 1)
Figure 1.

Key geometric parameters at TS (in red), geometry changes (in green) and charge transfers (in blue) from reactants to TS in (A) reaction 1; (B) reaction 2; (C) reaction 3; (D) reactions 4; (E) reaction 5.
Table 1.
Key Energy, Charge, and Geometry Results for Si-H Insertions
| TTN | ΔG‡ (kcal/ mol) |
ΔG° (kcal/ mol) |
QCT (e) |
QSiTS (e) |
RSiHTS (Å) |
ΔRSiH (Å) |
|
|---|---|---|---|---|---|---|---|
| 1 | 2520 | 9.60 | −62.22 | −0.223 | 1.573 | 1.534 | 0.043 |
| 2 | 5010 | 8.40 | −62.05 | −0.215 | 1.569 | 1.531 | 0.040 |
| 3 | 47 | 11.74 | −64.02 | −0.261 | 1.616 | 1.551 | 0.060 |
| 4 | 10.87 | −76.32 | −0.246 | 1.594 | 1.542 | 0.051 | |
| 5 | 11.24 | −49.36 | −0.260 | 1.591 | 1.547 | 0.056 |
Nonsynchronous bond formation and early transition state features.
Inspection of the geometric data suggest that the concerted TS(1) has a nonsynchronous feature. This conclusion is supported by a more pronounced elongation of the Fe-C bond by 0.108 Å compared to that of the Si-H bond (0.043 Å) from reactants to transition state, as illustrated by the data in green in Figure 1A. In addition, the C-Si bond formation is relatively late compared to the C-H bond formation as indicated by the longer C-Si bond length of ~3.1 Å vs. C-H bond length of ~1.7 Å at the transition state (see data in red in Figure 1A).
Furthermore, the small change of Si-H bond length (ΔRSiH, ~0.05 Å) and the small change (0.4°) in the bond angle between the two carbene substituents at TS(1) compared to those in the reactants (see Table S7) suggest that TS(1) has an early transition state character. These features indicate that the carbene carbon in the transition state is still largely sp2-hydridized as in the reactant, rather than exhibiting sp3 geometry as in the product. Indeed, as illustrated in Figure 2A, the key interactions between the carbene α-carbon and the Si-H bond involve the carbon p orbital which has π bonding and anti-bonding interactions with iron’s dπ orbital in the occupied and unoccupied frontier molecular orbitals, together with the σ(Si-H) bond. The pπ-dπ bonding interaction in this transition state is similar to and originates from the pπ-dπ bonding interaction in the IPC reactant (see Figure 2B), which further supports its early transition state character. The presence of only one interaction between Fe dπ orbital and the carbene α-carbon pπ orbital and iron orbital population scheme of (dxy)2(dxz)2(dyz)2(dz2)0(dx2-y2)0 are the same as reported previously for other IPCs.[9a] These results indicate that the unique pπ-dπ bonding pattern in IPC is maintained in the TS investigated here and the pπ orbital of the carbene α-carbon interacts with the Si-H bond in this TS, suggesting an important role of this pπ-dπ bonding for catalysis.
Figure 2.

Frontier metal-containing molecular orbitals of TS(1) and R1(1). (A) from low to high are HOMO-2 and LUMO+2 orbitals with contour values of ±0.01 and 0.02 au; (B) from low to high are HOMO-5, HOMO-3, HOMO-2, LUMO+4, LUMO+9 with contour values of ±0.04 au. The navy blue dashed line separates the regions of occupied and unoccupied MOs. Red boxes show pπ-dπ interaction.
Substrate and carbene effects
We next studied the substrate and carbene substituent effects to provide a better theoretical understanding of the reactivity trends observed experimentally.[1c] In the cytochrome c-catalyzed Si-H insertion reactions reported by Arnold and coworkers, a total number of turnovers (TTN) of 2520 was measured with Ph(Me)2SiH and a :C(Me)CO2Et carbene (modelled in reaction 1, Scheme 1). The introduction of an electron-donating groups (EDG) in the phenyl ring of the silane substrate was found to enhance the yield of this reaction (e.g., 4-acetylenyl-Ph)Me2SiH: 5010 TTN (modelled in reaction 2, Scheme 1). On the other hand, a change of the α-substituent on the carbene from Me to Et was reported to cause a dramatic decrease in TTN (47) with the electron-rich silane substrate 4-acetylenyl-Ph)Me2SiH (modelled in reaction 3, Scheme 1).[1c] As shown in Table 1 and Figure 3, the calculated barriers for the corresponding reactions 1, 2 and 3 agree very well with the experimentally observed reactivity trend (i.e., 2 > 1 > 3). These results indicate that the main reactivity features observed for these biocatalytic reactions are reproduced by the calculations, which suggests that the relative experimental reactivity trend of different substrates and carbenes is influenced by the heme core part, although the protein environment can also affect the overall reactivity and stereoselectivity of the biocatalyst through residues surrounding the heme group.[8c, 17]
Figure 3.

Schematic free energy diagram for Si-H insertion reactions 1–5.
Interestingly, among reactions 1–3 (Scheme 1), the least efficient reaction 3 (based on TTN) is characterized by the largest charge and geometry changes (e.g. |QCT|: 3 (0.261 e) > 1 (0.223 e) > 2 (0.215 e); ΔRSiH: 3 (0.060 Å) > 1 (0.043 Å) > 2 (0.040 Å), see Table 1). This results in the largest energy cost for the early TS and thus in the highest energy barrier among these reactions. In addition, as shown in Figure 1A–C, the major electronic driving force in all these reactions is the charge transfer from substrate to carbene. This finding is consistent with the beneficial effect of the EDG on the silane substrate (reaction 2) toward reducing the barrier compared to the unsubstituted dimethyl(phenyl)silane substrate (reaction 1), as observed experimentally.[1c] These results provide insights into the mechanistic basis for the reactivity trends observed in the presence of electron-rich and electron-deficient silane substrates in biocatalytic heme carbene Si-H insertions.[1c]
Effect of proximal ligand substitutions
Modification of the axial ligand coordinating the heme cofactor has provided an effective strategy for modulating the reactivity of hemoproteins as biocatalysts for carbene transfer reactions.[1h, 2b, c, 2h, 9d] More recently, we established that substitution of the conserved histidine axial ligand (His93) with an Asp residue is well tolerated in the myoglobin scaffold and results in a functional biocatalyst for olefin cyclopropanation.[18] To assess the impact of this structural change on the Si-H insertion reactivity, the activity of Mb(H64V,V68A) and its proximal ligand variants Mb(H64V,V68A,H93D) and Mb(H64V,V68A,H93F) were compared in the model reaction with dimethyl(phenyl)silane and EDA. As shown by the results in Table 2, the Asp-containing variant Mb(H64V,V68A,H93D) was found to exhibit comparable (slightly lower) Si-H insertion reactivity to the His-containing counterpart, whereas a comparatively lower reactivity was observed for the variant lacking axial coordination (i.e., Mb(H64V,V68A,H93F).
Table 2.
Si-H carbene insertion activity of Mb(H64V,V68A) and proximal ligand variants thereof.a
 | ||||
|---|---|---|---|---|
| Entry | Protein | Proximal Ligand |
Yieldb | TON |
| 1 | Mb(H64V,V68A) | His | 69% | 345 |
| 2 | Mb(H64V,V68A,H93D) | Asp | 66% | 330 |
| 3 | Mb(H64V,V68A,H93F) | Phe | 50% | 250 |
Reaction conditions: 10 mM 1a, 10 mM EDA, 20 μM Mb variant in 50 mM phosphate buffer (pH 7), 1 hour, r.t., in anaerobic chamber.
Based on GC conversion using calibration curves with isolated 3a.
To examine the effect of related proximal ligand substitutions on the reactivity of the heme carbene toward Si-H insertion, reaction 4 (where Asp is as the axial ligand) and reaction 5 (no axial ligand) were computationally compared with reaction 1. As shown in Table 1, the His-to-Asp substitution is accompanied by a 1.27 kcal/mol increase in the ΔG‡ of the reaction (i.e. reaction 4 vs. 1). However, both of these reactions have a lower ΔG‡ compared to the reaction with no axial ligand (reaction 5). For these systems, detailed calculations included in SI (see Table S4) show that the singlet TS is again the most favorable TS compared to other spin states, justifying its use for these analyses. The higher reactivity of histidine- and aspartate-ligated heme carbene toward Si-H insertion compared to the no axial ligand case may arise from a weaker Fe-C bond (more elongated by ~0.05 Å at R1 and by ~0.07 Å at TS, Table S7) due to their trans effects,[19] thus facilitating carbene transfer. Overall, the reactivity trend predicted for these reactions (1 > 4 > 5) using the heme core part with :C(Me)CO2Et is in qualitative agreement with the reactivity trend observed experimentally with the Mb-based proximal ligand variants (His >≈ Asp > Phe; Table 2) using a similar carbene :C(H)CO2Et.
Insights into structural factors to improve catalytic reactivity
Interestingly, good correlations between ΔG‡ and QCT/RSiHTS (ΔRSiH also has R2=0.9415) were found across all the Si-H insertion reactions studied here (see Figure 4). These relationships further highlight the importance of carbene electrophilicity in this reaction and the early TS feature, since smaller charge transfers and geometry changes are associated with lower energy barriers.
Figure 4.

ΔG‡ vs. (A) QCT and (B) RSiHTS in Si-H insertions.
These results suggest that the introduction of stronger electron-withdrawing substituents on the carbene group, porphyrin, or axial ligand could facilitate the charge transfer from the silane substrate to the carbene (the major electronic driving force found here), resulting in improved catalytic performance, in particular for the more challenging electron-deficient substrates. These insights may inspire future experimental approaches for sustainable biocatalytic Si-H insertion reactions.
Conclusions
This work provides the first computational and experimental data to compare different mechanistic possibilities to determine the basic mechanism of biocatalytic hemoprotein-catalyzed Si-H insertion reactions, as well as the first theoretical insights into effects of experimental reactivity trends due to modifications on substrates, carbenes, and catalysts. Collectively, our results indicate an FeII-based, concerted, hydride transfer mechanism involving nonsynchronous Si-C/C-H bond formation and an early transition state, as supported by (a) a detailed analysis of different reaction systems, spin state and conformation effects and (b) an overall good agreement between the computational predictions with experimental KIEs and proximal ligand effects obtained with Mb variants here as well as structure-activity trends for carbene and substrate substituents previously reported with engineered cytochrome c variants.[1c] Changes in the geometric properties and charge transfers around the reaction center along the computed reaction pathways were helpful to understand the effect of modifications of the substrate, carbene, and axial ligand on Si-H insertion reactivity. In particular, the reactivity changes could be associated to effects on the electronic driving force and early transition state feature, as indicated by the quantitative correlation between reaction barriers and geometry/charge properties. Based on these mechanistic analyses, suggestions were given to help the future development of efficient biocatalytic methods for Si-H insertion reactions with challenging substrates.
Experimental Section
All calculations were performed using the program Gaussian 09.[20] All models investigated in this work were subject to full geometry optimizations without any symmetry constraints using the PCM method[21] with a dielectric constant of 4.0 to simulate the protein environment effect as done previously[22] in this computational study of heme carbenoid Si-H insertions, which focuses on the basic mechanism and core part of the protein. The frequency analysis was used to verify the nature of the stationary points on respective potential energy surfaces and to provide zero-point energy corrected electronic energies (EZPE’s), enthalpies (H’s), and Gibbs free energies (G’s) at 1 atm and experimental reaction temperatures, i.e. room temperature (r.t.). The atomic charges and spin densities reported here are from the Natural Population Analysis (NPA) and Mulliken schemes respectively, as implemented in Gaussian 09. Relative Gibbs free energies and selected geometry, charge, and spin density results were discussed in the main text, while all absolute values of electronic energies (E’s), zero-point energy corrected electronic energies (EZPE’s), enthalpies (H’s), Gibbs free energies (G’s), key geometric parameters, charges, spin densities, 3D structures and coordinates of optimized structures of the most favorable conformations and spin states as well as other details are in SI.
All calculations were done using a range-separated hybrid DFT method with dispersion correction, ωB97XD,[23] based on its excellent performance on heme carbenes and other catalytic systems from several methodological studies.[9a–c, 24] This ωB97XD method was found to yield accurate predictions of various experimental spectroscopic properties, structural features, and reactivity results of IPCs compared to some other functionals.[9] The basis set includes the effective core potential (ECP) basis LanL2DZ[25] for iron and the triple-zeta basis 6–311G(d) for all other elements, which was found to provide accurate predictions of various experimental reaction properties of heme carbenes.[9b–d] The use of a much larger 6–311++G(2d,2p) basis for all non-metal atoms was found to yield similar results for heme carbene reactions[9c] and thus further support the efficient use of the current basis set here. The use of an ECP basis for metal here is common in many reaction studies involving transition metal carbenoids, such as Ir porphyrin carbene,[7f] Ru porphyrin carbene,[26] Rh carbene.[27] The advantage of an ECP basis is the inclusion of relativistic effect basically absent in an all-electron basis set. In addition, it is available for all transition metals, which may allow direct comparisons of effects of a vast amount of metal centers. The alternative use of an all-electron basis for the metal center[9d] was recently found to yield qualitatively same conclusions of geometric, electronic, and energetic features for heme carbene reactions, and therefore supports the use of LanL2DZ basis here, which may help direct comparisons with late transition metals in future studies, for which ECP basis is more readily available and commonly used.
The KIE calculations with tunneling effect correction (KIEW) were done using the following formulae reported recently:[28]
| (1) |
| (2) |
| (3) |
where h is Planck’s constant, υ is the imaginary frequency of the transition state, k is Boltzmann’s constant, T is temperature, and subscripts of H and D indicate hydrogen and deuterium respectively.
Protein Expression.
The Mb variants were expressed from pET-based vectors in E. coli BL21(DE3) as described previously.[2e, f] After transformation, cells were grown in TB medium (ampicillin, 100 mg L−1) at 37 °C (200 rpm) until OD600 reached 0.6. Cells were then induced with 0.25 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, cultures were shaken at 180 rpm and 27 °C and harvested after 20 h by centrifugation at 4,000 rpm at 4 °C. After cell lysis by sonication, the cell lysate was loaded on a Ni-NTA column equilibrated with Ni-NTA lysis buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH 8.0). After washing with 50 mL Ni-NTA lysis buffer and 50 mL of Ni-NTA wash buffer (50 mM KPi, 250 mM, NaCl, 20 mM imidazole, pH 8.0), the protein was eluted with Ni-NTA elution buffer (50 mM KPi, 250 mM, NaCl, 250 mM histidine, pH 7.0). The protein solution was buffer exchanged against potassium phosphate buffer (50 mM, pH 7.0), and the protein concentration was determined using ε410 = 156 mM−1 cm−1.
Mb-catalyzed Si—H insertion reactions.
Under standard reaction conditions, 500 μL scale reactions were carried out using 0.2−20 μM Mb variant, 10 mM dimethyl(phenyl)silane, 10 mM EDA, and 10 mM sodium dithionite, at room temperature and at the indicated reaction time. In a typical procedure, a solution containing the desired myoglobin variant in potassium phosphate buffer (50 mM, pH 7.0) with sodium dithionite was prepared in an anaerobic chamber. Reactions were initiated by addition of 10 μL of dimethylphenylsilane followed by the addition of 10 μL of EDA from 0.5 M stock solutions, and the reaction mixtures were stirred in the chamber for 1 h at room temperature. For reactions in sealed vials, a solution containing sodium dithionite (100 mM stock solution) in potassium phosphate buffer (KPi 50 mM, pH 7.0) was degassed by bubbling argon into the mixture for 3 min in a sealed glass crimp vial. A buffered solution containing myoglobin was degassed in a similar manner in a separate glass crimp vial. The two solutions were then mixed together via cannulation. Reactions were initiated by addition of 10 μL of dimethyl(phenyl)silane (from a 0.5 M stock solution in ethanol), followed by the addition of 10 μL of EDA (from a 0.5 M stock solution in ethanol) with a syringe, and the reaction mixture was stirred for the indicated time at room temperature, under positive argon pressure. The reactions were analyzed using a Shimadzu GC-2010 gas chromatograph equipped with an FID detector and a Chiral Cyclosil-B column (30 m × 0.25 mm × 0.25 μm film). Separation method: 1 μL injection, injector temp: 200 °C, detector temp: 300 °C. Gradient: column temperature set at 120 °C for 3 min, then to 150 °C at 0.8 °C/min, then to 245 °C at 25 °C/min. Total run time was 28.60 min. Calibration curve for quantification of the Si-insertion product was constructed using authentic standard prepared synthetically as described in the SI. GC/MS analyses were performed on a Shimadzu GCMS-QP2010 equipped with a RTXXLB column (30 m × 0.25 mm × 0.28 μm) and a quadrupole mass analyzer.
Supplementary Material
Acknowledgements
This work was supported by the U.S. National Science Foundation grant CHE-1300912 to YZ and the U.S. National Institute of Health grant GM098628 to RF.
References
- [1].a) Sreenilayam G, Fasan R, Chem. Commun 2015, 51, 1532–1534; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tyagi V, Bonn RB, Fasan R, Chem. Sci 2015, 6, 2488–2494; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kan SBJ, Lewis RD, Chen K, Arnold FH, Science 2016, 354, 1048–1051; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Key HM, Dydio P, Clark DS, Hartwig JF, Nature 2016, 534, 534–537; [DOI] [PubMed] [Google Scholar]; e) Dydio P, Key HM, Nazarenko A, Rha JY-E, Seyedkazemi V, Clark DS, Hartwig JF, Science 2016, 354, 102–106; [DOI] [PubMed] [Google Scholar]; f) Kan SBJ, Huang X, Gumulya Y, Chen K, Arnold FH, Nature 2017, 552, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Vargas DA, Tinoco A, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2018, 57, 9911–9915; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Moore EJ, Steck V, Bajaj P, Fasan R, J. Org. Chem 2018, 83, 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Coehlo PS, Brustad EM, Kannan A, Arnold FH, Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]; b) Coehlo PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM, Nat. Chem. Biol 2013, 9, 485–487; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang ZJ, Renata H, Peck NE, Farwell CC, Coehlo PS, Arnold FH, Angew. Chem.-Int. Ed 2014, 53, 6810–6813; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, Brustad EM, ChemBioChem 2016, 17, 394–397; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Bordeaux M, Tyagi V, Fasan R, Angew. Chem.-Int. Ed 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bajaj P, Sreenilayam G, Tyagi V, Fasan R, Angew. Chem.-Int. Ed 2016, 55, 16110–16114; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Tinoco A, Steck V, Tyagi V, Fasan R, J. Am. Chem. Soc 2017, 139, 5293–5296; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Sreenilayam G, Moore EJ, Steck V, Fasan R, Adv. Synth. Cat 2017, 359, 2076–2089; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Knight AM, Kan SBJ, Lewis RD, Brandenberg OF, Chen K, Arnold FH, ACS Cent. Sci 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tyagi V, Fasan R, Angew. Chem.-Int. Ed 2016, 55, 2512–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, Fasan R, Angew. Chem.-Int. Ed 2016, 55, 13562–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ponomarenko SA, Kirchmeyer S, Adv. Polym. Sci 2011, 235, 33–110. [Google Scholar]
- [6].a) Showell GA, Mills JS, Drug Discov. Today 2003, 8, 551–556; [DOI] [PubMed] [Google Scholar]; b) Franz AK, Wilson SO, J. Med. Chem 2013, 52, 388–405. [DOI] [PubMed] [Google Scholar]
- [7].a) Sambasivan R, Ball ZT, J. Am. Chem. Soc 2010, 132, 9289–9291; [DOI] [PubMed] [Google Scholar]; b) Chen D, Zhu D-X, Xu M-H, J. Am. Chem. Soc 2016, 138, 1498–1501; [DOI] [PubMed] [Google Scholar]; c) Yasutomi Y, Suematsu H, Katsuki T, J. Am. Chem. Soc 2010, 132, 4510–4511; [DOI] [PubMed] [Google Scholar]; d) Zhang Y-Z, Zhu S-F, Wang L-X, Zhou Q-L, Angew. Chem. Int. Ed 2008, 47, 8624–8626; [DOI] [PubMed] [Google Scholar]; e) Hyde S, Veliks J, Liegault B, Grassi D, Taillefer M, Gouverneur V, Angew. Chem.-Int. Ed 2016, 55, 3785–3789; [DOI] [PubMed] [Google Scholar]; f) Wang J-C, Xu Z-J, Guo Z, Deng Q-H, Zhou C-Y, Wan X-L, Che C-M, Chem. Comm 2012, 48, 4299–4301. [DOI] [PubMed] [Google Scholar]; g) Keipour H, Carrerasa V, Ollevier T, Org. Biomol. Chem 2017, 15, 5441–5456. [DOI] [PubMed] [Google Scholar]
- [8].a) Su M-D, Chem. Eur. J 2004, 10, 6073–6084; [DOI] [PubMed] [Google Scholar]; b) Ito K, Hara Y, Mori S, Kusama H, Iwasawa N, Chem. Eur. J 2009, 15, 12408–12416; [DOI] [PubMed] [Google Scholar]; c) Lewisa RD, Garcia-Borràsb M, Chalkleyc MJ, Bullerc AR, Houk KN, Kan SBJ, Arnold FH, Proc. Natl. Acad. Sci. U. S. A 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Khade RL, Fan W, Ling Y, Yang L, Oldfield E, Zhang Y, Angew. Chem.-Int. Ed 2014, 53, 7574–7578; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khade RL, Zhang Y, J. Am. Chem. Soc 2015, 137, 7560–7563; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Khade RL, Zhang Y, Chem. Eur. J 2017, 23, 17654–17658 ; [DOI] [PubMed] [Google Scholar]; d) Wei Y, Tinoco A, Steck V, Fasan R, Zhang Y, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Dakin LA, Ong PC, Panek JS, Staples RJ, Stavropoulos P, Organometallics 2000, 19, 2896–2908; [Google Scholar]; b) Landais Y, Parra-Rapado L, Planchenault D, Weber V, Tetrahedron Lett. 1997, 38, 229–232. [Google Scholar]
- [11].Meunier B, de Visser SP, Shaik S, Chem. Rev 2004, 104, 3947–3980. [DOI] [PubMed] [Google Scholar]
- [12].Debrunner PG, in Porphyrins Iron, Vol. 3 (Eds.: Lever ABP, Gray HB), VCH Publishers, New York, 1989, pp. 139–234. [Google Scholar]
- [13].a) Li Y, Huang JS, Zhou ZY, Che CM, You XZ, J. Am. Chem. Soc 2002, 124, 13185–13193; [DOI] [PubMed] [Google Scholar]; b) Guerin P, Battioni JP, Chottard JC, Mansuy D, J. Organomet. Chem 1981, 218, 201–209. [Google Scholar]
- [14].Mansuy D, Battioni JP, Lavallee DK, Fischer J, Weiss R, Inorg. Chem 1988, 27, 1052–1056. [Google Scholar]
- [15].Liu Y, Xu W, Zhang J, Fuller W, Schultz CE, Li J, J. Am. Chem. Soc 2017, 139, 5023–5026. [DOI] [PubMed] [Google Scholar]
- [16].Li K, Xue D, J. Phys. Chem. A 2006, 110, 11332–11337. [DOI] [PubMed] [Google Scholar]
- [17].Tinoco A, Wei Y, Bacik JP, Moore EJ, Ando N, Zhang Y, Fasan R, ACS Catal. 2019, 9, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moore EJ, Fasan R, Tetrahedron 2019, 75, 2357–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Khade RL, Yang Y-W, Shi Y-L, Zhang Y, Angew. Chem.-Int. Ed 2016, 55, 15058–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery Jr., J. A.; Peralta J; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ; Gaussian 09, Revision B.01 ed., Gaussian, Inc., Wallingford CT, 2010. [Google Scholar]
- [21].Mennucci B, Tomasi J, J. Chem. Phys 1997, 106, 5151–5158. [Google Scholar]
- [22].Torres RA, Lovell T, Noodleman L, Case DA, J. Am. Chem. Soc 2003, 125, 1923–1936. [DOI] [PubMed] [Google Scholar]
- [23].Chai J-D, Head-Gordon M, Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- [24].Yang K, Zheng J, Zhao Y, Truhlar DG, J. Chem. Phys 2010, 132, 164117. [DOI] [PubMed] [Google Scholar]
- [25].Hay PJ, Wadt WR, J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- [26].Chan K-H, Guan X, Lo VK-Y, Che C-M, Angew. Chem.-Int. Ed 2014, 53, 2982–2987. [DOI] [PubMed] [Google Scholar]
- [27].Nakamura E, Yoshikai N, Yamanaka M, J. Am. Chem. Soc 2002, 124, 7181–7192. [DOI] [PubMed] [Google Scholar]
- [28].Tahsini L, Bagherzadeh M, Nam W, de Visser SP, Inorg. Chem 2009, 48, 6661–6669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.