

Abstract
1-Deoxy-d-xylulose 5-phosphate synthase (DXPS) uses thiamine diphosphate (ThDP) to convert pyruvate and d-glyceraldehyde 3-phosphate (d-GAP) into 1-deoxy-d-xylulose 5-phosphate (DXP), an essential bacterial metabolite. DXP is not utilized by humans; hence, DXPS has been an attractive antibacterial target. Here, we investigate DXPS from Deinococcus radiodurans (DrDXPS), showing that it has similar kinetic parameters Kmd-GAP and Kmpyruvate (54 ± 3 and 11 ± 1 μm, respectively) and comparable catalytic activity (kcat = 45 ± 2 min−1) with previously studied bacterial DXPS enzymes and employing it to obtain missing structural data on this enzyme family. In particular, we have determined crystallographic snapshots of DrDXPS in two states along the reaction coordinate: a structure of DrDXPS bound to C2α-phosphonolactylThDP (PLThDP), mimicking the native pre-decarboxylation intermediate C2α-lactylThDP (LThDP), and a native post-decarboxylation state with a bound enamine intermediate. The 1.94-Å-resolution structure of PLThDP-bound DrDXPS delineates how two active-site histidine residues stabilize the LThDP intermediate. Meanwhile, the 2.40-Å-resolution structure of an enamine intermediate-bound DrDXPS reveals how a previously unknown 17-Å conformational change removes one of the two histidine residues from the active site, likely triggering LThDP decarboxylation to form the enamine intermediate. These results provide insight into how the bi-substrate enzyme DXPS limits side reactions by arresting the reaction on the less reactive LThDP intermediate when its cosubstrate is absent. They also offer a molecular basis for previous low-resolution experimental observations that correlate decarboxylation of LThDP with protein conformational changes.
Keywords: conformational change, pyruvate, decarboxylase, crystallography, bacterial metabolism, 1-deoxy-D-xylulose 5-phosphate synthase (DXPS), conformational dynamics, DXP synthase, methylerythritol phosphate (MEP) pathway, thiamine diphosphate (ThDP)
Introduction
1-Deoxy-d-xylulose 5-phosphate synthase (DXPS),3 an essential enzyme in bacterial central metabolism, catalyzes the formation of DXP, which is an intermediate in isoprenoid, pyridoxal phosphate, and thiamine diphosphate (ThDP) biosyntheses (1–5). DXP is not a biosynthetic intermediate utilized by humans; therefore, DXPS is an attractive anti-infective target. DXPS catalyzes the decarboxylation of pyruvate and subsequent carboligation to d-glyceraldehyde 3-phosphate (d-GAP) using a unique ThDP-dependent mechanism that can be selectively targeted (5–10). Unlike related pyruvate decarboxylases such as the E1 subunit of pyruvate dehydrogenase (E1-PDH) (11–17), the enzyme-bound pre-decarboxylation intermediate C2α-lactylThDP (LThDP; Fig. 1) on DXPS is stable in the absence of a decarboxylation trigger (15, 18–21). Upon binding, d-GAP acts as a trigger of LThDP decarboxylation to form the enamine, which subsequently reacts with d-GAP in its second role as acceptor substrate to form DXP (Fig. 1) (15). Besides d-GAP, DXPS utilizes an array of triggers and acceptor substrates, such as O2, which induces LThDP decarboxylation in the absence of d-GAP and accepts two electrons from the C2α-carbanion to form peracetate (Fig. S1A) (22–24).
Figure 1.

Mechanism of DXPS-catalyzed DXP formation and inhibition of DXP formation by MAP. The enzyme conformation (open, closed, or unknown) that is associated with the reaction intermediate state is indicated. E, DXP synthase; R, 4′-amino-2-methyl-5-pyrimidyl; R′, β-hydroxyethyldiphosphate; IP, 1′,4′-iminopyrimidine.
Although the mechanisms by which DXPS stabilizes LThDP and coordinates with d-GAP to induce LThDP decarboxylation are unknown, these steps are hypothesized to be driven by conformational changes on DXPS (21, 25). Hydrogen–deuterium exchange MS (HDX-MS) studies of DXPS from Escherichia coli (EcDXPS) indicate that, in the absence of ligand, EcDXPS displays EX1 exchange kinetics as indicated by the bimodal HDX pattern in three regions near the active site (residues 42–58, 183–199, and 278–298). According to the EX1 exchange mechanism, a bimodal distribution of two distinct states in equilibrium, undeuterated and deuterated, is observed when backbone H–D exchange occurs faster than refolding, such that regions are deuterated simultaneously when present in an open, solvent-exposed conformation. For EcDXPS, the distribution between undeuterated and deuterated states is influenced by substrate binding, implying that unligated EcDXPS exists in (at least) two stable, interconverting conformations (25). In the presence of the stable pyruvate mimic methylacetylphosphonate (MAP), HDX is reduced in these regions, suggesting that the C2α-phosphonolactylThDP (PLThDP)-bound enzyme complex, or the LThDP-bound enzyme complex by extension, adopts a closed conformation (Fig. 1) (25). In contrast, when incubated with d-GAP or DXP, EcDXPS shows increased HDX in these active-site regions, suggesting that d-GAP triggers a conformational change to an open conformation, which presumably lowers the barrier to LThDP decarboxylation (25). This distinctive mechanism in ThDP-dependent enzymology offers opportunities for selective targeting of unique conformations along the reaction coordinate.
Additional structural information is required to understand the molecular mechanisms of DXPS conformational changes and their roles in catalysis. Only two crystal structures of DXPS are available: the 2.4-Å-resolution structure of proteolyzed EcDXPS and the 2.9-Å-resolution structure of Deinococcus radiodurans DXPS (DrDXPS), both obtained with ThDP bound (26). These structures reveal features of DXPS that distinguish it from its closest homologs, such as E1-PDH (Fig. S2), including a unique domain arrangement that positions the active sites of DXPS between domains I and II within each monomer of the active dimer, compared with the active sites of E1-PDH, which are positioned at the dimer interface (26). Additionally, Freel Meyers and co-workers (23) previously demonstrated that the active site of DXPS is 2-fold larger than that of E1-PDH, presumably to accommodate ternary complex formation on DXPS. However, neither DXPS structure was obtained in the presence of substrate, substrate analog, or inhibitor; consequently, a molecular understanding of LThDP stabilization and d-GAP–induced LThDP decarboxylation has been elusive.
In this study, we report the first crystal structures of DXPS in the presence of a pre-decarboxylation intermediate mimic, PLThDP, and the native post-decarboxylation enamine intermediate. These structures of DXPS from D. radiodurans were determined under anoxic conditions to prevent unwanted side reactions (24). Conformational rearrangements of the protein are observed near the active site in these bound states, which is consistent with previous HDX-MS results (25) and provide insights into the molecular basis by which protein conformational flexibility dictates LThDP reactivity. We further provide a full kinetic characterization of DrDXPS under anoxic conditions, which along with our structural data allows us to re-examine the mechanism of DXPS.
Results
DrDXPS is mechanistically similar to other DXPS enzymes
Two key differences between DXPS and other ThDP-dependent enzymes include its ability to stabilize the pre-decarboxylation intermediate, LThDP, and the requirement for ternary complex formation in catalysis, enabled in part by a large active site (15, 23). Several DXPS enzymes have been shown to follow a unique random sequential mechanism (15, 18, 21, 48). Similar to other DXPS enzymes, DrDXPS displays DXP-forming activity (Table S1 and Fig. S3) and is inhibited by the pyruvate mimic MAP (Ki = 2.8 ± 0.2 μm; Table S1 and Fig. S4–S6). Circular dichroism (CD) analysis confirms stabilization of LThDP on DrDXPS, evidenced by accumulation of a positive CD signal at 316 nm (Fig. S7). This behavior is similar to EcDXPS (15) and distinct from other ThDP-dependent pyruvate decarboxylases that follow ping–pong kinetics and do not stabilize the LThDP intermediate. In addition, our results confirm pyruvate-competitive inhibition of DrDXPS by butylacetylphosphonate (BAP; Ki = 5.7 ± 0.5 μm; Table S1 and Fig. S4–S6), a sterically demanding alkylacetylphosphonate that targets the large DXPS active site required for ternary complex formation and is a poor inhibitor of other ThDP-dependent enzymes (6, 8). Finally, we show that DrDXPS displays oxygenase activity (Fig. S1B). Given our previous findings that O2 acts as a trigger of LThDP decarboxylation on EcDXPS, we reason that O2 contributes to conformational flexibility that may impede crystallization. Thus, crystallization studies reported here were conducted under anoxic conditions.
Structure of DrDXPS in the presence of MAP provides a high-resolution view of PLThDP-bound DXPS
DrDXPS was incubated and crystallized under anoxic conditions with excess MAP, and the structure was solved to 1.94-Å resolution (Tables S2 and S3) with one DrDXPS homodimer in the asymmetric unit (ASU). Each monomeric subunit of DrDXPS is composed of ThDP and three domains (Fig. 2). Domain I (residues 8–319) binds the pyrophosphate moiety of ThDP, domain II (residues 320–495) binds the pyrimidine moiety of ThDP, and domain III (residues 496–629) forms part of the dimer interface. Interestingly, DXPS is the only ThDP-dependent enzyme besides 2-oxoacid:ferredoxin oxidoreductases that utilize domains from the same protein chain to bind ThDP (27–30); other ThDP-dependent enzymes anchor one ThDP molecule using domains from two protein chains, which positions the active site at the dimer interface. This structure is the highest-resolution structure of DXPS solved to date, allowing for the generation of a structural model with improved refinement statistics, including a lower clash score, fewer Ramachandran outliers, and fewer side-chain outliers (Table S4). Despite differences in side-chain positions, the backbone atoms align well, with overall root-mean-square deviation (r.m.s.d.) values between our 1.94-Å-resolution structure and the 2.9-Å-resolution DrDXPS structure (1031 Cα atoms in a dimer; PDB code 2O1X) (26) and the 2.4-Å-resolution proteolyzed EcDXPS structure (821 Cα atoms in a dimer; PDB code 2O1S) (26) of 0.22 and 0.90 Å, respectively.
Figure 2.
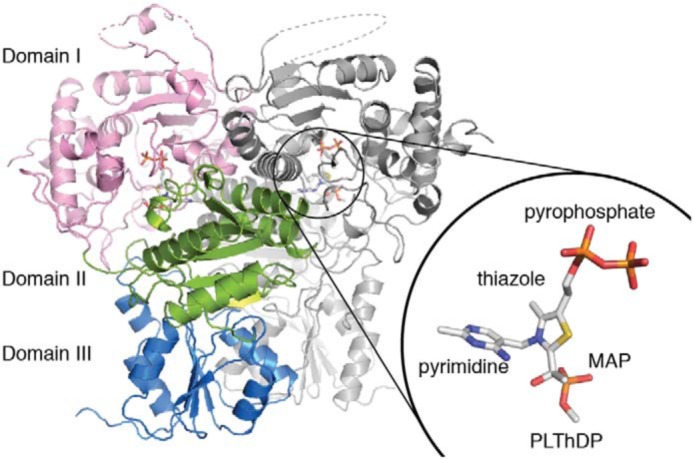
The overall structure of PLThDP-bound DrDXPS. Domain I (pink; residues 8–319) binds the pyrophosphate moiety of ThDP, domain II (green; residues 320–495) binds the pyrimidine moiety of ThDP, and domain III (blue; residues 496–629) forms part of the dimer interface. PLThDP is drawn in sticks and shown in the inset with each moiety labeled. The dashed lines represent disordered regions not seen in the structure.
PLThDP is observed in the active site, consistent with biochemical studies indicating that MAP reacts with ThDP to form a stable pre-decarboxylation intermediate mimic (Figs. 2, inset; 3A; and S8A). His-434, predicted to be involved in pyruvate binding (48, 49), and N4′ of ThDP are positioned to form hydrogen bonds with the C2α-hydroxyl of PLThDP, and Phe-398 stacks against the pyrimidine ring of the ThDP. His-51 and His-304, also predicted to be critical for catalysis in DXPS enzymes (48–50), are positioned to make favorable electrostatic interactions with the phosphonate of PLThDP (Fig. 3A). Because the phosphonate is a mimic of the carboxylate moiety of LThDP (31–34), we predict that His-51 and His-304 stabilize the carboxylate of LThDP.
Figure 3.

Active-site comparisons between DXPS and PDH in the presence of PLThDP. A, the active site of PLThDP-bound DXPS. His-51 and His-304 form hydrogen bonds with the phosphonate, but at 5.7 Å, Tyr-395 is too far away to hydrogen bond. The distance between N4′ and C2α-hydroxyl is 2.7 Å. The structure is colored in the same color scheme as Fig. 2. Tyr-395, Arg-423, and Arg-480, which have been implicated in d-GAP binding (20), are labeled. B, the active site of PLThDP-bound PDH (PDB code 2G25) (34) contains similar residues. His-106 and His-407 are within hydrogen-bond distance of the phosphonate of PLThDP as is Tyr-599 at 2.8 Å. The distance between N4′ and C2α-hydroxyl is 2.5 Å. The pyrophosphate-binding domain is colored pink, and the pyrimidine-binding domain is colored green. The structure is positioned at the same orientation and colored in a similar color scheme as DXPS in A for comparison. C, a stereo figure of structures of DXPS (colored as in Fig. 2) and PDH (gray) in the presence of PLThDP. Structures are aligned by the five atoms that form the thiazole ring. D, a side view of the active site of PLThDP-bound DXPS. His-51 and His-304 are 5.4 and 4.8 Å away, respectively, from C2 of ThDP. The P–C2α–C2 bond angle of PLThDP is 115°. The C2α–C2–S1–C5 dihedral angle is 164°, indicating that the phosphonolactyl moiety is moderately out of the aromatic plane of the thiazole ring. The structure is colored in the same color scheme as A. E, a side view of the active site of PLThDP-bound PDH (PDB code 2G25) (34) The figure is in the same view as D. His-106 and His-407 are closer to C2 of ThDP at 5.2 and 4.3 Å (shifting rightward in the figure), respectively, than the corresponding histidines are in DXPS. The P–C2α–C2 bond angle of PLThDP is 104°. The C2α–C2–S1–C5 dihedral angle is 163°, indicating that the phosphonolactyl group is moderately out of the aromatic plane of the thiazole ring. The structure is colored in the same color scheme as B.
The active site of PLThDP-bound DrDXPS is very similar to that of PLThDP-bound E1-PDH (PDB code 2G25; Figs. 3B and S9) (34, 35). In both cases, the C–P bond of PLThDP is approximately perpendicular to the thiazolium ring, and the C2α-hydroxyl of PLThDP interacts with a His residue (His-434 in DrDXPS and His-640 in E1-PDH) and with the ThDP N4′ (Fig. 3, A and B). Also, in both cases, a Phe residue stacks against the pyrimidine ring of the ThDP. Additionally, both enzymes have two histidine residues providing favorable electrostatic contacts with the phosphonate moiety of PLThDP. Although the residues in contact with PLThDP are the same, there are subtle differences in distances for these two enzymes. For example, the distance from N4′ of ThDP to the C2α-hydroxyl moiety of PLThDP is slightly longer in DrDXPS (2.7 Å in DXPS and 2.5 Å in PDH). The stacking Phe residues are somewhat displaced as are the histidine residues that contact the phosphonate moiety (Fig. 3C).
Although these differences are subtle, it has been suggested that minor variations in the geometry of LThDP may substantially influence decarboxylation rates (34). In particular, it was noted that although one would expect tetrahedral geometry for the LThDP, the angle observed in PLThDP-bound E1-PDH was compressed 104°, indicating a strained conformation (Fig. 3E). Given that this strained conformation would be relieved through decarboxylation, the authors suggested that the strain imposed by active-site composition and geometry could explain facile decarboxylation, which is estimated to be faster than 100 s−1 (36). In contrast, the decarboxylation rate in DXPS is low in the absence of d-GAP (15, 18). The conformation of PLThDP in DXPS refines with a larger angle of 115°, which is consistent with a more stable LThDP intermediate (Fig. 3D). However, variations in PLThDP occupancies, refinement protocols, and resolution warrant caution in drawing strong conclusions from small differences in geometries of these PLThDP-bound states.
Previous biochemical studies of EcDXPS revealed that Arg-420 (Arg-423 in DrDXPS), Arg-478 (Arg-480 in DrDXPS), and Tyr-390 (Tyr-395 in DrDXPS) are important for d-GAP binding in carboligation (20). In the PLThDP-bound DrDXPS structure, Arg-423, Arg-480, and Tyr-395 are 6.9, 8.5, and 10.6 Å away from the phosphonate moiety of PLThDP, respectively (Fig. 3A). Despite many efforts, we have not been able to obtain a structure with d-GAP bound, but the PLThDP-bound DXPS structure indicates that there is room in the active site for d-GAP to bind without a conformational change (Fig. S10).
Structure of DrDXPS cocrystallized with pyruvate reveals a new protein conformation that removes an active-site histidine residue from the active site
A structure of DrDXPS cocrystallized anoxically with pyruvate was solved to 2.40-Å resolution (Tables S2 and S3). The ASU contains one monomeric subunit of DrDXPS, which forms the physiological dimer by crystallographic symmetry. Residues 186–208, 217–224, 244–246, and 292–306 are not visible in this structure, although they were visible in the structure of PLThDP-bound DrDXPS (Table S3). SDS-PAGE analysis of reproduced protein crystals shows no sign of proteolysis (Fig. S11), indicating that the absent residues were disordered instead of proteolyzed in situ. The remaining residues have an r.m.s.d. of 0.27 Å (of 467 Cα in a monomer) to the structure of one monomer of PLThDP-bound DrDXPS.
Near C2 of ThDP, electron density is present that is of an appropriate size for a three-atom adduct (Fig. S8B). Given that DXPS forms a three-carbon, enamine intermediate upon LThDP decarboxylation, it appears we have captured DXPS in a post-decarboxylation state. The protonation and oxidation state of the adduct cannot be determined at this resolution; thus, the intermediate could be either enamine or the less reactive acetylThDP. We will refer to this intermediate as the enamine with the caveat that the enamine may also have converted to the less reactive acetylThDP state during the time frame of the crystallization experiments (37, 38). Notably, cocrystallization of other ThDP-dependent enzymes with substrates (39) has also led to structures of post-decarboxylation enzymatic states, consistent with our observations here.
We found that the C2α-hydroxyl of the enamine makes contacts similar to those made by the same moiety in PLThDP-bound DrDXPS; in both cases, hydrogen bonds are made to His-434 and the N4′ of ThDP (Fig. 4). However, due to a change in geometry following decarboxylation, contact distances are longer in the enamine structure: 3.2 (enamine) versus 2.8 Å (PLThDP) for His-434 and 3.0 (enamine) versus 2.7 Å (PLThDP) for N4′. Instead of having tetrahedral geometry around C2 and thus deviating from the plane of the thiazole ring, the enamine has trigonal planar geometry and is in the plane of the thiazole ring, as expected (34). Also as predicted, the tighter packing between His and N4′ of ThDP with the C2α-hydroxyl of the tetrahedral PLThDP intermediate is relaxed in the case of enamine-bound DXPS (34). This relief of close packing has been proposed to be a driving force for decarboxylation in E1-PDH (34).
Figure 4.
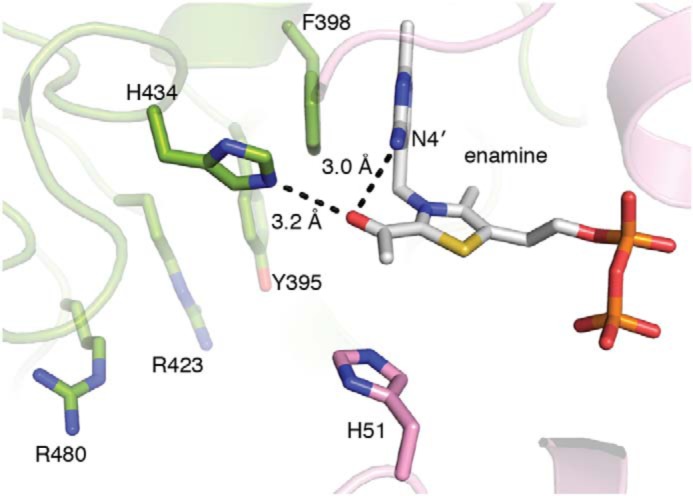
Active site of DrDXPS cocrystallized with pyruvate. A three-atom adduct is found in the active site of the DrDXPS structure cocrystallized with pyruvate, suggesting that decarboxylation of the pyruvate has occurred. The C2α-hydroxyl of the enamine forms hydrogen bonds with His-304 and N4′ of ThDP. Tyr-395, Arg-423, and Arg-480, which have been implicated in d-GAP binding (20), are labeled.
Further comparison of the two DXPS structures (PLThDP versus enamine) reveals a key difference in the active site (Fig. 5). His-304, which was positioned to make favorable electrostatic contacts with the phosphonate of PLThDP, is missing from the active site in the structure with enamine, suggesting that decarboxylation is either preceded by or accompanied by removal of a positive charge. The source of the His-304 displacement is a major conformational change in the position of two structural motifs that connect domains I and II: residues 292–306 that contains active-site residue His-304 and residues 307–319 that form a β-hairpin. Based on the shape of the β-hairpin and the conformational changes observed, we name this region (residues 307–319), the spoon motif, and name the preceding region (residues 292–306), the fork motif. In the PLThDP-bound structure, the fork motif is positioned to contribute His-304 to the active site, and the spoon motif is filling in much of the active-site cleft (Figs. 5A, 6A, and S12A). However, in the enamine structure, the spoon motif has undergone a large conformational change and is positioned on one side of the active-site cleft where it adopts a bent conformation and hydrogen bonds to a glycine-rich region in domain II (residues 350–358), which had no previously assigned function (Figs. 5B, 6B, and S12B). This movement of the spoon motif appears to “tug” on the fork motif, causing it to become disordered, such that residues 292–306 are no longer visible in the enamine structure (Fig. 5B, dashed line). Alternatively, an order-to-disorder transition of the fork motif might initiate movement of the spoon motif. Regardless, the movements of these two motifs appear to be coupled.
Figure 5.

Active-site rearrangements in DrDXPS. A, the structure of DrDXPS in the presence of PLThDP, highlighting the positions of the fork motif (residues 292–306) and spoon motif (residues 307–319). The structures are colored in the same color scheme as Fig. 2 except for the fork and the spoon motifs, which are colored cyan and orange, respectively. B, the structure of DrDXPS cocrystallized with pyruvate. The spoon motif (orange) is shifted by 17 Å from its location in A and bent toward the pocket formed by residues 354–357 (Figs. 6 and 7). The cyan dashed lines represent the fork motif (including His-304), for which there is no density in this structure. Tyr-395, Arg-423, and Arg-480, which have been implicated in d-GAP binding (20), are labeled. Cα atoms of Gly-290, Gly-292, and Ala-307 (if present in structures) are shown as spheres to highlight the conformational changes. The structural overlay of the two structures is shown in Fig. S13.
Figure 6.

Zoomed-in views of the spoon motif in structures of DXPS. A, in the structure of PLThDP-bound DXPS, the spoon motif resides near domain I in the active-site cavity. The hydrogen bonds are formed between the backbone of the spoon motif and Ser-46, Tyr-295, and Tyr-302 of domains I and II, which stabilize this conformation of the spoon motif. B, in the structure of enamine-bound DXPS, the spoon motif is bent toward residues 354–357 of domain II. The hydrogen bonds formed by the backbone atoms of Ala-312–Pro-311 of the spoon motif and Val-356–Glu-357 of domain II stabilize this conformation of the spoon motif. The structures are colored in the same color scheme as Fig. 5A.
In PLThDP-bound DXPS, both the spoon and fork motifs are fully ordered (Fig. 5A). Near the beginning of the fork motif are Gly-290 and Gly-292, two residues in the left-handed helix region of the Ramachandran plot, which offer structural flexibility to this region of the protein, and the flexibility does appear to be substantial. The r.m.s.d. between the two structures in this region is high, 17.4 Å (for 13 Cα), and the first residue of the spoon motif, Ala-307, moves by an impressive 13.1 Å (Figs. 5 and S13). Although His-304 is not visible in the enamine structure, the repositioning of Ala-307 away from the active site indicates that His-304 must move away from the active site and therefore can no longer interact with catalytic intermediates. Together, these structures suggest that movements of the fork and spoon motifs can modulate the reactivity of ThDP-bound intermediates.
Discussion
The work presented here provides three-dimensional snapshots of two different conformational states of DXPS. These structural snapshots provide a framework for the interpretation of the previous HDX research on EcDXPS that first revealed that DXPS has both “open” and “closed” states and contribute the first views of substrate/reaction intermediate-bound states for DXPS.
Previous work showed that when EcDXPS is incubated with MAP, a pyruvate mimic, the HDX rates of three regions near the active site (residues 44–58, 185–201, and 283–303 in DrDXPS) decreased (Fig. 7A). The state was thus termed the closed state as the reduced exchange rates indicated less solvent accessibility. In the PLThDP-bound structure reported here, all three of these previously identified regions are buried by the fork and the spoon motifs (Fig. 7B), explaining the reduced HDX rates observed. The structure of PLThDP-bound DrDXPS provides a view of DXPS in this closed conformation. When DXPS is incubated with d-GAP, the HDX rates of the same three peptides mentioned above are accelerated by 20–60-fold compared with the rates with PLThDP-bound DXPS. Although we do not have a structure of d-GAP-bound DXPS, the enamine structure provides a view of a structural state in which the active site is open, and we found that when the spoon motif is bent, all three HDX regions are exposed to solvent (Fig. 7C). In this apparent open conformation, the solvent-accessible surface areas of residues 44–58, 185–201, and 283–291 are increased by 110–220 Å2 in each segment. Hence, the structure with the bent spoon motif is consistent with the open conformation observed by HDX.
Figure 7.

Regions of DXPS that show changes in hydrogen–deuterium exchange upon ligand binding. A, graphical representation of domain I residues of DrDXPS. Segments colored are the fork motif (cyan), the spoon motif (orange), and regions with significantly altered HDX rates in the full-length EcDXPS when incubated with either d-GAP or MAP (yellow; by sequence alignment) (25). The length of each segment is proportional to the number of residues. Residues 11–40 and 61–180 are omitted because of the limited space. The third HDX region (residues 283–303) partially overlaps with the fork motif (residues 292–306). B, in the structure of PLThDP-bound DXPS, the fork and spoon motifs pack against all three HDX regions, residues 44–58 (1), 185–201 (2), and 283–291 (located behind the fork motif; not labeled). His-51 and His-304 form hydrogen bonds with the phosphonate moiety of PLThDP. C, in the structure of DXPS in the presence of the enamine, the spoon motif is found in a different conformation (not visible at this orientation; see Fig. 5 for the spoon motif). The conformational changes expose all three HDX regions, residues 44–58 (1), 185–201 (partially resolved; 2), and 283–291 (3). The structures are colored in the same color scheme as Fig. 5A.
Combining the structural insights gained in this study with previous structural findings, we propose a more detailed description of the role of conformational dynamics in DXP formation. At the beginning of the reaction cycle before pyruvate binds, DXPS exists in a bimodal distribution of open and closed conformations according to HDX studies (25). Upon pyruvate binding and formation of LThDP, DXPS adopts a closed conformation (25). Our structure of PLThDP-bound DrDXPS suggests that the closed conformation is stabilized through the interaction of the LThDP carboxylate with His-304, which is part of the mobile region. In addition to stabilizing a closed active-site conformation, His-304 and His-51 stabilize the LThDP intermediate itself through interactions with the C2α-hydroxyl. A mutagenesis study of EcDXPS conducted concurrently with this study has provided additional structural and biochemical evidence to support a role of His-49 and His-299 (His-51 and His-304 in DrDXPS) in coordinating the closed conformation, which is critical for efficient LThDP formation.4
Following LThDP formation, d-GAP binds, forming the ternary complex and triggering the adoption of an open state (25). Our structural data suggest that His-304 is displaced from the active site when the enzyme is in the open conformation. It is plausible that removal of this positively charged histidine from the active site destabilizes the LThDP intermediate, triggering decarboxylation.
His-407 in E1-PDH (analogous to His-304 in DrDXPS) is also part of a flexible region that adopts an ordered conformation in the presence of PLThDP (34, 35). However, there is no evidence that His-407 is displaced from the active site following decarboxylation in that enzyme. There is no post-decarboxylation structure of E1-PDH, but it has been proposed that conformational changes occur to create the binding site for the E2 subunit of PDH (34, 36) and to protect the reactive enamine prior to E2-PDH binding (41). There are, of course, several ways to “protect” a reactive intermediate in an enzyme that utilizes two substrates. An enzyme can keep the intermediate from forming in the absence of the cosubstrate (a gated mechanism), or it can form the intermediate but keep the intermediate sequestered in the absence of the cosubstrate. DXPS appears to use the former approach, and E1-PDH uses the latter.
Like DXPS, ThDP-dependent pyruvate:ferredoxin oxidoreductase also has a gated step: CoA binding triggers an electron transfer event. In pyruvate:ferredoxin oxidoreductase, the introduction of the negatively charged thiolate moiety of CoA into the active site is believed to facilitate the electron transfer (29). It does not appear, however, that the negative charge of d-GAP is responsible for its ability to trigger LThDP decarboxylation on DXPS as biochemical studies suggest that decarboxylation can be triggered in the absence of the phosphate moiety by d-glyceraldehyde (20). Instead, the decarboxylation mechanism proposed for DXPS is more similar to the “bait-and-switch” mechanism (39) of oxalate oxidoreductase in which altering charge distribution within the active site plays a central role in decarboxylation. Likewise, removing the positively charged His-304 from the active site of DXPS is expected destabilize the carboxylate of LThDP.
In summary, the structures presented here, obtained under anoxic conditions, provide long-sought insights into intermediate conformations of DXPS and shed light on previous biochemical and HDX characterizations that have indicated a critical role for conformational dynamics in the unique mechanism of DXPS. The structure of PLThDP-bound DXPS in the closed state suggests possible ways that DXPS may stabilize LThDP. Conversely, attempts to cocrystallize pyruvate with DrDXPS unexpectedly provided a post-decarboxylation intermediate-bound structure of DXPS in an open conformation. The structure of the open conformation reveals a major conformational change of the spoon motif, which is consistent with previous HDX-MS studies and provides a higher-resolution description of DXPS conformational dynamics. Overall, our study contributes new knowledge to our understanding of the unique mechanism of this ThDP-dependent enzyme and provides a foundation for future structural studies of DXPS and rational DXPS inhibitor design.
Experimental procedures
Preparation of DrDXPS overexpression construct
D. radiodurans dxs was amplified from genomic DNA with the following primers (Integrated DNA Technologies): 5′-TAT TGC CAT ATG AAC GAA CTT CCC-3′ and 5′-GCA ATA AAG CTT CTA CAC CTC AAT CGG-3′. The PCR was set up as follows: 98 °C for 1 min followed by 30 cycles of 98 °C for 10 s, an annealing temperature of 64 °C for 30 s, and 72 °C for 75 s followed by a final 10-min elongation at 72 °C. GC Buffer (New England Biolabs) was required to obtain the desired PCR product. The purified PCR product and empty pET28b(+) were digested with NdeI and HindIII (New England Biolabs) at 37 °C for 3 h. The digested PCR product was gel-purified and ligated with the linearized vector at 16 °C overnight. The ligated construct was transformed into Escherichia coli TOP10 competent cells for amplification. The purified dxs-pET28b(+) plasmid was fully sequenced by GENEWIZ to confirm the presence of WT D. radiodurans dxs and N-His6 tag.
Overexpression and purification of WT DrDXPS
DrDXPS was overexpressed in E. coli BL21 (DE3) cells harboring dxs-pET28b(+) (26) and purified as described previously (42) with some modifications. Cells were lysed via microfluidizer (Microfluidics Corp.) by passing the resuspended cells through a chilled microfluidizer two times. Fractions containing DXPS following purification by ion exchange were combined and then concentrated to less than 12 ml. Particulates were removed from the protein solution by centrifugation at 21,000 × g at 4 °C for 30 min. The supernatant was loaded onto a G200 26/60 HiLoad gel filtration column (GE Healthcare) and eluted over 1 column volume (320 ml) in gel-filtration buffer (10 mm HEPES, pH 8, 5% (v/v) glycerol, 200 mm NaCl, and 0.5 mm TCEP) at 4 °C. DXPS eluted as a single peak consistent with a dimer of DXPS proteins. Fractions containing DXPS were combined, and ThDP and MgCl2 were added to the protein solution to a final concentration of 1 mm each. The protein was concentrated to >25 mg/ml, and then particulates were removed by centrifugation at 21,000 × g at 4 °C. The supernatant was flash frozen in liquid nitrogen and then stored at −80 °C.
Anoxic characterization of DXP formation
The E. coli methylerythritol-phosphate synthase (IspC)-coupled enzyme assay described previously (9, 19) was utilized to monitor DXP formation with minor modifications. All experiments were performed in the anoxic chamber (Coy Laboratory Products) with an N2/H2 environment at room temperature (25–28 °C). The assay mixture contained DXPS (200 nm), IspC (4 μm), NADPH (300 μm), d-GAP, and pyruvate in DXPS reaction buffer (50 mm HEPES, pH 8, 100 mm NaCl, 2 mm MgCl2, and 1 mm ThDP). DXP formation was initiated upon simultaneous addition of d-GAP and pyruvate to the reaction solution to a final volume of 200 μl. NADPH depletion was monitored by absorption at 340 nm using a 96-well Infinite M Nano UV/visible plate reader (Tecan). d-GAP was held constant at 500 μm when pyruvate was the varied substrate (7.5–1000 μm), and pyruvate was held constant at 1 mm when d-GAP was the varied substrate (7.5–1000 μm). All experiments were conducted in triplicate.
Detection of oxygenase activity
The oxygen-consuming activity of DrDXPS was monitored to determine whether this enzyme catalyzes an oxygenase side reaction as described previously (24) with minor modifications. Pyruvate (1 mm) was added to DXPS (5 μm) in DXPS reaction buffer, and O2 was monitored over time with an Oxytherm+ respiration oxygen monitoring system (Hansatech Instruments Ltd.). All reactions were carried out in triplicate.
Detection of LThDP accumulation on DrDXPS by CD spectroscopy
The ability of DrDXPS to stabilize LThDP was determined using CD spectroscopy to monitor LThDP formation upon titration DXPS with pyruvate as described previously (15) with minor modifications. Pyruvate (12.5–500 μm) was titrated onto DXPS (50 μm) in DXPS reaction buffer (described above but Tris, pH 8, instead of HEPES). CD spectra from 280 to 500 nm with a 1-nm step and 3.5-s averaging time were collected at 4 °C between each addition.
Characterization of inhibitory activity of MAP and BAP against DrDXPS
The determination of inhibitory constant (Ki) and mode of inhibition (MOI) with respect to pyruvate of MAP and BAP were conducted using the IspC-coupled enzyme assay as described above with the following deviations. For Ki determination, MAP or BAP and pyruvate were added simultaneously to the rest of the reaction solution. MAP (0.1–75 μm) or BAP (1–100 μm) was varied, whereas pyruvate and d-GAP were held constant at 108 (2 × Km) and 250 μm (10 × Km), respectively. Plots of initial rate of DXP formation versus concentration of inhibitor were fit to the Morrison equation (43) using GraphPad Prism to determine Ki. To determine MOI, IC50 values for each inhibitor against DXPS at 2 × Kmpyruvate and 10 × Kmpyruvate were determined as described above and plotted against [pyruvate]/Kmpyruvate. The slope of this line was used to determine MOI with respect to pyruvate. All experiments were conducted in triplicate.
Cocrystallization of DrDXPS with MAP
The initial crystallization condition was identified using crystallization screens dispensed by a Mosquito liquid-handling robot (TPP Labtech) in an MBRAUN anoxic chamber with an N2 environment at room temperature. The condition was optimized using the sitting-drop crystallization method in the same environment. 1.0 μl of 27.0 mg/ml DrDXPS in storage buffer containing 1.2 eq of MAP and 5.7 mm d-GAP was mixed with 1.0 μl of well solution (0.056 m BES (Acros), 0.044 m triethanolamine, pH 7.5, 15% (w/v) PEG 3000 (Rigaku), 20% (v/v) 1,2,4-butanetriol (Alfa Aesar), 1% (w/v) nondetergent sulfobetaines (NDSB)-256 (Hampton), 25 mm l-arginine, 25 mm l-threonine, 25 mm l-histidine, and 25 mm trans-4-hydroxy-l-proline) to make a 2-μl sitting drop in a sealed well with 500 μl of well solution. Transparent plate crystals grew in 12–24 h. The crystal used to determine the structure was looped from the drop directly and flash cooled in liquid nitrogen. All chemicals, except specified otherwise, were from Sigma-Aldrich.
Cocrystallization of DrDXPS with pyruvate
DrDXPS with pyruvate was crystallized using the sitting-drop crystallization method dispensed by a Mosquito liquid-handling robot in an MBRAUN anoxic chamber with an N2 environment at room temperature. 50 nl of 28.0 mg/ml DrDXPS in storage buffer containing 50 mm pyruvate (Sigma-Aldrich) was mixed with 150 nl of well solution (0.069 m MOPSO, 0.031 m Bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane(Bis-Tris) pH 6.5, 12.5% (w/v) PEG 4000, 20% (w/v) 1,2,6-hexanetriol, 20 mm dl-arginine, 20 mm dl-threonine, 20 mm dl-histidine, 20 mm dl-5-hydroxylysine, and 20 mm trans-4-hydroxy-l-proline) to make a 200-nl sitting drop in a sealed well with 75 μl of well solution. Transparent rod crystals grew in 14–21 days. The crystal used to determine the structure was looped from the drop directly and flash cooled in liquid nitrogen. 10–20 crystals were collected for SDS-PAGE sample preparation. Each crystal was washed in 500 nl of well solution, harvested from the wash solution, and dissolved in 4× Laemmli sample buffer (Bio-Rad) with 2-mercaptoethanol (Sigma-Aldrich). All chemicals, except specified otherwise, were from Molecular Dimensions.
Data collection and processing
Data were collected at the Advanced Photon Source on Northeastern Collaborative Access Team beamline 24-ID-C on a Pilatus 6MF detector. All data were indexed and scaled in HKL2000 (44) with a CC1/2 of ∼0.8 used as the indicator of where to trim the high-resolution data. CC1/2 is the Pearson's correlation coefficient. Data statistics are listed in Table S2.
Structure determination and refinement
The structure of DrDXPS cocrystallized MAP was determined to 1.94-Å resolution by molecular replacement (MR) with Phaser (45) implemented in PHENIX (46) using the published structure of DrDXPS (PDB code 2O1X (26)) as a search model. An MR solution containing a DrDXPS homodimer per ASU was found with log-likelihood gain of 25,827, translation function Z-score of 136.4, and initial Rwork/Rfree of 30.5%/30.5%. Rigid-body refinement was performed, and atomic coordinates and B-factors were then iteratively refined in phenix.refine (46) with model building and manual adjustments in Coot (47). Final cycles of refinements include TLS parameterizations with one TLS group for the protein monomer. Water molecules were added manually using Fo − Fc electron density contoured to 3.0σ as the criterion. Noncrystallographic symmetry restraints were used throughout refinement.
The structure of DrDXPS cocrystallized pyruvate was determined to 2.40-Å resolution by MR using the published structure of DrDXPS (PDB code 2O1X (26)) as a search model. An MR solution containing a DrDXPS monomer per ASU was found with log-likelihood gain of 5,589, translation function Z-score of 63.9, and initial Rwork/Rfree of 29.8%/30.0%. The DrDXPS monomer forms a physiological homodimer by crystallographic symmetry. The refinement steps were the same as for the 1.94-Å-resolution structure described above except noncrystallographic symmetry restraints were not applicable with only one copy of DrDXPS in the ASU.
Restraints for the enamine adduct were based on previous calculations performed for pyruvate:ferredoxin oxidoreductase from Moorella thermoacetica (29). Restraints for PLThDP complex were from Grade Web Server (Global Phasing). Composite-omit electron density maps calculated by phenix.composite_omit_map (46) were used to verify all three models. The refinement statistics are in Table S2, and residues built into each chain are listed in Table S3. All structure figures were rendered in PyMOL. The software used was compiled by SBGrid (40).
Author contributions
P. Y.-T. C., A. A. D., C. L. F. M., and C. L. D. conceptualization; P. Y.-T. C., A. A. D., C. L. F. M., and C. L. D. formal analysis; P. Y.-T. C., A. A. D., C. L. F. M., and C. L. D. investigation; P. Y.-T. C. visualization; P. Y.-T. C., A. A. D., C. L. F. M., and C. L. D. writing-original draft; P. Y.-T. C., A. A. D., C. L. F. M., and C. L. D. writing-review and editing; A. A. D. validation; C. L. F. M. and C. L. D. supervision; C. L. F. M. and C. L. D. funding acquisition.
Supplementary Material
Acknowledgments
We thank Lauren Bambarger for assistance in synthesizing MAP and Sara Sanders for assistance in synthesizing BAP. We thank the Center for Molecular Biophysics at The Johns Hopkins University for access to the CD spectrometer. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by NIGMS, National Institutes of Health (NIH) Grant P30 GM124165. The Pilatus 6M detector on 24-ID-C beam line is funded by NIH Office of Research Infrastructure High-End Instrumentation Grant S10 RR029205. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grants R35 GM126982 (to C. L. D.) and R01 GM084998 (C. L. F. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S13 and Tables S1–S4.
The atomic coordinates and structure factors (codes 6OUV and 6OUW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
A. A. DeColli, X. Zhang, K. L. Heflin, F. Jordan, and C. L. Freel Meyers, manuscript under review.
- DXPS
- 1-deoxy-d-xylulose 5-phosphate synthase
- ThDP
- thiamine diphosphate
- d-GAP
- d-glyceraldehyde 3-phosphate
- DXP
- 1-deoxy-d-xylulose 5-phosphate
- E1-PDH
- E1 subunit of pyruvate dehydrogenase
- Dr
- D. radiodurans
- PLThDP
- C2α-phosphonolactylThDP
- LThDP
- C2α-lactylThDP
- Ec
- E. coli
- PDB
- Protein Data Bank
- HDX
- hydrogen–deuterium exchange
- MAP
- methylacetylphosphonate
- BAP
- butylacetylphosphonate
- ASU
- asymmetric unit
- r.m.s.d.
- root-mean-square deviation
- MOI
- mode of inhibition
- TCEP
- tris(2-carboxyethyl)phosphine
- BES
- 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid
- MOPSO
- 3-morpholino-2-hydroxypropanesulfonic acid
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MR
- molecular replacement
- TLS
- translation, libration, screw.
References
- 1. Sprenger G. A., Schörken U., Wiegert T., Grolle S., de Graaf A. A., Taylor S. V., Begley T. P., Bringer-Meyer S., and Sahm H. (1997) Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-d-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc. Natl. Acad. Sci. U.S.A. 94, 12857–12862 10.1073/pnas.94.24.12857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lois L. M., Campos N., Putra S. R., Danielsen K., Rohmer M., and Boronat A. (1998) Cloning and characterization of a gene from Escherichia coli encoding a transketolase-like enzyme that catalyzes the synthesis of d-1-deoxyxylulose 5-phosphate, a common precursor for isoprenoid, thiamin, and pyridoxol biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 95, 2105–2110 10.1073/pnas.95.5.2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Du Q., Wang H., and Xie J. (2011) Thiamin (vitamin B1) biosynthesis and regulation: a rich source of antimicrobial drug targets? Int. J. Biol. Sci. 7, 41–52 10.7150/ijbs.7.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mukherjee T., Hanes J., Tews I., Ealick S. E., and Begley T. P. (2011) Pyridoxal phosphate: biosynthesis and catabolism. Biochim. Biophys. Acta 1814, 1585–1596 10.1016/j.bbapap.2011.06.018 [DOI] [PubMed] [Google Scholar]
- 5. Wang X., and Dowd C. S. (2018) The methylerythritol phosphate pathway: promising drug targets in the fight against tuberculosis. ACS Infect. Dis. 4, 278–290 10.1021/acsinfecdis.7b00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith J. M., Vierling R. J., and Meyers C. F. (2012) Selective inhibition of E. coli 1-deoxy-d-xylulose-5-phosphate synthase by acetylphosphonates. Medchemcomm 3, 65–67 10.1039/C1MD00233C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith J. M., Warrington N. V., Vierling R. J., Kuhn M. L., Anderson W. F., Koppisch A. T., and Freel Meyers C. L. (2014) Targeting DXP synthase in human pathogens: enzyme inhibition and antimicrobial activity of butylacetylphosphonate. J. Antibiot. 67, 77–83 10.1038/ja.2013.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanders S., Vierling R. J., Bartee D., DeColli A. A., Harrison M. J., Aklinski J. L., Koppisch A. T., and Freel Meyers C. L. (2017) Challenges and hallmarks of establishing alkylacetylphosphonates as probes of bacterial 1-deoxy-d-xylulose 5-phosphate synthase. ACS Infect. Dis. 3, 467–478 10.1021/acsinfecdis.6b00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartee D., and Freel Meyers C. L. (2018) Targeting the unique mechanism of bacterial 1-deoxy-d-xylulose-5-phosphate synthase. Biochemistry 57, 4349–4356 10.1021/acs.biochem.8b00548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartee D., and Freel Meyers C. L. (2018) Toward understanding the chemistry and biology of 1-deoxy-d-xylulose 5-phosphate (DXP) synthase: a unique antimicrobial target at the heart of bacterial metabolism. Acc. Chem. Res. 51, 2546–2555 10.1021/acs.accounts.8b00321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jordan F., Zhang Z., and Sergienko E. (2002) Spectroscopic evidence for participation of the 1′,4′-imino tautomer of thiamin diphosphate in catalysis by yeast pyruvate decarboxylase. Bioorg. Chem. 30, 188–198 10.1006/bioo.2002.1249 [DOI] [PubMed] [Google Scholar]
- 12. Nemeria N., Baykal A., Joseph E., Zhang S., Yan Y., Furey W., and Jordan F. (2004) Tetrahedral intermediates in thiamin diphosphate-dependent decarboxylations exist as a 1′,4′-imino tautomeric form of the coenzyme, unlike the Michaelis complex or the free coenzyme. Biochemistry 43, 6565–6575 10.1021/bi049549r [DOI] [PubMed] [Google Scholar]
- 13. Jordan F., and Nemeria N. S. (2005) Experimental observation of thiamin diphosphate-bound intermediates on enzymes and mechanistic information derived from these observations. Bioorg. Chem. 33, 190–215 10.1016/j.bioorg.2005.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nemeria N. S., Chakraborty S., Balakrishnan A., and Jordan F. (2009) Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J. 276, 2432–3446 10.1111/j.1742-4658.2009.06964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel H., Nemeria N. S., Brammer L. A., Freel Meyers C. L., and Jordan F. (2012) Observation of thiamin-bound intermediates and microscopic rate constants for their interconversion on 1-deoxy-d-xylulose 5-phosphate synthase: 600-fold rate acceleration of pyruvate decarboxylation by d-glyceraldehyde-3-phosphate. J. Am. Chem. Soc. 134, 18374–18379 10.1021/ja307315u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel H., Nemeria N. S., Andrews F. H., McLeish M. J., and Jordan F. (2014) Identification of charge transfer transitions related to thiamin-bound intermediates on enzymes provides a plethora of signatures useful in mechanistic studies. Biochemistry 53, 2145–2152 10.1021/bi4015743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jordan F., and Nemeria N. S. (2014) Progress in the experimental observation of thiamin diphosphate-bound intermediates on enzymes and mechanistic information derived from these observations. Bioorg. Chem. 57, 251–262 10.1016/j.bioorg.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 18. Eubanks L. M., and Poulter C. D. (2003) Rhodobacter capsulatus 1-deoxy-d-xylulose 5-phosphate synthase: steady-state kinetics and substrate binding. Biochemistry 42, 1140–1149 10.1021/bi0205303 [DOI] [PubMed] [Google Scholar]
- 19. Brammer L. A., Smith J. M., Wade H., and Meyers C. F. (2011) 1-Deoxy-d-xylulose 5-phosphate synthase catalyzes a novel random sequential mechanism. J. Biol. Chem. 286, 36522–36531 10.1074/jbc.M111.259747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Basta L. A. B., Patel H., Kakalis L., Jordan F., and Meyers C. L. F. (2014) Defining critical residues for substrate binding to 1-deoxy-d-xylulose 5-phosphate synthase—active site substitutions stabilize the predecarboxylation intermediate C2α-lactylthiamin diphosphate. FEBS J. 281, 2820–2837 10.1111/febs.12823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Battistini M. R., Shoji C., Handa S., Breydo L., and Merkler D. J. (2016) Mechanistic binding insights for 1-deoxy-d-xylulose-5-phosphate synthase, the enzyme catalyzing the first reaction of isoprenoid biosynthesis in the malaria-causing protists, Plasmodium falciparum and Plasmodium vivax. Protein Expr. Purif. 120, 16–27 10.1016/j.pep.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brammer L. A., and Meyers C. F. (2009) Revealing substrate promiscuity of 1-deoxy-d-xylulose 5-phosphate synthase. Org. Lett. 11, 4748–4751 10.1021/ol901961q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morris F., Vierling R., Boucher L., Bosch J., and Freel Meyers C. L. (2013) DXP synthase-catalyzed C-N bond formation: nitroso substrate specificity studies guide selective inhibitor design. Chem. Bio. Chem. 14, 1309–1315 10.1002/cbic.201300187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeColli A. A., Nemeria N. S., Majumdar A., Gerfen G. J., Jordan F., and Freel Meyers C. L. (2018) Oxidative decarboxylation of pyruvate by 1-deoxy-d-xyulose 5-phosphate synthase, a central metabolic enzyme in bacteria. J. Biol. Chem. 293, 10857–10869 10.1074/jbc.RA118.001980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou J., Yang L., DeColli A., Freel Meyers C., Nemeria N. S., and Jordan F. (2017) Conformational dynamics of 1-deoxy-d-xylulose 5-phosphate synthase on ligand binding revealed by H/D exchange MS. Proc. Natl. Acad. Sci. U.S.A. 114, 9355–9360 10.1073/pnas.1619981114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiang S., Usunow G., Lange G., Busch M., and Tong L. (2007) Crystal structure of 1-deoxy-d-xylulose 5-phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J. Biol. Chem. 282, 2676–2682 10.1074/jbc.M610235200 [DOI] [PubMed] [Google Scholar]
- 27. Costelloe S. J., Ward J. M., and Dalby P. A. (2008) Evolutionary analysis of the TPP-dependent enzyme family. J. Mol. Evol. 66, 36–49 10.1007/s00239-007-9056-2 [DOI] [PubMed] [Google Scholar]
- 28. Gibson M. I., Chen P. Y.-T., and Drennan C. L. (2016) A structural phylogeny for understanding 2-oxoacid oxidoreductase function. Curr. Opin. Struct. Biol. 41, 54–61 10.1016/j.sbi.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen P. Y.-T., Aman H., Can M., Ragsdale S. W., and Drennan C. L. (2018) Binding site for coenzyme A revealed in the structure of pyruvate:ferredoxin oxidoreductase from Moorella thermoacetica. Proc. Natl. Acad. Sci. U.S.A. 115, 3846–3851 10.1073/pnas.1722329115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen P. Y.-T., Li B., Drennan C. L., and Elliott S. J. (2019) A reverse TCA cycle 2-oxoacid:ferredoxin oxidoreductase that makes C-C bonds from CO2. Joule 3, 595–611 10.1016/j.joule.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kluger R., and Pike D. C. (1977) Active site generated analogs of reactive intermediates in enzymic reactions. Potent inhibition of pyruvate dehydrogenase by a phosphonate analog of pyruvate. J. Am. Chem. Soc. 99, 4504–4506 10.1021/ja00455a052 [DOI] [PubMed] [Google Scholar]
- 32. O'Brien T. A., Kluger R., Pike D. C., and Gennis R. B. (1980) Phosphonate analogues of pyruvate. Probes of substrate binding to pyruvate oxidase and other thiamin pyrophosphate-dependent decarboxylases. Biochim. Biophys. Acta 613, 10–17 10.1016/0005-2744(80)90186-2 [DOI] [PubMed] [Google Scholar]
- 33. Turano A., Furey W., Pletcher J., Sax M., Pike D., and Kluger R. (1982) Synthesis and crystal structure of an analog of 2-(α-lactyl) thiamin, racemic methyl 2-hydroxy-2-(2-thiamin)ethylphosphonate chloride trihydrate. A conformation for a least-motion, maximum-overlap mechanism for thiamin catalysis. J. Am. Chem. Soc. 104, 3089–3095 10.1021/ja00375a024 [DOI] [Google Scholar]
- 34. Arjunan P., Sax M., Brunskill A., Chandrasekhar K., Nemeria N., Zhang S., Jordan F., and Furey W. (2006) A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 281, 15296–15303 10.1074/jbc.M600656200 [DOI] [PubMed] [Google Scholar]
- 35. Arjunan P., Nemeria N., Brunskill A., Chandrasekhar K., Sax M., Yan Y., Jordan F., Guest J. R., and Furey W. (2002) Structure of the pyruvate dehydrogenase multienzyme complex E1 component from Escherichia coli at 1.85 Å resolution. Biochemistry 41, 5213–5221 10.1021/bi0118557 [DOI] [PubMed] [Google Scholar]
- 36. Balakrishnan A., Nemeria N. S., Chakraborty S., Kakalis L., and Jordan F. (2012) Determination of pre-steady-state rate constants on the Escherichia coli pyruvate dehydrogenase complex reveals that loop movement controls the rate-limiting step. J. Am. Chem. Soc. 134, 18644–18655 10.1021/ja3062375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menon S., and Ragsdale S. W. (1996) Unleashing hydrogenase activity in carbon monoxide dehydrogenase/acetyl-CoA synthase and pyruvate:ferredoxin oxidoreductase. Biochemistry 35, 15814–15821 10.1021/bi9615598 [DOI] [PubMed] [Google Scholar]
- 38. Reed G. H., Ragsdale S. W., and Mansoorabadi S. O. (2012) Radical reactions of thiamin pyrophosphate in 2-oxoacid oxidoreductases. Biochim Biophys Acta 1824, 1291–1298 10.1016/j.bbapap.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gibson M. I., Chen P. Y.-T., Johnson A. C., Pierce E., Can M., Ragsdale S. W., and Drennan C. L. (2016) One-carbon chemistry of oxalate oxidoreductase captured by X-ray crystallography. Proc. Natl. Acad. Sci. U.S.A. 113, 320–325 10.1073/pnas.1518537113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Cutting edge: collaboration gets the most out of software. eLife 2, e01456 10.7554/eLife.01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kale S., Arjunan P., Furey W., and Jordan F. (2007) A dynamic loop at the active center of the Escherichia coli pyruvate dehydrogenase complex E1 component modulates substrate utilization and chemical communication with the E2 component. J. Biol. Chem. 282, 28106–28116 10.1074/jbc.M704326200 [DOI] [PubMed] [Google Scholar]
- 42. Nemeria N. S., Shome B., DeColli A. A., Heflin K., Begley T. P., Meyers C. F., and Jordan F. (2016) Competence of thiamin diphosphate-dependent enzymes with 2′-methoxythiamin diphosphate derived from bacimethrin, a naturally occurring thiamin anti-vitamin. Biochemistry 55, 1135–1148 10.1021/acs.biochem.5b01300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Copeland R. A. (2005) Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 46, 1–265 [PubMed] [Google Scholar]
- 44. Otwinowski Z., and Minor W. (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Handa S., Dempsey D. R., Ramamoorthy D., Cook N., Guida W. C., Spradling T. J., White J. K., Woodcock H. L., and Merkler D. J. (2018) Mechanistic studies of 1-deoxy-d-xylulose-5-phosphate synthase from Deinococcus radiodurans. Biochem. Mol. Biol. J. 4, 1–11 10.21767/2471-8084.100051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. White J. K., Handa S., Vankayala S. L., Merkler D. J., and Woodcock H. L. (2016). Thiamin diphosphate activation in 1-deoxy-d-Xylulose 5-phosphate synthase: insights into the mechanism and underlying intermolecular interactions. J. Phys. Chem. B. 120, 9922–9934 10.1021/acs.jpcb.6b07248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Querol-Audí J., Boronat A., Centelles J. J., and Imperial S. (2014). Catalytically important residues in E. coli 1-deoxy-d-xylulose 5-phosphate synthase. J. Biosci. Med. 2, 30–35 10.4236/jbm.2014.24006 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.