

Abstract
Protein-tyrosine phosphatases (PTPs) counteract protein tyrosine phosphorylation and cooperate with receptor-tyrosine kinases in the regulation of cell signaling. PTPs need to undergo oxidative inhibition for activation of cellular cascades of protein-tyrosine kinase phosphorylation following growth factor stimulation. It has remained enigmatic how such oxidation can occur in the presence of potent cellular reducing systems. Here, using in vitro biochemical assays with purified, recombinant protein, along with experiments in the adenocarcinoma cell line A431, we discovered that bicarbonate, which reacts with H2O2 to form the more reactive peroxymonocarbonate, potently facilitates H2O2-mediated PTP1B inactivation in the presence of thioredoxin reductase 1 (TrxR1), thioredoxin 1 (Trx1), and peroxiredoxin 2 (Prx2) together with NADPH. The cellular experiments revealed that intracellular bicarbonate proportionally dictates total protein phosphotyrosine levels obtained after stimulation with epidermal growth factor (EGF) and that bicarbonate levels directly correlate with the extent of PTP1B oxidation. In fact, EGF-induced cellular oxidation of PTP1B was completely dependent on the presence of bicarbonate. These results provide a plausible mechanism for PTP inactivation during cell signaling and explain long-standing observations that growth factor responses and protein phosphorylation cascades are intimately linked to the cellular acid–base balance.
Keywords: phosphatase, bicarbonate, epidermal growth factor receptor (EGFR), thioredoxin reductase, peroxiredoxin, oxidative inactivation, peroxymonocarbonate, protein-tyrosine phosphatase (PTP), redox regulation, redox signaling, phosphorylation cascade
Introduction
Receptor-tyrosine kinase (RTK)4 activation leads to transmission of downstream phosphorylation cascades upon growth factor stimulation, and has major importance in physiology and proliferative diseases such as cancer. Protein-tyrosine phosphatases (PTPs), including PTP1B, counteract protein tyrosine phosphorylation and thereby act together with RTKs to regulate cell signaling (1). PTP activity depends upon a conserved active-site Cys residue (1–4), which renders these enzymes susceptible to oxidative inactivation. This has physiological importance, because RTK activation triggers transient H2O2 production from NADPH oxidases (NOXs) (5), which in turn leads to reversible oxidation and inhibition of PTPs. Inactivation of PTPs is believed to be absolutely required for RTK signaling (4, 6). NOXs are membrane-localized (typically on the plasma, endosomal, or endoplasmic reticulum membrane) and use cytoplasmic NADPH to produce superoxide and H2O2 on the opposing membrane surface. The H2O2 must then enter the cell, facilitated through aquaporins (7, 8), to exert its signaling actions. During stimulation of platelet-derived growth factor and epidermal growth factor (EGF) receptor pathways, the catalytic Cys residue of PTP1B becomes reversibly oxidized and thus inhibited (4, 9). The initial oxidation product is a sulfenic acid (-SOH) (10, 11), which can condense to an internal sulfenylamide (12–14) or undergo glutathionylation (15, 16). Reversibly oxidized PTP1B can be reactivated by the thioredoxin (Trx) system (thioredoxin reductase 1 (TrxR1) and NADPH with or without Trx1 or thioredoxin-related protein of 14 kDa (TRP14) (10, 17–19), thus enabling the Trx system to modulate cellular RTK signaling (17).
It is much debated exactly how oxidation of PTP1B occurs in a cellular environment, especially considering that members of the peroxiredoxin (Prx) family of thiol proteins, which are expressed at high levels in cells and also recycled predominantly by the Trx/TrxR/NADPH system, are many orders of magnitude more reactive and therefore likely to react with nearly all intracellular H2O2 (20–22). Some cytosolic thiol-containing proteins can be oxidized via Prx-mediated relays (23, 24), but this mechanism has not been demonstrated for PTP1B (25). With PTP1B being a therapeutic target with key roles in pathologies such as type 2 diabetes, obesity, and cancer (26, 27), as well as an archetypical example of oxidative inactivation in redox signaling, it is important that the mechanism of this process becomes understood.
We recently characterized the effect of H2O2 on PTP1B activity and found that a reconstituted Prx2/Trx1/TrxR1/NADPH system prevents PTP1B inactivation through both the removal of H2O2 and recycling of the oxidized PTP1B (25). It thus remains unclear how PTP1B can be inactivated by H2O2 in the presence of a functional Prx/Trx system. One possibility is that oxidation could be mediated by the more reactive peroxymonocarbonate (HCO4−), which is spontaneously formed in a reversible reaction between H2O2 and CO2/bicarbonate (HCO3−) (28, 29).
The X-ray structure of peroxymonocarbonate shows a true hydroperoxide (30), and it has been structurally and kinetically characterized using 13C NMR (28). Bicarbonate at physiological concentrations accelerates the reaction of H2O2 with thiols, thioethers, amines, and other small molecules such as GSH and albumin via formation of peroxymonocarbonate (29, 31, 32). However, of particular note is the finding by Gates and co-workers (33) that isolated PTP1B is especially reactive and reacts 7000 times faster with HCO4− than with H2O2 itself. Although the impact of this mechanism is modulated by the equilibrium shown above, it was suggested as a possible route for PTP1B oxidation during cell signaling (20), but this proposal has not been tested experimentally. Importantly, detection of the peroxymonocarbonate species in vitro requires up to 0.1–1 m concentrations, making it virtually impossible to detect directly in cellular systems. It is a highly reactive, short-lived chemical species that is in equilibrium with H2O2, which gives the same reaction products as hydrogen peroxide. Thus, there are no specific probes or reagents that would distinguish peroxymonocarbonate from H2O2. However, recent studies have identified links between regulation of bicarbonate levels, cell growth, and cancer progression (34). Signaling through the EGF receptor, which is an important event therapeutically targeted for cancer treatment (35–37), has been directly linked to bicarbonate-regulating enzymes. Notably, carbonic anhydrase IX, a glycoprotein with an extracellular domain that catalyzes hydration of CO2 (CO2 + H2O → HCO3− +H+), becomes activated upon EGF ligand stimulation (38). Carbonic anhydrase IX also localizes at the leading edge of focal adhesions, and its overexpression increases the rate of adhesion and spreading (39). Electroneutral sodium and bicarbonate cotransporter proteins (NBCs), such as NBCn1, facilitate chloride-HCO3− exchange with a Na+/HCO3− stoichiometry of 2:1. These membrane-bound proteins are important regulators of intracellular bicarbonate levels and help to control cellular pH (40). In MCF7 breast cancer cells, NBCn1 expression is highly up-regulated upon HER2 expression (41) and is correlated with tumor progression and metastasis (42). Furthermore, genetic or pharmaceutical disruption of several NBCs (SLCA4, SLC4A7, and SLC4A9) suppresses tumor growth of breast cancer spheroids and murine xenografts (43, 44), and chemical inhibition of bicarbonate influx using S0859 decreases spheroid growth of HCT116 cells (45). Considering these observations, we asked whether bicarbonate can directly facilitate PTP1B oxidation in the presence of a peroxiredoxin-recycling system and in a cellular setting. Surprisingly, our results reveal that bicarbonate is an obligate component of cellular PTP1B oxidation and EGF responsiveness.
Results
Bicarbonate potently facilitates H2O2-dependent inactivation of PTP1B
Using a direct activity assay with pure recombinant PTP1B, we first confirmed the previously described facilitation of H2O2-mediated inactivation of PTP1B by bicarbonate (33). Compared with the inefficient dose- and time-dependent direct inactivation of PTP1B by H2O2 (Fig. 1A), inclusion of a physiological concentration of 25 mm bicarbonate potently increased inactivation (Fig. 1B). Notably, bicarbonate alone had no effect (Fig. 1B, control). We next examined whether inclusion of bicarbonate facilitated H2O2-mediated inactivation of PTP1B in the presence of a functional Trx system.
Figure 1.

Bicarbonate potentiates H2O2-dependent inactivation of PTP1B. A, recombinant PTP1B (600 nm) was treated with increasing concentrations of H2O2, 0 μm buffer control (●), 3.4 μm (○), 6.3 μm (♦), 12.5 μm (▾) 25 μm (▴), 50 μm (■) (20 mm HEPES, 100 mm NaCl buffer, pH 7.4, containing 0.1 mm diethylenetriaminepentaacetic acid, 0.05% BSA, 1 mm sodium azide) and then assayed for PTP activity at the indicated times. PTP1B activity is given in min−1 (mol of product/mol of enzyme/min). Data points represent means ± S.D. (error bars) (n = 3). B, PTP1B was treated as in A but in the presence of 25 mm bicarbonate (final concentration after adding H2O2/bicarbonate and assay buffer). Data points represent means ± S.D. (n = 4). A representative run is presented in Fig. S1.
H2O2 and bicarbonate in combination can overcome protection of PTP1B activity by the Trx/Prx system
As reported previously, in the absence of bicarbonate, Prx2/Trx1/TrxR1/NADPH (at 10, 2, 0.5, and 200 μm, respectively) fully protects PTP1B against inactivation by 100 μm H2O2 (25). This is due to a combination of H2O2 removal by the peroxidase activity of Prx2 and reduction of the sulfenic acid and sulfenylamide forms of PTP1B by TrxR1 and Trx1, as illustrated in Fig. 2A. In the presence of bicarbonate (25 mm), where HCO4− can cause faster PTP1B inactivation, Prx2/Trx1/TrxR1/NADPH at the same concentrations still gave almost complete protection (Fig. 2B, 1 μm TrxR1). However, by lowering the TrxR1 concentration (Fig. 2B) or Trx1 concentration (Fig. 2C), it was possible to obtain conditions where PTP1B was inactivated for the duration of the experiment or inactivated at early time points, followed by a time-dependent regain in activity during the assay. The latter pattern is reminiscent of the transient PTP1B inactivation expected to occur in cells during an oxidative burst after growth factor stimulation. A higher concentration of pH-adjusted bicarbonate (45 mm, pH 7.4) was also able to fully overcome the complete protection of PTP1B against 50 μm H2O2 provided by the cycling Prx/Trx system, as seen by a rapid and sustained loss of PTP1B activity (Fig. 2D). At intermediate concentrations of bicarbonate, maintaining the same pH, H2O2 caused transient PTP1B inactivation followed by recovery (Fig. 2D). These results show that there is an interplay between bicarbonate, PTP1B, and the Trx/Prx redox cycling systems and that there are conditions whereby bicarbonate, presumably through the spontaneous production of peroxymonocarbonate (33), promotes transient inactivation of PTP1B by H2O2. To rule out possible inhibitory effects of bicarbonate and H2O2 on the Trx system, NADPH consumption was measured using insulin as a substrate (46). The results showed no difference in consumption by bicarbonate alone or in combination with H2O2 as compared with buffer control (Fig. S2).
Figure 2.

Modulation of bicarbonate- and H2O2-dependent inactivation of PTP1B in the presence of a Prx2/TrxR1/Trx1 redox system. A, scheme showing relevant reactions in the experimental PTP1B/Prx2/thioredoxin system. Sox of PTP1B could be either the sulfenic acid or sulfenylamide species, with the former also reducible by TrxR/NADPH in the absence of Trx. B, PTP1B (600 nm) was treated with 50 μm H2O2 together with 25 mm bicarbonate in the presence of Trx1 (5 μm), Prx2 (10 μm), NADPH (300 μm), and increasing TrxR1 concentrations of 0 nm (■), 2 nm (▴), 10 nm (▾), 50 nm (♦), 250 nm (○), and 1 μm (□) and with 1 μm TrxR1 using buffer-treated control without H2O2 (●). PTP activity was measured after the indicated times. Data points represent means ± S.D. (error bars) (n = 3). C, analyses were performed as in B with TrxR1 (1 μm) and increasing Trx1 concentration, 0 μm (♦), 0.5 μm (▾), 1 μm (▴), 2 μm (■), and 5 μm (●) (n = 1). D, analyses performed as in A with TrxR1 (15 nm) and increasing concentrations of bicarbonate as indicated, 0 mm (■), 5 mm (▴), 10 mm (▾), 15 mm (♦), 25 mm (○), 35 mm (□), and 45 mm (▵) (n = 1).
Inhibition of bicarbonate cotransporters in A431 cells decreases EGF-induced phosphorylation cascades
We next addressed whether the mechanism identified with the purified enzymes applies to cellular systems, by examining the effect of bicarbonate on protein phosphorylation cascades triggered by growth factor stimulation. We used human A431 epidermal squamous carcinoma cells, as these cells are known to inactivate PTP1B by oxidation in response to treatment with EGF (10, 47). Extracellularly produced bicarbonate is transported into cells through membrane-bound cotransporter proteins, NBCs, that control intracellular bicarbonate levels and thereby cellular pH (40). We thus stimulated A431 cells with EGF ligand for 2, 4, and 6 min with or without 1-h pretreatment with S0859, a well-characterized inhibitor of NBCs (48). As shown in Fig. 3 (A and B), EGF ligand induced a rapid increase in total protein tyrosine phosphorylation, primarily associated with the ∼198 kDa band representing the EGF receptor (Fig. 3A). This phosphorylation was notably inhibited when bicarbonate influx was blocked with S0859. The EGFR phosphorylation site Tyr-992 (pY992), previously shown to be regulated by PTP1B (49), was also significantly inhibited at all time points by S0859 (Fig. 3, C and D).
Figure 3.

Pharmacological inhibition of bicarbonate cotransporters in A431 cells decreases EGF ligand-induced phosphotyrosine formation. Overnight starved A431 cells in regular bicarbonate-containing DMEM 0.1% FCS) were pretreated with the NBC inhibitor S0859 (100 μm) or DMSO control for 1 h prior to EGF stimulation for 2, 4, and 6 min. Cell extracts were separated by SDS-PAGE and Western blotted (IB) for total phosphotyrosine and EGF receptor (A) and specific phosphorylation of the EGF receptor Tyr-992 (pY992) residue (C). Bar diagrams show densitometry ratios for total signal over all phosphotyrosine bands (B) or the specific pTyr-992 signal (D), relative to the immunoblot staining for the EGF receptor. Every ratio is compared with that in unstimulated control, which was set to 1. Results are mean ± S.D. (error bars) for three independent experiments with filled circles showing individual results. Statistically significant differences are indicated (*, p < 0.05). The membranes in the figure stained with Ponceau for total protein loading are shown in Fig. S3.
Lactic acid pretreatment of A431 cells decreases EGF-induced phosphorylation
We next investigated whether protein tyrosine phosphorylation patterns were affected by physiologically relevant lactic acid concentrations, which decrease cellular bicarbonate as well as pH at events of metabolic acidosis. Lactic acid (up to 40 mm) was added to the growth medium of A431 cells 2 min prior to the addition of EGF ligand. Measurements made after 5-min stimulation showed that lactic acid concentrations above 10 mm resulted in a progressive decrease in total protein tyrosine phosphorylation (Fig. 4A). Time course measurements showed that the addition of 15 mm lactic acid slowed the onset of EGF-dependent phosphorylation, whereas 30 mm gave very strong inhibition at all time points (Fig. 4B).
Figure 4.

Treatment of A431 cells with lactic acid prior to EGF ligand stimulation results in less phosphorylation. A, concentration dependence. Serum-starved A431 cells in regular DMEM containing bicarbonate were pretreated for 2 min with 0, 5, 10, 20, and 40 mm lactic acid and subsequently stimulated with EGF (100 ng/ml) for 5 min. Lysates were analyzed for total phosphotyrosine. B, time course. A431 cells were pretreated for 2 min with 0, 15, and 30 mm lactic acid and subsequently stimulated with EGF (100 ng/ml) for 2, 4, and 6 min. Lysates were analyzed for total phosphotyrosine. The right-hand panels in A and B show densitometry analyses as in Fig. 3 with filled circles showing individual results (n = 3; mean ± S.D. (error bars); *, p < 0.05). The membranes in this figure stained with Ponceau for total protein loading are shown in Fig. S4.
Bicarbonate depletion at constant pH decreases EGF-induced protein tyrosine phosphorylation in A431 cells
As the results in Figs. 3 and 4 could be attributed to effects of bicarbonate or to general pH effects, we next assessed whether lowering cellular levels of bicarbonate without changing the pH would still affect protein tyrosine phosphorylation patterns. A431 cells were cultured in regular DMEM and subsequently starved overnight in low-serum bicarbonate-free HEPES-buffered (50 mm) medium, with the addition of 0, 40, or 60 mm bicarbonate, adjusted to pH 7.4, and equilibrated in air with varying percentages of corresponding CO2 levels (0, 5, and 10%, respectively). The A431 cells were stimulated with EGF ligand after 1 day of these culture conditions and harvested 2, 4, and 6 min after the EGF addition. Analyses of total protein phosphotyrosine revealed increases in phosphorylation at the higher bicarbonate concentration, most pronounced for a band (not identified) at about 28 kDa seen at longer exposures (Fig. 5). Thus, increased cellular bicarbonate levels augment EGFR-linked protein phosphorylation in A431 cells independently of pH-derived effects.
Figure 5.
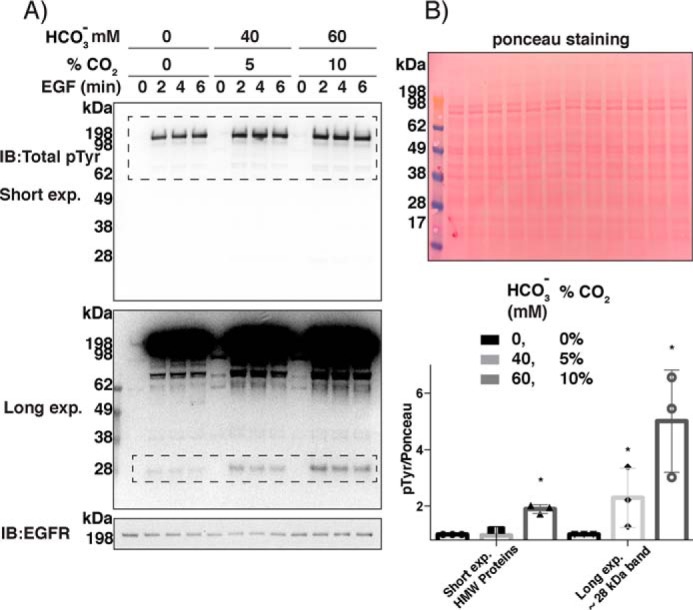
Bicarbonate increases total phosphorylation in A431 cells after EGF ligand stimulation. A, A431 cells were grown and incubated overnight in low-serum HEPES-buffered (50 mm) DMEM at pH 7.4 with 0, 40, and 60 mm bicarbonate added and equilibrated with 0, 5, and 10% CO2, respectively, as indicated. The cells were then stimulated with EGF and analyzed for total phosphotyrosine. B, densitometry for three independent experiments, of short- and long-exposed membranes, for high-molecular weight (HMW) phosphorylated proteins, and ∼28 kDa band, respectively (dashed rectangles in A) quantified in relation to Ponceau staining with 2-, 4-, and 6-min time points combined (n = 3; mean ± S.D. (error bars); *, p < 0.05). Symbols indicate individual results. IB, immunoblotting.
Oxidation of PTP1B in A431 cells during EGF stimulation depends on the presence of bicarbonate
The observed bicarbonate-dependent increases seen in EGF-triggered phosphorylation are consistent with oxidative inactivation of PTPs. However, this is an indirect readout, and other explanations are possible. We therefore looked for direct evidence of oxidative PTP1B inactivation using a cysteine-labeling assay that detects reversible thiol oxidation (50, 51). The basis of the assay is to use a biotin tag that labels only reversibly oxidized thiol proteins and isolates the labeled proteins with streptavidin. Immunoblotting against the protein of interest will give a positive response only if it has been oxidized. When this procedure was applied to A431 cells maintained in regular bicarbonate-containing DMEM, we detected a time-dependent increase in oxidized PTP1B peaking at 2 min after the EGF addition (Fig. 6A). This is in agreement with earlier reports (10). Most strikingly, pretreatment with the NBC inhibitor S0859 completely abrogated the EGF-dependent PTP1B oxidation at the 2-min time point (Fig. 6A). Furthermore, PTP1B oxidation was not seen when the cells were treated with EGF in HEPES-buffered bicarbonate-free DMEM. Finally, the addition of pH-equilibrated bicarbonate to the bicarbonate-free cells before EGF stimulation brought oxidation of PTP1B back to the level seen in bicarbonate-containing DMEM (Fig. 6A). Thus, the presence of bicarbonate in cells is a crucial and apparently obligate component for EGF-dependent phosphorylation cascades that require oxidation and inactivation of PTP1B.
Figure 6.
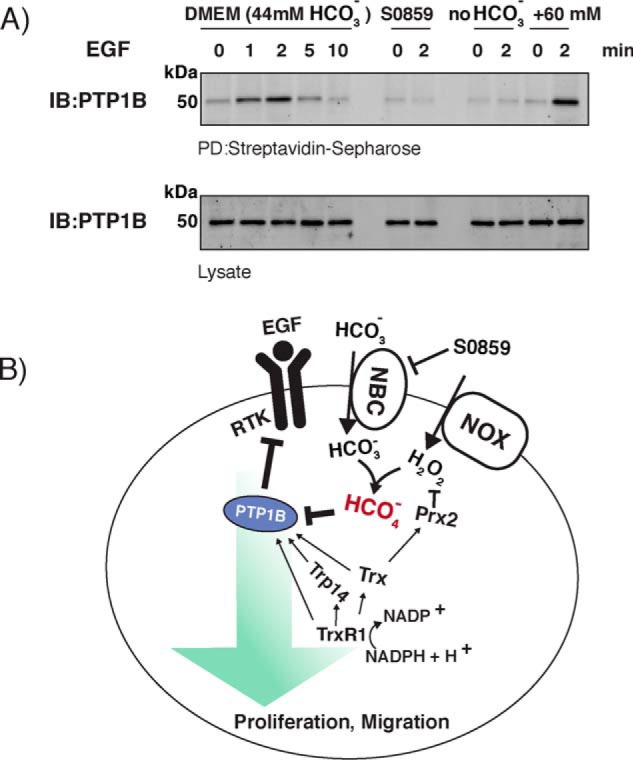
A, bicarbonate is required for EGF-dependent reversible PTP1B oxidation in A431 cells. A431 cells were incubated overnight either in low-serum regular DMEM-containing bicarbonate or in HEPES-buffered DMEM (50 mm, pH 7.4). Pretreatment of the cells in bicarbonate-containing DMEM with the NBC inhibitor S0859 (50 μm) was for 1 h prior to stimulation. Bicarbonate (60 mm) was added to cells in HEPES-buffered DMEM and subsequently stimulated. At the indicated times after EGF stimulation, cells were subjected to the cysteinyl-labeling assay using biotinylated iodoacetyl-PEG2-biotin for analysis of reversible PTP1B oxidation. Biotinylated proteins were purified on streptavidin-Sepharose beads, resolved by SDS-PAGE, and visualized using antibodies against PTP1B. PTP1B control levels were determined from total cell lysate by SDS-PAGE and blotting (IB) against PTP1B. Representative Western blotting of three independent experiments is shown. B, model for regulation of PTP1B activity during growth factor signaling. EGFR activation induces a transient burst of H2O2, which reacts with bicarbonate to give PTP1B oxidation via peroxymonocarbonate (red) and activation of phosphorylation pathways (green arrow). Prxs compete for the H2O2, and the Trx system decreases the availability of H2O2 by supporting the Prx cycle and also acts by reactivating oxidized PTP1B. See “Discussion” for further details.
Discussion
Reversible oxidative inhibition of PTPs is a key regulatory event during growth factor signaling (52). The molecular mechanisms leading to PTP oxidation have remained unclear, although it is well-known that RTK activation involves cellular production of H2O2 derived from NOX enzymes (5). Several models for PTP inactivation have been proposed, such as the “floodgate” hypothesis in which inactivation of Prxs enables oxidation of less-reactive proteins (53) or redox relay–mediated oxidation via Prxs (23, 24). Our results add another dimension and reveal that a concerted action of H2O2 together with bicarbonate is required for inactivation of PTP1B and the initiation of protein phosphorylation cascades.
The results with the recombinant proteins clearly showed that H2O2 triggered inactivation of PTP1B in the presence of a fully active and cycling Prx system only when bicarbonate was present. We used all reactants at physiologically relevant concentrations and were able to vary conditions within these to see transient inactivation and reactivation, reminiscent of a signaling step. We propose a mechanism to explain these results in which H2O2 can either be scavenged by Prx2 or react with bicarbonate to generate peroxymonocarbonate, which is responsible for PTP1B inactivation (Fig. 2A). Increasing the bicarbonate favors the latter, whereas regeneration of reduced Prx2 by the thioredoxin system enhances H2O2 removal and is protective. It is possible that peroxymonocarbonate could also react with Prx2 in this system. However, we have recently found that oxidation of the reduced protein is unaffected by the presence of bicarbonate.5 Although hyperoxidation was enhanced, this would cause only minor inactivation of the Prx2 under the conditions of our current study. Nevertheless, in other situations, this could amplify the effect on PTP1B. Complementing these oxidative reactions are the reductive steps involving reduction of Prx2 disulfide to maintain its scavenging activity as well as recycling of PTP1B, either through a reaction of the sulfenic acid intermediate directly with TrxR1 (25) or recycling of the sulfenylamide by TrxR1 together with Trx1 or TRP14 (17). The balance between these steps determines the extent to which PTP1B is initially inactivated and subsequently reactivated as the H2O2 is consumed and the reducing mechanisms dominate.
To test the relevance of this mechanism of PTP1B inactivation during EGF-triggered signaling context, we modulated the cellular bicarbonate levels using three different approaches. Lowering bicarbonate uptake with the NBC inhibitor S0859 resulted in less EGFR-dependent protein tyrosine phosphorylation in A431 cells. Likewise, the addition of lactic acid, which lowers bicarbonate levels by a shift in acid–base equilibrium, also decreased phosphotyrosine levels following EGF ligand stimulation in a dose-dependent manner. These observations are compatible with the notion that lower intracellular levels of bicarbonate give less inhibition of cellular PTP activity, but other effects of acidification could not be excluded. However, the experiments with cells grown in pH-controlled HEPES-buffered DMEM revealed signaling effects of bicarbonate not related to changes in pH.
Our results collectively suggest a molecular mechanism by which physiological changes in bicarbonate levels affect growth factor signaling responses. This conclusion is substantiated by the finding that the cysteinyl-labeling assay detected EGF-dependent oxidation of PTP1B only in the presence of bicarbonate. Thus, bicarbonate seems to be an obligate factor for EGF-dependent PTP1B oxidation. We hence propose a model for redox regulation of PTP1B during growth factor signaling, in which the growth factor initiates transient production of H2O2, which must act jointly with bicarbonate to achieve PTP1B inactivation and initiate phosphorylation cascades. For the reasons given in the Introduction, we believe that, although it is not possible to validate experimentally, this mechanism most likely occurs through the formation of peroxymonocarbonate (Fig. 6B).
Our investigation so far has focused on PTP1B, but it is possible that the redox state of other thiol proteins could be regulated by bicarbonate. Gates and co-workers (33) showed that SHP1 is also highly reactive with peroxymonocarbonate, but this was not a general property of all of the thiol proteins they studied. Thus, there appears to be selectivity, and our findings give credence to the possibility that peroxymonocarbonate has a wider, yet unexplored, role in H2O2-mediated redox signaling. They also provide a plausible explanation for how some thiol proteins that react slowly with H2O2 in isolation (when bicarbonate is typically not present) appear to be oxidant-sensitive during cell signaling.
Another important consideration is that intracellular bicarbonate-regulating enzymes such as CA II, IX, XII, and NBCs have been shown to contribute do disease pathology, such as promotion of tumor development, through a more alkaline intracellular milieu (34, 38, 39, 54). Although most focus has been on pH effects of alkalinization of the intracellular milieu, we here propose an alternative and yet unexplored mechanism whereby redox changes associated with growth factor responses, such as the inhibition of PTP1B, are dependent specifically upon bicarbonate.
Experimental procedures
Expression of recombinant proteins
The catalytic domain (residues 1–322) of PTP1B was subcloned into pD441-H6 vector using PCR with primers 5′-GTAGGTCTCGGTGGTATGGAGATGGAAAAGGAGTTC-3′ and 5′-GTAGGTCTCTTATTACCCATTGTGTGGCTCCAGG-3′ and Eco31I and DpnI restriction sites. The PTP1B protein was expressed and purified under reducing conditions according to previously described procedures, including subsequent tag removal (25, 55). Prx2 protein was expressed and purified with a His tag, which was cleaved off as described previously (56). Recombinant human Trx1 and rat TrxR1 WT were expressed and purified as described previously (55). PTP1B was subjected to buffer exchange to remove reductant before all experiments, using Zeba Spin Desalting Columns (Thermo Scientific catalog no. 87766). Protein concentrations were determined using Bradford reagents.
Treatment of PTP1B with H2O2 and bicarbonate
Reduced PTP1B (600 nm) was pre-incubated for 20 min in 20 mm HEPES, 100 mm NaCl buffer, pH 7.4, containing 0.1 mm EDTA, 0.05% BSA, 1 mm sodium azide, with the indicated concentrations of Trx1, TrxR1, NADPH (N7505, Sigma-Aldrich), and Prx2. Sodium azide was used to inhibit any trace amounts of catalase. Each reaction mixture was exposed to H2O2 with and without bicarbonate for the indicated times. Bicarbonate and H2O2 were premixed prior to treatment. Subsequent to treatment, measurement of PTP activity was performed.
PTP activity assay
PTP activity was assessed using 15 mm chromogenic substrate 4-nitrophenyl phosphate (P4744, Sigma-Aldrich) as described previously (57). Rates of absorbance increase were measured at 410 nm at 22 °C using an Infinite M200 Pro plate reader (Tecan) and calibrated using a 4-nitrophenol standard curve.
Treatment of PTP1B with H2O2
Buffer-exchanged reduced PTP1B was exposed to H2O2 and different components of the Trx system at the indicated time points followed by the addition of substrate and measurement of activity. We initially observed that incubation of PTP1B alone resulted in some time-dependent inactivation that was partially prevented in the presence of BSA, so BSA (0.05%) was added to the buffer. The activity after each H2O2 treatment was related to the activity of untreated PTP1B incubated for the same time. A 0–24% loss of activity was seen in control conditions at 30 min.
Insulin-coupled TrxR assay
NADPH consumption was measured through incubation of the Trx system (1 μm TrxR1, 20 μm Trx1, and 300 μm NADPH) together with 0.16 mm insulin in 20 mm HEPES, 100 mm NaCl buffer, pH 7.4, containing 0.1 mm EDTA, 0.05% BSA with and without 25 mm bicarbonate and 100 μm H2O2.
Cell culture conditions and treatments
Adenocarcinoma cell line A431 cells (ATCC) were typically, unless stated otherwise, cultured in DMEM (44 mm sodium bicarbonate, 5% CO2) + 10% (v/v) fetal bovine serum, 2 mm l-glutamine, penicillin, and streptomycin. Cells were grown until 90% confluence and then starved for 24 h in 0.1% fetal bovine serum. Pretreatment of starved cells prior to ligand stimulation was performed with the indicated concentrations of either lactic acid (Sigma-Aldrich, catalog no. L6402), S0859 (Sigma-Aldrich, catalog no. SML0638), or sodium bicarbonate (Merck, catalog no. 9018415). Ligand stimulations were performed with 100 ng/ml EGF ligand (R&D Systems, catalog no. 236-EG-200). For cell culture conditions with varying concentrations of bicarbonate, bicarbonate-free DMEM (D5648) (0.1% (v/v) FBS, 2 mm l-glutamine, penicillin, and streptomycin) was supplemented with 50 mm HEPES and 0, 40, and 60 mm bicarbonate, set to pH 7.4, and pre-equilibrated in 0, 5, and 10% CO2, respectively, for 3 days prior to the addition to cells, as stated above.
SDS-PAGE and analysis of protein tyrosine phosphorylation
Treated cells were washed with ice-cold PBS (pH 7.4) and lysed with lysis buffer (0.5% Triton X-100, 0.5% sodium deoxycholate salt/deoxycholic acid, 150 mm NaCl, 20 mm Tris (pH 7.5), 10 mm EDTA, and 30 mm sodium pyrophosphate (pH 7.5), supplemented with 200 μm sodium orthovanadate and a protease inhibitor mixture (Roche Applied Science)). Protein concentration of lysates was determined using the Bradford assay. Equal amounts of lysate protein were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore), blocked with 5% milk in TBS, and immunoblotted for total phosphotyrosine 4G10 (Merck, catalog no. 05-321) (1:1000) and EGFR pTyr-992 (Cell Signaling). The total amount of loading was verified using Ponceau staining of blotted membranes or EGFR intensities (AF231, R&D Systems). Protein phosphotyrosine intensities were quantified using ImageJ and shown as a ratio of loading control, as described.
Detection of reversible PTP oxidation in cells
The cysteinyl-labeling assay of reversibly oxidized PTP1B was performed as described previously (50, 51). The lysis buffer (50 mm sodium acetate, pH 5.5, 150 mm NaCl, 10% glycerol, 1% Surfact-Amp Nonidet P-40, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 50 units/ml superoxide dismutase, 50 units/ml catalase, 10 mm iodoacetic acid) was degassed and placed on ice into a hypoxic glove-box station equilibrated with 100% argon. Cells cultured in 100-mm dishes were treated and then transferred from a 37 °C/5% CO2 environment into a hypoxic glove-box station, medium was carefully removed, and cells were rapidly lysed with 800 μl of ice-cold lysis buffer. Lysates were transferred to amber-colored microcentrifuge tubes and shaken for 1 h at room temperature to alkylate reduced thiols. Protein concentrations were determined, and 1 mg of lysate was applied to a spin column to remove excess iodoacetic acid. Tris(2-carboxyethyl)phosphine (1 mm) was then added to reduce reversibly oxidized protein thiols. Following this step, 5 mm EZ-link iodoacetyl-PEG2-biotin probe (Pierce) was added to the lysate to label the reactivated thiols. Labeled proteins were then pulled down by streptavidin-Sepharose beads, which were washed with lysis buffer (pH 5.5), resuspended in 20 μl of 4× Laemmli sample buffer, and heated at 90 °C for 90 s. Samples were resolved by SDS-PAGE and blotted with anti-PTP1B. Immunoblots of total cell lysates were run as controls.
Statistical analyses
Analyses of data were performed using GraphPad Prism with two-way analysis of variance followed by Bonferroni post hoc tests for multiple comparisons.
Author contributions
M. D. performed a majority of the experiments and analysis of data and contributed to writing the paper. Q. C. expressed and purified TrxR1 and Trx1. S. H. M. R. performed the CLA assay. P. E. P. expressed Prx2. B. B. performed the CLA assay and provided intellectual input. C. C. W. designed experiments, analyzed data, provided essential intellectual input, and contributed to writing the paper. E. S. J. A. was responsible for the project overall and contributed to design of experiments, analyzed data, provided intellectual input, and wrote the paper.
Supplementary Material
Acknowledgment
We thank Dr. Mark Hampton for helpful discussions.
This work was supported by the Karolinska Institutet, the Knut and Alice Wallenberg Foundations, the Swedish Cancer Society, the Karolinska Institutet Research Foundations (to E. S. J. A.), Swedish Research Council Grant (to E. S. J. A. and M. D.) 537-2014-360, Swedish Society of Medicine Grant SLS-786841, Åke Wiberg Stiftelse Grants M17-0101 and M18-0141 (to M. D.), National Institutes of Health Grant HL138605, American Heart Association Grant 17GRNT33700265 (to B. B.), and the New Zealand Marsden Fund (to C. C. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S4.
A. V. Peskin and C. C. Winterbourn, unpublished observations.
- RTK
- receptor-tyrosine kinase
- PTP
- protein-tyrosine phosphatase
- EGF
- epidermal growth factor
- Trx
- thioredoxin
- Prx
- peroxiredoxin
- NBC
- sodium and bicarbonate exchanger protein
- NOX
- NADPH oxidase.
References
- 1. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 10.1038/nrm2039 [DOI] [PubMed] [Google Scholar]
- 2. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., and Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 10.1016/j.cell.2004.05.018 [DOI] [PubMed] [Google Scholar]
- 3. Andersen J. N., Mortensen O. H., Peters G. H., Drake P. G., Iversen L. F., Olsen O. H., Jansen P. G., Andersen H. S., Tonks N. K., and Møller N. P. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 21, 7117–7136 10.1128/MCB.21.21.7117-7136.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frijhoff J., Dagnell M., Godfrey R., and Ostman A. (2014) Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid. Redox Signal. 20, 1994–2010 10.1089/ars.2013.5643 [DOI] [PubMed] [Google Scholar]
- 5. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 10.1038/nri1312 [DOI] [PubMed] [Google Scholar]
- 6. Ostman A., Frijhoff J., Sandin A., and Böhmer F. D. (2011) Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem. 150, 345–356 10.1093/jb/mvr104 [DOI] [PubMed] [Google Scholar]
- 7. Bertolotti M., Farinelli G., Galli M., Aiuti A., and Sitia R. (2016) AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukoc. Biol. 100, 1071–1079 10.1189/jlb.2AB0116-045R [DOI] [PubMed] [Google Scholar]
- 8. Medraño-Fernandez I., Bestetti S., Bertolotti M., Bienert G. P., Bottino C., Laforenza U., Rubartelli A., and Sitia R. (2016) Stress regulates aquaporin-8 permeability to impact cell growth and survival. Antioxid. Redox Signal. 24, 1031–1044 10.1089/ars.2016.6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanner J. J., Parsons Z. D., Cummings A. H., Zhou H., and Gates K. S. (2011) Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid. Redox Signal. 15, 77–97 10.1089/ars.2010.3611 [DOI] [PubMed] [Google Scholar]
- 10. Lee S. R., Kwon K. S., Kim S. R., and Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 10.1074/jbc.273.25.15366 [DOI] [PubMed] [Google Scholar]
- 11. Frijhoff J., Dagnell M., Augsten M., Beltrami E., Giorgio M., and Östman A. (2014) The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 68, 268–277 10.1016/j.freeradbiomed.2013.12.022 [DOI] [PubMed] [Google Scholar]
- 12. van Montfort R. L., Congreve M., Tisi D., Carr R., and Jhoti H. (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777 10.1038/nature01681 [DOI] [PubMed] [Google Scholar]
- 13. Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., and Barford D. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 10.1038/nature01680 [DOI] [PubMed] [Google Scholar]
- 14. Sivaramakrishnan S., Cummings A. H., and Gates K. S. (2010) Protection of a single-cysteine redox switch from oxidative destruction: on the functional role of sulfenyl amide formation in the redox-regulated enzyme PTP1B. Bioorg Med. Chem. Lett. 20, 444–447 10.1016/j.bmcl.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rinna A., Torres M., and Forman H. J. (2006) Stimulation of the alveolar macrophage respiratory burst by ADP causes selective glutathionylation of protein tyrosine phosphatase 1B. Free Radic. Biol. Med. 41, 86–91 10.1016/j.freeradbiomed.2006.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barrett W. C., DeGnore J. P., König S., Fales H. M., Keng Y. F., Zhang Z. Y., Yim M. B., and Chock P. B. (1999) Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38, 6699–6705 10.1021/bi990240v [DOI] [PubMed] [Google Scholar]
- 17. Dagnell M., Frijhoff J., Pader I., Augsten M., Boivin B., Xu J., Mandal P. K., Tonks N. K., Hellberg C., Conrad M., Arnér E. S., and Östman A. (2013) Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-β receptor tyrosine kinase signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 13398–13403 10.1073/pnas.1302891110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwertassek U., Haque A., Krishnan N., Greiner R., Weingarten L., Dick T. P., and Tonks N. K. (2014) Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 281, 3545–3558 10.1111/febs.12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parsons Z. D., and Gates K. S. (2013) Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 52, 6412–6423 10.1021/bi400451m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antunes F., and Brito P. M. (2017) Quantitative biology of hydrogen peroxide signaling. Redox Biol. 13, 1–7 10.1016/j.redox.2017.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Denu J. M., and Tanner K. G. (1998) Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 10.1021/bi973035t [DOI] [PubMed] [Google Scholar]
- 22. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 10.1038/nchembio.85 [DOI] [PubMed] [Google Scholar]
- 23. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 10.1038/nchembio.1695 [DOI] [PubMed] [Google Scholar]
- 24. Stöcker S., Maurer M., Ruppert T., and Dick T. P. (2018) A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14, 148–155 10.1038/nchembio.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dagnell M., Pace P. E., Cheng Q., Frijhoff J., Östman A., Arnér E. S. J., Hampton M. B., and Winterbourn C. C. (2017) Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 292, 14371–14380 10.1074/jbc.M117.793745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krishnan N., Bonham C. A., Rus I. A., Shrestha O. K., Gauss C. M., Haque A., Tocilj A., Joshua-Tor L., and Tonks N. K. (2018) Harnessing insulin- and leptin-induced oxidation of PTP1B for therapeutic development. Nat. Commun. 9, 283 10.1038/s41467-017-02252-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krishnan N., Koveal D., Miller D. H., Xue B., Akshinthala S. D., Kragelj J., Jensen M. R., Gauss C. M., Page R., Blackledge M., Muthuswamy S. K., Peti W., and Tonks N. K. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 10.1038/nchembio.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bakhmutova-Albert E. V., Yao H., Denevan D. E., and Richardson D. E. (2010) Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 49, 11287–11296 10.1021/ic1007389 [DOI] [PubMed] [Google Scholar]
- 29. Trindade D. F., Cerchiaro G., and Augusto O. (2006) A role for peroxymonocarbonate in the stimulation of biothiol peroxidation by the bicarbonate/carbon dioxide pair. Chem. Res. Toxicol. 19, 1475–1482 10.1021/tx060146x [DOI] [PubMed] [Google Scholar]
- 30. Adam A., and Mehta M. (1998) KH(O2)CO2H2O2: an oxygen-rich salt of monoperoxocarbonic acid. Angew. Chem. Int. Ed. Engl. 37, 1387–1388 [DOI] [PubMed] [Google Scholar]
- 31. Richardson D. E., Regino C. A., Yao H., and Johnson J. V. (2003) Methionine oxidation by peroxymonocarbonate, a reactive oxygen species formed from CO2/bicarbonate and hydrogen peroxide. Free Radic. Biol. Med. 35, 1538–1550 10.1016/j.freeradbiomed.2003.08.019 [DOI] [PubMed] [Google Scholar]
- 32. Balagam B., and Richardson D. E. (2008) The mechanism of carbon dioxide catalysis in the hydrogen peroxide N-oxidation of amines. Inorg. Chem. 47, 1173–1178 10.1021/ic701402h [DOI] [PubMed] [Google Scholar]
- 33. Zhou H., Singh H., Parsons Z. D., Lewis S. M., Bhattacharya S., Seiner D. R., LaButti J. N., Reilly T. J., Tanner J. J., and Gates K. S. (2011) The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J. Am. Chem. Soc. 133, 15803–15805 10.1021/ja2077137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gorbatenko A., Olesen C. W., Boedtkjer E., and Pedersen S. F. (2014) Regulation and roles of bicarbonate transporters in cancer. Front. Physiol. 5, 130 10.3389/fphys.2014.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hynes N. E., and MacDonald G. (2009) ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 21, 177–184 10.1016/j.ceb.2008.12.010 [DOI] [PubMed] [Google Scholar]
- 36. Grandis J. R., and Sok J. C. (2004) Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol. Ther. 102, 37–46 10.1016/j.pharmthera.2004.01.002 [DOI] [PubMed] [Google Scholar]
- 37. Wang X., Batty K. M., Crowe P. J., Goldstein D., and Yang J. L. (2015) The potential of panHER inhibition in cancer. Front. Oncol. 5, 2 10.3389/fonc.2015.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dorai T., Sawczuk I. S., Pastorek J., Wiernik P. H., and Dutcher J. P. (2005) The role of carbonic anhydrase IX overexpression in kidney cancer. Eur. J. Cancer 41, 2935–2947 10.1016/j.ejca.2005.09.011 [DOI] [PubMed] [Google Scholar]
- 39. Svastova E., and Pastorekova S. (2013) Carbonic anhydrase IX: a hypoxia-controlled “catalyst” of cell migration. Cell Adh. Migr. 7, 226–231 10.4161/cam.23257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aalkjaer C., Boedtkjer E., Choi I., and Lee S. (2014) Cation-coupled bicarbonate transporters. Compr. Physiol. 4, 1605–1637 10.1002/cphy.c130005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gorbatenko A., Olesen C. W., Mørup N., Thiel G., Kallunki T., Valen E., and Pedersen S. F. (2014) ErbB2 upregulates the Na+,HCO3−-cotransporter NBCn1/SLC4A7 in human breast cancer cells via Akt, ERK, Src, and Kruppel-like factor 4. FASEB J. 28, 350–363 10.1096/fj.13-233288 [DOI] [PubMed] [Google Scholar]
- 42. Boedtkjer E., Moreira J. M., Mele M., Vahl P., Wielenga V. T., Christiansen P. M., Jensen V. E., Pedersen S. F., and Aalkjaer C. (2013) Contribution of Na+,HCO3−-cotransport to cellular pH control in human breast cancer: a role for the breast cancer susceptibility locus NBCn1 (SLC4A7). Int. J. Cancer 132, 1288–1299 10.1002/ijc.27782 [DOI] [PubMed] [Google Scholar]
- 43. McIntyre A., Hulikova A., Ledaki I., Snell C., Singleton D., Steers G., Seden P., Jones D., Bridges E., Wigfield S., Li J. L., Russell A., Swietach P., and Harris A. L. (2016) Disrupting hypoxia-induced bicarbonate transport acidifies tumor cells and suppresses tumor growth. Cancer Res. 76, 3744–3755 10.1158/0008-5472.CAN-15-1862 [DOI] [PubMed] [Google Scholar]
- 44. Lee S., Axelsen T. V., Andersen A. P., Vahl P., Pedersen S. F., and Boedtkjer E. (2016) Disrupting Na+, HCO3−-cotransporter NBCn1 (Slc4a7) delays murine breast cancer development. Oncogene 35, 2112–2122 10.1038/onc.2015.273 [DOI] [PubMed] [Google Scholar]
- 45. Hulikova A., Vaughan-Jones R. D., and Swietach P. (2011) Dual role of CO2/HCO3− buffer in the regulation of intracellular pH of three-dimensional tumor growths. J. Biol. Chem. 286, 13815–13826 10.1074/jbc.M111.219899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arner E. S., and Holmgren A. (2001) Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. Chapter 7, Unit 7.4 [DOI] [PubMed] [Google Scholar]
- 47. Lou Y. W., Chen Y. Y., Hsu S. F., Chen R. K., Lee C. L., Khoo K. H., Tonks N. K., and Meng T. C. (2008) Redox regulation of the protein tyrosine phosphatase PTP1B in cancer cells. FEBS J. 275, 69–88 10.1111/j.1742-4658.2007.06173.x [DOI] [PubMed] [Google Scholar]
- 48. Ch'en F. F., Villafuerte F. C., Swietach P., Cobden P. M., and Vaughan-Jones R. D. (2008) S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br. J. Pharmacol. 153, 972–982 10.1038/sj.bjp.0707667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milarski K. L., Zhu G., Pearl C. G., McNamara D. J., Dobrusin E. M., MacLean D., Thieme-Sefler A., Zhang Z. Y., Sawyer T., and Decker S. J. (1993) Sequence specificity in recognition of the epidermal growth factor receptor by protein tyrosine phosphatase 1B. J. Biol. Chem. 268, 23634–23639 [PubMed] [Google Scholar]
- 50. Boivin B., Zhang S., Arbiser J. L., Zhang Z. Y., and Tonks N. K. (2008) A modified cysteinyl-labeling assay reveals reversible oxidation of protein tyrosine phosphatases in angiomyolipoma cells. Proc. Natl. Acad. Sci. U.S.A. 105, 9959–9964 10.1073/pnas.0804336105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boivin B., Yang M., and Tonks N. K. (2010) Targeting the reversibly oxidized protein tyrosine phosphatase superfamily. Sci. Signal. 3, pl2 10.1126/scisignal.3137pl2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meng T. C., Buckley D. A., Galic S., Tiganis T., and Tonks N. K. (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J. Biol. Chem. 279, 37716–37725 10.1074/jbc.M404606200 [DOI] [PubMed] [Google Scholar]
- 53. Wood Z. A., Poole L. B., and Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 10.1126/science.1080405 [DOI] [PubMed] [Google Scholar]
- 54. Parks S. K., Chiche J., and Pouyssegur J. (2011) pH control mechanisms of tumor survival and growth. J. Cell. Physiol. 226, 299–308 10.1002/jcp.22400 [DOI] [PubMed] [Google Scholar]
- 55. Xu J., Eriksson S. E., Cebula M., Sandalova T., Hedström E., Pader I., Cheng Q., Myers C. R., Antholine W. E., Nagy P., Hellman U., Selivanova G., Lindqvist Y., and Arnér E. S. (2015) The conserved Trp114 residue of thioredoxin reductase 1 has a redox sensor-like function triggering oligomerization and crosslinking upon oxidative stress related to cell death. Cell Death Dis. 6, e1616 10.1038/cddis.2014.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., and Winterbourn C. C. (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 286, 18048–18055 10.1074/jbc.M111.232355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Montalibet J., Skorey K. I., and Kennedy B. P. (2005) Protein tyrosine phosphatase: enzymatic assays. Methods 35, 2–8 10.1016/j.ymeth.2004.07.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.