

Abstract
Activation of ionotropic P2X receptors increases free intracellular Ca2+ ([Ca2+]i) by initiating a transmembrane cation flux. We studied the “a” and “k” splice variants of the rat purinergic P2X7 receptor (rP2X7aR and rP2X7kR) to exhibit a significant difference in Ca2+ flux through this channel. This difference is surprising because the variants share absolute sequence identity in the area of the pore that defines ionic selectivity. Here, we used patch-clamp fluorometry and chimeric receptors to show that the fraction of the total current carried by Ca2+ is a function of the primary sequence of the cytoplasmic N terminus. Using scanning mutagenesis, we identified five sites within the N terminus that respond to mutagenesis with a decrease in fractional calcium current and an increase in permeability to the polyatomic cation, N-methyl-d-glucamine (NMDG+), relative to Na+ (PNMDG/PNa). We tested the hypothesis that these sites line the permeation pathway by measuring the ability of thiol-reactive MTSET+ to alter the current of cysteine-substituted variants, but we detected no effect. Finally, we studied the homologous sites of the rat P2X2 receptor (rP2X2R) and observed that substitutions at Glu17 significantly reduced the fractional calcium current. Taken together, our results suggest that a change in the structure of the N terminus alters the ability of an intra-pore Ca2+ selectivity filter to discriminate among permeating cations. These results are noteworthy for two reasons: they identify a previously unknown outcome of mutagenesis of the N-terminal domain, and they suggest caution when assigning structure to function for truncated P2X receptors that lack a part of the N terminus.
Keywords: ion channel, purinergic receptor, calcium transport, patch clamp, permeability, calcium channel, fractional calcium current, ligand-gated, P2X 7 (P2X7) P2RX7, patch-clamp fluorometry, splice variant
Introduction
P2X receptors (P2XRs)2 are ligand-gated ion channels activated by extracellular ATP (1, 2). Although each of the seven family members (P2X1R–P2X7R) is worthy of study, the P2X7R draws the most attention because of its essential roles in inflammation, immunity, and neuropathic pain (3–7). P2X7Rs are expressed by a host of central and peripheral immune cells, including monocytes, macrophages, lymphocytes, neutrophils, dendritic cells, astrocytes, and microglia (8). In macrophages and microglia, stimulation of P2X7R channels by the high concentrations of extracellular ATP that accompany tissue damage promotes inflammasome-mediated caspase-1 activation and the subsequent release of proinflammatory cytokines (9–11). In lymphocytes, P2X7Rs are involved in autocrine regulation of T-cell activation (12) and promote changes in lymphocyte cell volume and translocation of phosphatidylserine, two events that precede lysis (13).
Several splice variants of mammalian P2X7Rs are currently identified (14, 15). Of these, only two, P2X7aR and P2X7kR, show significant functional activation as defined by ATP-stimulated generation of membrane current (16, 17). The P2X7aR is the predominate variant, being widely expressed across cell types, including macrophages and microglia. They are low-affinity purinergic receptors that require millimolar concentrations of ATP to fully activate (18). In contrast, P2X7kRs are found in lymphocytes and are fully activated by micromolar concentrations of ATP. Interestingly, P2X7aRs but not P2X7kRs transduce a significant transmembrane Ca2+ flux capable of triggering downstream signaling cascades (19). This last detail is particularly noteworthy because the two splice variants share absolute sequence identity in the domains thought to be responsible for ion selection within the channel pore.
In this paper, we use a combination of molecular biology and patch-clamp fluorimetry to identify N-terminal residues responsible for the higher fractional Ca2+ current of the rat P2X7aR (rPX7aR) by comparison to the rat P2X7kR (rP2X7kR). We suggest that the composition and geometry of the cytoplasmic N terminus influences the dimensions of the channel in a manner that determines the ease with which Ca2+ moves through the pore. These data define a previously unknown function of the N terminus and suggest caution when interpreting structural information from crystals of truncated P2X7Rs.
Results
Compared with rP2X7aRs, rP2X7kRs are more sensitive to BzATP and ADP-ribosylation, show a slower rate of deactivation, and display intrinsically elevated permeabilities to large organic cations (20, 21). We recently reported that rP2X7kRs also transduce significantly less Ca2+ as a fraction of total current than their rP2X7aR partners (19). To set the foundation for this report and to introduce the fractional calcium current method, we reproduced this finding in a new set of experiments. rP2X7aR and rP2X7kR were transiently expressed in HEK293T cells and studied using an intracellular (pipette) solution containing 2 mm K5-fura-2. Fig. 1, A–D, shows representative traces and plots of membrane current and fura-2 fluorescence evoked by applying 100 μm BzATP to cells expressing one of the two splice variants. In both cases, BzATP triggered inward membrane current and a decrease in ΔF380, as expected for activation of an ion channel permeable to Ca2+. HEK293T cells express metabotropic P2Y receptors capable of mobilizing Ca2+ from internal stores in response to ATP (22). However, as reported previously (19, 23), we found that untransfected cells failed to display ligand-gated currents and fluorescence changes in response to the application of BzATP or ATP, suggesting that the dialysis of the cell that occurs during whole-cell recording disrupts the signaling pathway responsible for the metabotropic response. From results obtained over several years, we found that the fractional calcium current of the rP2X7kR (2.8 ± 0.3%, n = 34) was significantly smaller (p < 0.0001) than the fractional calcium current of the rP2X7aR (7.2 ± 0.2%, n = 134). This difference is puzzling in light of past work on rat P2X2 and P2X4 receptors that point to the lateral portals and pore-lining second-transmembrane domain (TM2) as the primary determinates of Ca2+ permeability (24), areas in which the two rP2X7R splice variants share absolute sequence identity (Fig. S1).
Figure 1.

Fractional calcium current and PNMDG/PNa of two rP2X7R splice variants. A and C show raw and integrated data collected from voltage-clamped HEK293T cells transiently expressing either rP2X7aRs (A) or rP2X7kRs (C). Holding voltage = −60 mV. Membrane currents (nA) are shown as black traces, integrated currents (nC) as red traces, and fura-2 fluorescence (BU; 380-nm excitation and 510-nm emission) as gray traces. Applications of 100 μm BzATP (cyan bars) generated inward membrane current and decreases in fura-2 fluorescence. These data were used to plot integrated currents (QT) versus calibrated fluorescent signals (QCa) in B and D. The fractional calcium currents (Pf%) were calculated from the slopes of the linear fits to the plotted data. The fractional calcium current of the rP2X7kR is smaller than that of the rP2X7aR despite the fact that the two splice variants share sequence identity in the pore-forming domains. E and G show raw data of Erev measurements from the two WT splice variants bathed in the extracellular NMDG+ solution. Holding voltage = −40 mV, and voltage ramps ranged from −80 to 60 mV. ATP (500 μm) evoked outward current through the rP2X7aR (E), and inward current through the rP2X7kR (G). Ramp1 was used to measure leak current, and Ramp2 was used to measure total current in the presence of ATP. F and H, plot ATP-gated current, equal to Ramp2 − Ramp1, versus ramp voltage. PNMDG/PNa values were calculated from the values of the X-intercept (i.e. Erev) as described under “Experimental procedures.”
rP2X7aR and rP2X7kR display intrinsic differences in PNMDG/PNa unrelated to accumulation and depletion of intracellular ions
rP2X7aR and rP2X7kR also show dissimilar abilities to permeate large polyatomic cations, measured as differences in PNMDG/PNa values derived from reversal potential (Erev) measurements of agonist-gated currents (20). As a prelude to experiments on chimeric proteins (see below), we determined the Erev values of the ATP-gated currents of the WT splice variants by applying voltage ramps at 0.5 s after the start of applications of ATP to HEK293T cells transfected with genes encoding either the rP2X7aR or rP2X7kR (Fig. 1, E–H). We obtained a range of current amplitudes either by picking cells based on the level of fluorescence emitted by the co-transfected reporter protein, eGFP, or using a range of ATP concentrations (0.01–1.0 mm ATP). In these experiments, uncompensated series resistance equaled 4.1 ± 0.2 MΩ (range = 2.1–5.2, n = 23), and slope conductances ranged from ∼0.5 to 180 nS. In keeping with published reports (20), we found that the PNMDG/PNa of the rP2X7aR was 0.06 ± 0.00 (Erev = −70.2 ± 1.4; n = 14), and the PNMDG/PNa of the rP2X7kR was 0.30 ± 0.03 (Erev = −32.1 ± 3.0; n = 10), suggesting that the rP2X7kR splice displays a higher intrinsic permeability to NMDG+ than the rP2X7aR.
The difference in PNMDG/PNa of the two splice variants might reflect unintended changes in concentrations of intracellular ions, as reported recently for rP2X2Rs (25). To explore the possibility that intracellular NMDG+ accumulation and Na+ depletion underlie the striking differences in intrinsic PNMDG/PNa of the rP2X7aR and rP2X7kR, we used the Reservoir Model and the protocol of Li et al. (25) to estimate changes in the intracellular concentration of ions and the Erev values of the two splice variants. We used realistic values of access resistance (5 MΩ), channel density (100 nS), and our measured values of PNMDG/PNa to estimate the expected Erev values. The in silico protocol is shown in Fig. 2A for rP2X7aR and in Fig. 2D for rP2X7kR. In each simulation, a control I/V curve (50 ms, raw voltage and current data shown as red lines) was obtained from a model cell held at −90 mV and exposed to ATP (leftmost horizontal cyan bar in each panel of Fig. 2D). The ATP was removed; the membrane potential was jumped to −60 mV, and ATP was reapplied for a length of time (0.5 s, middle cyan bar of Fig. 2D) equal to the time at which we applied voltage ramps in our empirical measurements. Then, the membrane potential was returned to −90 mV, and a second I/V curve (raw voltage and current data shown as blue lines in Fig. 2D) was obtained in the presence of ATP to determine the effect of changes in intracellular ion concentrations on the Erev of the ATP-gated current. For the rP2X7aR, the model predicted an outward agonist-gated current (Fig. 2A), a modest change in [Na+]i and [NMDG+]i (Fig. 2B), and a small shift in the Erev (∼4 mV) (Fig. 2C). The Erev measured after the 0.5-s application of ATP was −67 mV, which was close to our empirical measurement of −70 mV obtained at the same time point. In the case of the rP2X7kR, ATP caused an inward current (Fig. 2D) and a larger change in [Na+]i and [NMDG+]i (Fig. 2E). However, like the P2X7aR, the shift in Erev was modest (∼5 mV) with a final value of −24 mV (Fig. 2F) which, again, was a reasonable match to our empirical measurement of −32 mV. The disparate starting points and modest shifts in the Erev values of rP2X7aR and rP2X7kR (see Fig. 2, C and F) do not support the hypothesis that changes in intracellular ion concentrations underlie the large differences in empirically measured Erev values of the two splice variants measured at the start of an application of ATP. Therefore, we conclude that disruption of ion gradients is not responsible for the stark differences in the intrinsic PNMDG/PNa values of rP2X7aRs and rP2X7kRs.
Figure 2.

In silico modeling of ATP-gated responses. A and D show the voltage protocols (top traces) and resulting currents (bottom traces) for rP2X7aRs or rP2X7kRs, respectively. ATP causes an immediate inward current through the rP2X7aR (A) and an immediate outward current through the rP2X7kR (D) at a holding voltage of −60 mV. B and E show the expected changes in intracellular concentrations of ions, and C and F show the expected shift in Erev caused by the ionic changes. The shifts are modest for both splice variants. G plots the results of empirical measurements of the slope conductances of the ATP-gated currents obtained near the Erev of the rP2X7aR (“A”) and rP2X7kR (“K”) for cells bathed in the NMDG+ bath solution.
We also considered the possibility that the difference in Erev values of the rP2X7aR and rP2X7kR reflected an experimental error that occurs when large access resistances cause significant voltage drops across the tips of the recording electrode. To determine whether access resistance impacted our measurements of Erev, we plotted slope conductance against the measured Erev values of our empirical measurements. The Erev values of currents through rP2X7aR and rP2X7kR were close to values predicted by the Reservoir Model (dotted lines of Fig. 2G) when slope conductances were <100 nS. In the case of the rP2X7kR, slope conductances of >100 nS resulted in Erev values that were more negative than their predicted value while remaining significantly more positive that the predicted Erev of the rP2X7aR. Because we saw no clear effect of conductance on the Erev of either splice variant when slope conductance was <100 nS, we conclude that the empirical differences we measured in PNMDG/PNa were not explained by an error in voltage-clamp recording.
Role of the N terminus in determining Ca2+ flux through P2X7Rs
Next, we sought to determine whether the unique domains encoded by exon 1 (i.e. the N terminus and part of TM1; Fig. 3A) were responsible for the divergent fractional calcium currents and PNMDG/PNa values of the rP2X7aR and rP2X7kR. To identify the responsible domain(s), we made chimeric proteins that contained parts of both the rP2X7aR and rP2X7kR (Fig. 3B). Specifically, we constructed two chimeras by replacing specific domains of rP2X7aR with their rP2X7kR counterparts. The first, called N-term(k), contains the N terminus of the rP2X7kR, and the second, called TM1(k), contains the TM1 of rP2X7kR. Accurate construction of each chimera was verified by sequencing, and then the constructs were transfected into HEK293T cells to measure fractional calcium current and PNMDG/PNa. Again, to minimize effects of changes in intracellular ion concentrations, we measured Erev values 0.5 s after the start of applications of ATP. In physiological Na+ solutions, both chimeras responded to ATP with inward currents that displayed unique phenotypes. We noticed that the rates of deactivation of membrane current following washout of BzATP fell between the faster rP2X7aR and the slower rP2X7kR, with the N-term(k) chimera deactivating faster than the TM1(k) chimera (Fig. 3C). We did not quantify this difference because deactivation was variable and a study of gating kinetics was not the focus of this work. The more important outcome was a difference in ionic selectivity. We found that the N-term(k) chimera closely resembled the WT rP2X7kR in having a small fractional calcium current (4.0 ± 0.4%, n = 15) (Fig. 3D) and a high intrinsic PNMDG/PNa (0.22 ± 0.01, n = 8) (Fig. 3E). In contrast, the PNMDG/PNa (0.05 ± 0.00, n = 9) and fractional calcium current (8.8 ± 0.5, n = 6) of the TM1(k) chimera were not statistically significantly different (p < 0.01) from those of the WT rP2X7aR. We draw three conclusions from these experiments. First, in keeping with past results on P2X2Rs (26), our data suggest that TM1 plays little or no role in regulating Ca2+ flux through rP2X7Rs. Second, the data point to the N terminus as the primary determinate of the difference in ionic selectivity of the chimeras. If so, then the N terminus most likely underlies the difference in selectivity of the WT rP2X7R splice variants, too. Third, we found a strong negative correlation between fractional calcium current and PNMDG/PNa, suggesting that a wider pore produces a smaller fractional calcium current.
Figure 3.

Chimeric receptors. A, exon 1 of rat and mouse P2X7aRs and P2X7kRs shows differences in primary sequence. All other parts are identical (see also Fig. S1). B, cartoon representations of the different chimeric rP2X7Rs. Domains contributed by rP2X7aRs are shown in black. Domains contributed by rP2X7kRs are shown in red. The shaded boxes are common to both splice variants. C, raw data traces of representative currents through WT and chimeric rP2X7Rs recorded at a holding voltage of −60 mV and normalized to their peak currents. D, box-and-whisker plots of fractional calcium currents of the two splice variants. The thick solid black lines demarcate median values; boxes are interquartile ranges, and whiskers are equal to standard deviations. Constructs that are significantly different from the WT rP2X7aR are denoted by red lettering in the x axis label. E, box-and-whisker plots of intrinsic PNMDG/PNa of the WT and chimeric receptors. Note that lower fractional calcium current (D) correlates with higher intrinsic PNMDG/PNa (E).
To further probe the role of the N terminus in setting fractional calcium current, we performed scanning mutagenesis of the rP2X7aR to identify individual site(s) that might in some way regulate Ca2+ flux through the pore. We generated 23 N-terminal mutant receptors, each with a single altered amino acid. In most cases, we substituted alanine for charged or polar amino acids to reduce polarity and/or charge. In cases where the existing residue was already neutral, we substituted the basic amino acid, arginine, to increase charge. Nineteen of the 23 mutant receptors responded to applications of 100 μm BzATP with large inward currents. Of these, most showed fractional calcium currents that were not significantly different from that of the WT rP2X7aR (Fig. 4A). The exceptions were Glu14 and Thr15. When replaced by alanine, these two mutants (E14A and T15A) showed fractional calcium currents that were significantly smaller (3.7 ± 0.7%, n = 10; and 4.06 ± 0.4%, n = 20, respectively) than the WT rP2X7aR (WT). Further increasing hydrophobicity by addition of tryptophan at position 15 (T15W) caused an even greater decrease in fractional calcium current to 1.1 ± 0.7% (n = 5; Fig. 4B). Thus, the T15W mutation shows that a single mutation in the N terminus of the rP2X7aR is sufficient to render the channel unfavorable for passage of Ca2+.
Figure 4.

Fractional calcium current of N-terminal P2X7aR mutants. A, amino acid sequence of the N terminus of the rP2X7aR was changed one residue at a time to yield 23 mutants. Nineteen of these gave measurable currents in response to 100 μm BzATP. Only E14A and T15A had fractional calcium currents that were significantly different (marked with red lettering the x axis label) from the WT rP2X7aR (wt). The four unresponsive mutants were regenerated in a rP2X7aR-YFP background (I21R*, S23A*, V24R*, and N25A*); of these, only V24A* failed to respond to BzATP. The fractional calcium currents (Pf%) of the other three were significantly smaller than the WT rP2X7aR-YFP (wt*). B, fractional calcium current was measured from a range of rP2X7aR mutants involving Thr15. Again, results significantly different from the WT rP2X7aR are denoted with red lettering.
Four of the 23 mutant receptors (Ile21, Ser23, Val24, and Asn25) showed no response to either 300 μm BzATP or 5 mm ATP. We previously found that we could recover activity from a mutated P2X2R by attaching YFP to its C terminus (26). Although we do not understand why this works, we used the same strategy in an attempt to recover function from the four silent rP2X7aR mutants described here. We tagged the WT rP2X7aR with YFP to give a rP2X7aR-YFP chimera, and then we used this construct as a template for mutagenesis to yield rP2X7aR-YFP-I21R, rP2X7aR-YFP-S23A, rP2X7aR-YFP-V24A, and rP2X7aR-YFP-N25A. Three of these (I21R*, S23A*, and N25A*) showed robust BzATP-gated currents that superficially resembled the rP2X7aR-YFP. However, all three mutants displayed fractional calcium currents that were significantly smaller (Fig. 4A). The reductions in fractional calcium current were not caused by the addition of the YFP tag because we measured no significant difference in the fractional calcium current of the template rP2X7aR-YFP (wt* of Fig. 4A) by comparison with the WT rP2X7aR (wt of Fig. 4A). Addition of YFP did not rescue the V24A mutant, and this mutant was not considered further.
Taken together, we used mutant receptors to identify two stretches of the N terminus that influence the ion selectivity of the pore. The first is made of the adjoining amino acids, Glu14 and Thr15, that sit in or near a consensus site for PKC phosphorylation (27). The second consists of juxtamembrane amino acids previously proposed to regulate P2X7R receptor gating (28).
Mimicking phosphorylation of the consensus N-terminal PKC site does not alter Ca2+ flux
Thr15 is conserved throughout the P2X receptor family and is thought to form a consensus TX(K/R) site for protein kinase C phosphorylation (16). To determine whether the different fractional calcium currents of the rP2X7aR, rP2X7kR, and the rP2X7aR-T15A/T15W mutants could be explained by a change in the phosphorylation state of the receptor, we performed additional rounds of mutagenesis on Thr15 (Fig. 4B). First, we swapped the threonine for serine to give a mutant T15S-rP2X7aR that contained the native amino acid of the rP2X7kR. This mutation tended to increase fractional calcium current to 9.7 ± 0.3% (n = 20), although the difference was not significant. The fact that this mutation did not decrease fractional calcium current demonstrates that the identity of the amino acid occupying this site does not by itself explain the lower fractional calcium current measured from the rP2X7kR splice variant. Next, we replaced Thr15 with glutamate to mimic the addition of a fixed negative charge that results from phosphorylation. The resulting T15E mutant was essentially impermeable to Ca2+ (fractional calcium current = 0.6 ± 0.1, n = 11), which leaves open the possibility that phosphorylation might indeed decrease fractional calcium current. However, this is unlikely to be true because swapping Thr15 with positively charged lysine (T15K) also decreased fractional calcium current (Fig. 4B). Thus, these data do not support the hypothesis that the negative charge resulting from phosphorylation of Thr15 regulates Ca2+ flux through rP2X7Rs.
Attempts to chemically modify cysteine-substituted N-terminal mutants fail to alter ATP-gated currents
How does the N terminus regulate Ca2+ through the channel? It could directly interact with permeating ions if it lined the pore. In fact, the N and C termini of some P2XRs assemble into a transient “cytoplasmic cap” that forms fenestrations for ion egress into the cytoplasm (29, 30). We sought to test the hypothesis that the five relevant N-terminal amino acids (orange residues of Fig. S1) line the innermost aspect of the rP2X7aR permeation pathway where they influence Ca2+ flux through direct electrostatic interactions. Thus, we constructed cysteine-substituted P2X7aR mutants and measured the effect of applying sulfhydryl-reactive MTSET+ on the ATP-gated current.
Four of the mutants (T15C, I21C, S23C, and N25C) responded to 2 mm ATP with significant inward current that superficially resembled the WT receptor in rates of activation and deactivation. The fifth mutant, rP2X7aR-E14C, showed a biphasic current with a rapidly desensitizing initial phase followed by a small plateau phase (upper middle panel of Fig. 5).
Figure 5.
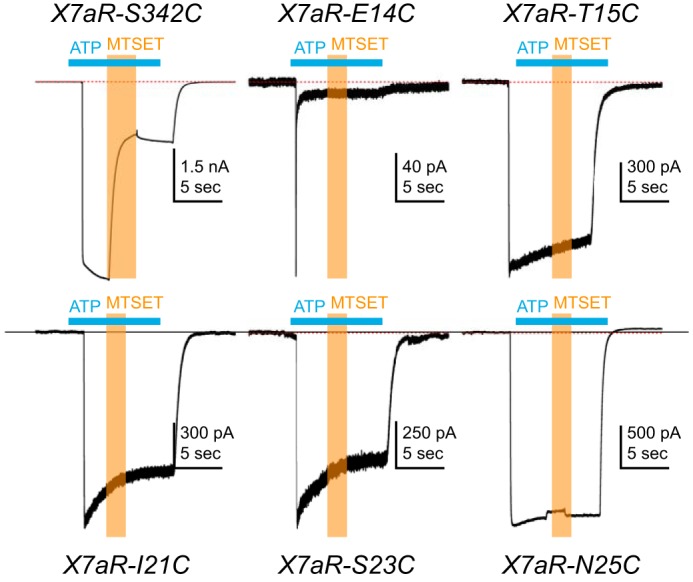
Cysteine-scanning mutagenesis. HEK293T cells expressing cysteine-substituted mutant rP2X7aRs were voltage-clamped at a holding potential of −60 mV. ATP (2 mm) was applied at times indicated by the horizontal cyan bars, and MTSET (1 mm) was co-applied at times indicated by the vertical orange bars. Co-application of MTSET caused a significant and irreversible inhibition of rP2X7aR-S342C mutant receptors but had no effect on any of the other five cysteine-substituted rP2X7aRs. Note that the rP2X7aR-E14C differed from the WT rP2X7aR and the other mutant receptors in showing a biphasic current. Although MTSET had no effect on the plateau phase of the rP2X7aR-E14C, the altered phenotype of the ATP-gated current suggests that caution be used in drawing firm conclusions from this mutant receptor.
Next, we looked to see whether modification of the cysteinyl side chains altered current flow through the channel. Previous work demonstrated accessibility of cysteine-substituted pore-lining residues of the rP2X7aR pore to water-soluble cysteinyl-reactive compounds (31). Therefore, as proof of concept, we measured the effect of 1 mm MTSET+ on one of these pore-lining mutants (rP2X7aR-342C). As reported previously (31), modification of rP2X7aR-342C significantly decreased ATP-gated current (upper left panel of Fig. 5). In stark contrast, co-application of MTSET+ had no effect on the magnitude of the ATP-gated responses of the four mutant rP2X7aRs (T15C, I21C, S23C, and N25C) with current properties that resembled the WT receptor (Fig. 5). Cd2+ (40 μm), another cysteine-reactive probe (32), similarly failed to alter current through these four mutant receptors. We also co-applied MTSET+ during the small plateau phase of the current through the rP2X7aR-E14A mutant and failed to see an effect. Although we are tempted to conclude that E14A does not line the pore, the drastically altered phenotype of the current response made firm conclusions impossible to draw. Nevertheless, the simplest hypothesis that explains the lack of effect of MTSET+ on E14A and the other four functional mutants is that these residues are either inaccessible to hydrophilic modifiers or do not block current when modified. In either case, our results argue against an impactful interaction of the residues and permeating Ca2+ in the channel pore. Instead, we favor the hypothesis that the mutations cause an indirect change in the structure of the pore that negatively influences rP2X7aR's preference for Ca2+, perhaps by disrupting an intrapore Ca2+-binding site.
Where is the intra-pore Ca2+ selectivity filter?
Mutating one of three amino acids (Thr336, Thr339, and Ser340; red residues of Fig. S1) in the TM2 of the rP2X2R significantly decreases Ca2+ permeability (33) and flux (23). Recent work suggests that the equivalent sites in the hP2X7aR (Ser339, Ser342, and Tyr343) line the channel pore and help define the monovalent cation selectivity filter (31). To determine whether these sites influence Ca2+ flux, we measured the fractional calcium current after site-directed mutagenesis of the TM2 of the rP2X7aR. First, we substituted glutamate with the belief that addition of fixed negative charge would increase Ca2+ flux, as expected from published results on rP2X2R (23). We measured a significant increase in fractional calcium current for two of the three mutants (S339E and Y343E; Fig. 6). The third mutant, S342E, failed to respond to BzATP (100 μm). Second, we mutated Ser339 and Ser342 to tyrosine, which conserves polarity but increases bulk. We found no effect of the S339Y mutation on fractional calcium current. In contrast, we measured a significant decrease in the S342Y mutant (Fig. 6), which supports the hypothesis that this residue sits at the narrowest part of the pore (33). Third, we mutated Tyr343 to phenylalanine to remove the hydroxyl group but conserve size and saw a large reduction in fractional calcium current (Y343F, Fig. 6). Taken together, our data support the hypothesis that the selectivity filter of the rP2X7aR sits in a narrow part of the transmembrane domain close to Ser342 (31) and suggest that homologous sites regulate the Ca2+ current across the P2XR family.
Figure 6.
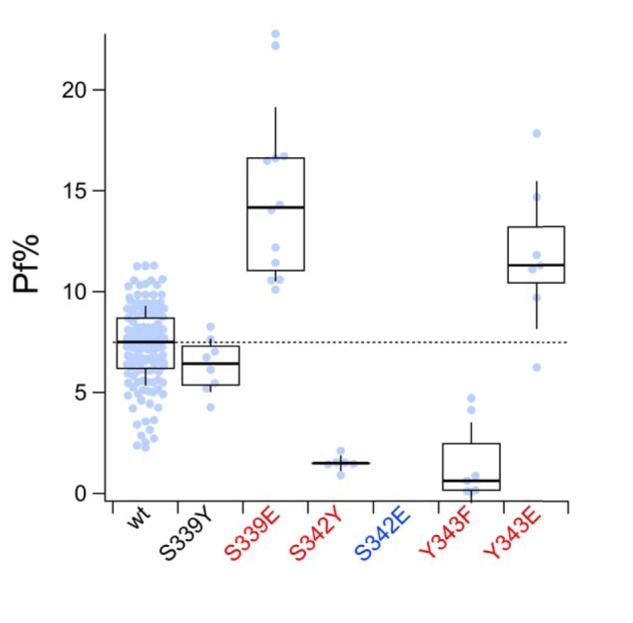
Pore-lining residues form the intra-pore Ca2+-binding site. Fractional calcium current (Pf%) was measured from HEK293T cells expressing rP2X7Rs with single mutations at sites in TM2 that are thought to line the pore (31). Mutants with fractional calcium currents that significantly differ from the WT P2X7aR (wt) are labeled with red lettering on the x axis. Only the S342E mutant (blue lettering) failed to respond to BzATP with measurable inward current. Other mutants (S339E, S342Y, Y343F, and Y343E) showed changes in fractional calcium current that suggest their involvement with permeating Ca2+ in the pore of the channel.
Small fractional calcium currents are a common feature of wide P2XRs
As mentioned previously, rP2X7kRs have a high intrinsic permeability to NMDG+ (20) and show little preference for Ca2+ (19). The unusually high intrinsic PNMDG/PNa suggests a wide pore. Does the wider pore explain the lower fractional calcium current of the rP2X7kR? It could if expanding the narrow region around Ser342 reduces the ability to manage Ca2+. To determine whether small fractional calcium current shows a positive correlation with high intrinsic NMDG+ permeability, we measured and compared the fractional calcium currents and PNMDG/PNa values of a collection of P2X7Rs. We included three constructs with fractional calcium currents that resembled the rP2X7aR (rP2X7aR-R20A of Fig. 4, TM1(k) of Fig. 3, and mouse P2X7aR (19)) and eight constructs that resembled the rP2X7kR (rP2X7aR-E14A of Fig. 4A, rP2X7aR-T15A,W,E,K of Fig. 4B, mouse P2X7kR (19), two C-terminal truncated rP2X2Rs (“Δ18” and “trunc”) (34, 35)), and N-term(k) of Fig. 3). Our results are plotted in Fig. 7. We found a negative correlation between fractional calcium current and PNMDG/PNa (Pearson's r = −0.836) with lower fractional calcium currents accompanying higher PNMDG/PNa values. These data support the hypothesis that the lower fractional calcium current of the WT rP2X7kR results from a wide pore and a disrupted Ca2+ selectivity filter and suggest that the open pore conformations of rP2X7aR and rP2X7kR, formed from identical TM2s, are somewhat different in geometry.
Figure 7.
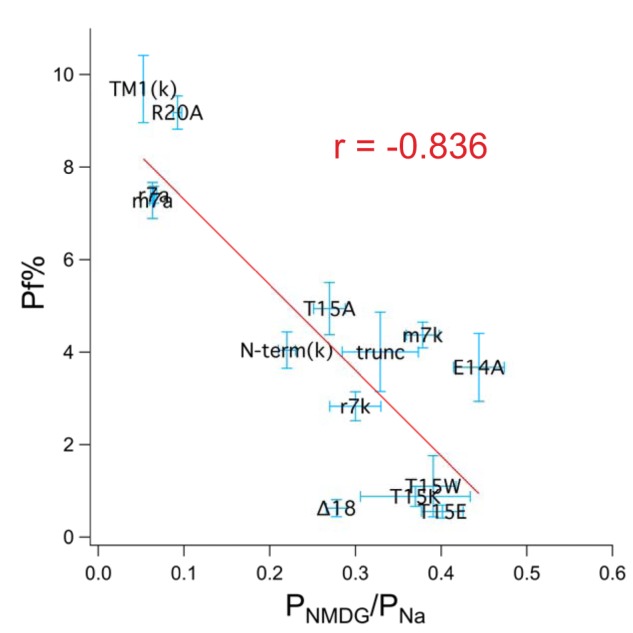
Fractional calcium current negatively correlates with PNMDG/PNa. The graph shows data obtained from rat, mouse, and human P2X7Rs. Fractional calcium current (Pf%) is plotted against PNMDG/PNa. The red line is the best linear fit to the data using the resident curve fitting algorithm of Igor Pro.
An N-terminal mutant of rP2X2R also shows a reduced fractional calcium current
Finally, we wondered whether our observations on rP2X7R represent a general trend across the receptor family. To gain insight, we mutated the five equivalent sites of the rP2X2R (Glu17, Thr18, Val24, Gln26, and Arg28; see green residues of Fig. S1) and measured fractional calcium current. As reported previously (27), we found that mutating Thr18 greatly accelerated desensitization, which made measurements of fractional calcium current impossible; after many unsuccessful attempts, we abandoned the study of this site. We substituted alanine or glutamine at the four other sites to reduce polarity; these mutations decreased the fractional calcium current of Glu17 mutants but had no effect at the other three sites (Fig. 8). Thus, although mutations in the N terminus of the rP2X2R are capable of reducing the Ca2+ flux, they are less effective than those placed in rP2X7R.
Figure 8.
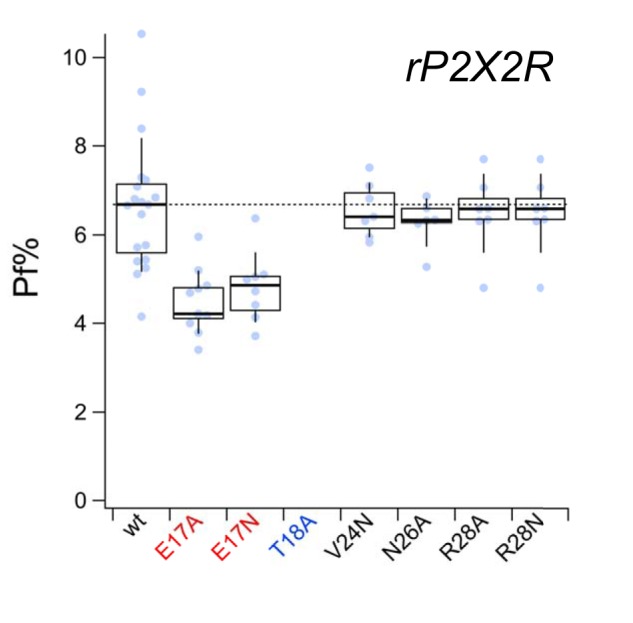
Fractional calcium current of N-terminal rP2X2R mutants. Fractional calcium current (Pf%) was measured from WT (wt) and N-terminal mutant rP2X2Rs as described under “Experimental procedures.” Although T18A (labeled with blue lettering on the x axis) showed an ATP-gated current, it desensitized too quickly to give a measurable change in the fractional calcium current. Mutants with fractional calcium currents that were significantly different from the WT rP2X2R are labeled with red lettering.
Discussion
The cytoplasmic N and C termini of all P2XRs are subject to the effects of post-translational modification, protein–protein interactions, and/or single-nucleotide polymorphisms on channel expression, trafficking, and gating. These outcomes are particularly well-documented for the C terminus of the P2X7R for which a wealth of data exist (14). Less is known about the shorter N terminus (36). In P2X1 receptors, the N terminus mediates partial agonist efficacy (37), desensitization (38), and the sensitivity of gating to cholesterol (39, 40) and phorbol ester (41). The N terminus regulates membrane targeting of the P2X6 receptor (42) and alters P2X7aR channel gating in a manner that prevents the facilitation of the membrane current seen with repeated or prolonged applications of ATP (28, 34). Facilitation is a hallmark property that sets P2X7aRs apart from other family members (43). Interestingly, replacing the N terminus of the human P2X2R with the cognate region of the human P2X7R does not impart facilitation to the chimera suggesting that the gating effect involves an interaction of the N terminus with another receptor domain (28). N-terminal mutagenesis of rP2X7aR also dramatically alters the permeability of the channel to large polyatomic cations (28, 34, 44), a result we use here to help explain the novel effect of mutagenesis on Ca2+ flux through the channel. Importantly, the Erev of the rP2X7kR did not result from a gradual change in the concentration of intracellular ions as demonstrated for rP2X2Rs, but rather reflects an intrinsic property of the pore. We draw this conclusion from an in silico reservoir model of whole-cell patch clamp that accurately predicts the time course and magnitude of the shift in Erev of ATP-gated current of rP2X2R measured under bi-ionic NMDG+out/Na+in conditions (25). The model predicts, and experimental studies confirm, that ATP gates an immediate outward current under bi-ionic conditions that resolves to an inward current over the course of a few seconds (25). The shift in the direction of the current results from a change in Erev that reflects a gradual increase in [NMDG+]i and a decrease in [Na+]i. In our empirical studies of rP2X7Rs, we sought to measure the intrinsic permeability of the receptors to NMDG+ before significant ion accumulation/depletion occurs by applying voltage ramps within the first second of ATP application. Under these conditions, ATP gates an outward current through the rP2X7aR with a PNMDG/PNa of 0.06. In contrast, current through the rP2X7kR was immediately inward with a PNMDG/PNa of 0.3. The polarity of the currents and the dissimilar PNMDG/PNa values support the hypothesis that the two WT splice variants express genuine differences in intrinsic permeability to polyatomic cations at the start of application of ligand. To further test this supposition, we used the reservoir model and the intrinsic PNMDG/PNa values obtained from our empirical measurements to predict the phenotypes of rP2X7aR and rP2X7kR currents during the first second of drug application. We found that the model accurately reproduced the empirical data. Current through the rP2X7aR remained outward throughout the 0.5-s application of ATP. In contrast, ATP immediately gated an inward current through rP2X7kR. These results strongly suggest that the rP2X7kR pore is constitutively dilated by comparison with the rP2X7aR. Thus, we conclude that the dissimilarity in PNMDG/PNa measured from rP2X7aR and P2X7kR primarily results from significant differences in intrinsic NMDG+ permeabilities of the two splice variants, most likely the result of distinct pore diameters.
We found the difference in the size of the fractional calcium current of the two functional splice variants of the rP2X7Rs could be explained by the amino acid composition of their N termini, a situation similar to that reported for ASIC1 channels (45). Furthermore, we find that the difference cannot be traced to a single amino acid. We do not know how the disparate amino acids influence function, although we hypothesize they act to change the structure of the pore; definitive evidence from full-length crystal structures of both splice variants is necessary to confirm this hypothesis. However, we posit that the higher intrinsic NMDG+ permeability of the P2X7kR splice variant reflects a wider pore by comparison with the weakly-permeable P2X7aR, an assumption that is supported by the negative correlation of PNMDG/PNa and fractional calcium current (see Fig. 6). The fact that Ca2+ flux through the P2X7aR (∼7%) exceeds the magnitude expected from the molar ratios of extracellular concentrations of Na+ and Ca2+ (∼4–5%, depending on whether or not ionic activities are considered) suggests that a selectivity filter within the permeation pathway shows a slight preference for Ca2+ at the expense of Na+, as suggested previously for rP2X2Rs (28). If so, then the wider pore of the rP2X7kR may place the residues responsible for selection farther apart and reduce their ability to coordinate Ca2+.
Does the fact that rP2X7kR has a smaller fractional calcium current than rP2X7aR mean that it transduces less Ca2+? Surprisingly, the answer may be no. Although the rP2X7kR has a smaller fractional calcium current because of its wider pore and weaker Ca2+ selectivity, it also has a slower deactivation time course. It is possible then that the small but sustained trickle of Ca2+ across the membrane through rP2X7kR may equal or exceed the bigger but shorter burst of Ca2+ flux through rP2X7aR in its ability to increase [Ca2+]i. Furthermore, the millimolar concentrations of ATP needed to activate the rP2X7aR could significantly chelate free Ca2+ near the extracellular opening of the channel and thus negatively impact the size of the Ca2+ current (19). In contrast, the much lower concentrations of ATP required to fully activate rP2X7kRs would have a negligible impact. We suggest that the lower fractional calcium current of the rP2X7kR evolved as a safety valve to prevent Ca2+ overload during the slow deactivation of inward current that follows even a short application of ATP (20) and thus prevents the deleterious effects of a large increase in [Ca2+]i on cell viability.
In summary, we describe a novel effect of the N terminus on regulation of Ca2+ flux through rP2X7Rs. Our data suggest that the structure of the N terminus plays a role in determining the diameter of the channel. We suggest that the width of the open pore at its narrowest dimension determines the strength of the electrostatic interaction of key residues of the ion selectivity filter with the permeating ions. Thus, the fractional calcium current of the rP2X7kR is smaller than that of the rP2X7aR because its wider pore weakens coordination of Ca2+. The fact that the structure of the N terminus within close proximity to TM1 can influence a key property of the distant selectivity filter of the pore-forming TM2 should be kept in mind when interpreting the crystal structures of P2XRs. Finally, our work leaves open the possibility that the N terminus could be a locus for real time regulation of P2X7R selectivity and flux by as yet undefined accessory proteins.
Experimental procedures
Construction of mutant and chimeric receptors
We constructed two chimeras, N-term(k) and TM1(k), using the PCR overlap technique (46). To construct N-term(k), we replaced the first 28 N-terminal amino acids of the rP2X7aR with the first 25 N-terminal amino acids of the rP2X7kR, yielding a chimera that resembled P2X7aR in all but the N-terminal tail. To construct TM1(k), we replaced the first 25 N-terminal amino acids of the rP2X7kR with the first 28 N-terminal amino acids of the rat rP2X7aR, effectively giving a chimeric protein in which the first transmembrane domain (TM1) of the rP2X7kR (amino acids 25–57) replaced the equivalent TM1 amino acids (amino acids 28–60) of rP2X7aR. Therefore, because both splice variants contain a common sequence upstream of TM1, TM1(k) resembles rP2X7aR in all but the TM1. Both constructs were verified by DNA sequencing (Retrogen, Inc., San Diego).
Point mutations were engineered using the QuikChange Lightening II Site-directed mutagenesis kit (Stratagene, La Jolla, CA) and were verified by sequencing at Retrogen.
Cell culture and transfection
HEK293T cells (CRL-3216, ATCC, Manassas, VA) were maintained in exponential growth in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FBS, 2 mm glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin (all culture reagents were from Thermo Fisher Scientific, Waltham, MA) and incubated in 75-cm2 Falcon tissue culture flasks (Corning Life Sciences, Oneonta, NY) at 37 °C in a humidified atmosphere containing 5% CO2. Upon reaching ∼70% confluence, the cells were enzymatically dissociated using 0.05% trypsin-EDTA (Gibco); then, aliquots of the cell suspension were transferred to 35-mm tissue culture dishes (Nunc, Roskilde, Denmark) where they were co-transfected with either WT, chimeric, or mutant P2X7Rs and fluorescent reporter plasmids using Effectene Transfection Reagents (Qiagen, Valencia, CA). Each dish contained ∼7.5 × 105 cells, 1 ml of DMEM/FBS, 0.2–1.0 μg of cDNA encoding the P2X7R target protein, 0.3 μg of cDNA encoding the fluorescent reporter (AsRed or eGFP), 4 μl of Enhancer, and 8 μl of Effectene. These cells were incubated for 12 h at 37 °C in a humidified incubator, after which the transfection medium was removed, and cells were maintained for an additional 24 h in 2 ml/dish of standard culture medium minus the transfection reagents. On the morning of the experiment, transfected cells were treated for 3 min with 0.05% trypsin-EDTA to facilitate detachment from the bottom of the dish, plated at low density (50,000 cells/ml) onto 35-mm culture dishes, and then left to rest in the tissue culture incubator for 1–10 h before resuspension and transfer to the recording chamber at the start of an experiment.
Measuring fractional calcium current
Detailed accounts of the fractional calcium current method are published (19, 23).
ATP is a low-affinity agonist that requires millimolar concentrations to fully activate rP2X7aRs (47). At these concentrations, ATP significantly chelates extracellular Ca2+, an unwanted effect that invalidates measurements of fractional calcium current (19). Therefore, we used the higher affinity agonist, BzATP (100 μm, except where noted), to gate channels in this set of experiments. At this concentration, BzATP causes negligible chelation, allowing fractional calcium current to be measured at physiological concentrations of extracellular Ca2+.
Membrane current from HEK293T cells transiently expressing rP2X7Rs was measured at room temperature using lightly fire-polished electrodes pulled from World Precision Instruments 1B150F glass capillaries (Sarasota, FL) using a P-97 Flaming/Brown Micropipette Puller (Sutter Instruments, Novato, CA) and Axon Instrument 200 series amplifiers (Molecular Devices, San Jose, CA). Data were filtered at 5 kHz using the internal circuitry of the amplifier and digitized at 10 kHz with 16-bit accuracy using ITC-16 analog-to-digital boards (Heka Instruments, Holliston, MA), iMac computers (Apple Computer, Cupertino, CA), and AxoGraph software (Axograph Scientific, Australia). The electrodes had open-tip resistances of 1.5–3.0 MΩ when measured in the extracellular bath solution. The intracellular solution contained (in mm): 140 CsCl, 10 tetraethylammonium-Cl, 10 HEPES, 2 mm fura-2 K5 (Thermo Fisher Scientific, Waltham, MA) brought to pH 7.3 with CsOH. The extracellular solution contained 150 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, brought to pH 7.4 with ∼4 mm NaOH.
To measure Ca2+ influx, cells were loaded with fura-2 by passive diffusion for 10 min through the tip of the recording electrode (23, 48). BzATP was applied once every 3 min for the durations indicated in the figure legends, and changes in the fura-2 signal were measured using a Nikon TE2000-S inverted microscope equipped with epifluorescence illumination and a photomultiplier tube. Specifically, light emitted from the 100-watt xenon lamp was passed through a 380-nm bandpass filter and directed through the microscope objective (HMC ×40 ELWD Plan Fluor; Modulation Optics, Inc.) into the recording chamber by reflection from the surface of a 400-nm dichroic long pass mirror. Light emitted by fura-2 was gathered by the objective, passed through the dichroic mirror and a 510-nm bandpass filter, and recorded using the analog output of a Photomultiplier Detection System (Horiba Scientific, Edison, NJ). Fluorescence was quantified as multiples of a bead unit (BU), where one BU equaled the average fluorescence of seven Carboxyl Bright Blue 4.6-μm microspheres (Polysciences, Warrington, PA) measured on the morning of the experiment. Dye saturation was avoided by minimizing the duration of drug application, which varied from 0.5 to 6 s; as shown previously, fractional calcium current does not vary with the length of drug application (23).
To calculate the fractional calcium current, agonist-gated current was integrated to give the total charge transfer across the cell membrane (QT, in coulombs). The decrease in 510-nm emission of fura-2 excited by the 380-nm light (ΔF380) was measured in calibrated BU allowing ΔF380 to be converted to coulombs of Ca2+ charge (QCa). Then, the fraction of total membrane current carried by Ca2+ (Pf) is equal to QCa/QT, and the percent fractional Ca2+ current (fractional calcium current) is Pf·100%. Experiments in which plots of QCa versus QT were nonlinear indicated saturation of fura-2 by the ATP-gated influx of Ca2+; these experiments were discarded.
To calibrate the beads, ΔF380 was measured from HEK293T cells transiently or stably expressing the rat P2X2 receptor (rP2X2R) and bathed in an extracellular solution containing 110 mm Ca2+ and no Na+. We calibrated with rP2X2R because, unlike rP2X7Rs, it is not fully inhibited by high concentrations of extracellular Ca2+ (49, 50). Under these conditions, QT equals QCa. Plotting ΔF380 versus QT gives a straight line with a slope equal to ΔF380/ΔQT, called the Fmax. Once Fmax is determined, then QCa (in nC) can be derived by dividing ΔF380 (in BU) by Fmax (in BU/nC). We recalibrated Fmax on a regular basis to overcome problems with changes in the efficiency of the light source and the inherent signal strength of the fluorescent microspheres that weakened over time. In the experiments described here, the Fmax ranged from 0.03 to 0.05 BU/nC.
Measuring PNMDG/PNa
We used ATP to activate rP2X7Rs in experiments measuring NMDG+ permeability because ATP is significantly less expensive than BzATP, and because the problem of unwanted chelation of extracellular Ca2+ was avoided by using a bath solution devoid of divalent cations. We typically used 0.1 and 2.0 mm ATP to activate rP2X7kRs and rP2X7aRs, respectfully, except where noted otherwise. Whole-cell currents were measured at room temperature from single detached cells held at −40 mV using electrodes with open tip resistances of 1.0–2.5 MΩ. Pipettes contained (in mm) 122 NaCl, 32 NaOH, 10 EGTA, and 10 HEPES (pH 7.3). We used a 2 m KCl agar bridge positioned downstream of the cells to reduce liquid junction potentials of the bath solution and the Ag/AgCl ground wire. In all experiments, uncompensated series resistances were <6 MΩ, which was compensated by 75% using the internal circuitry of the amplifier except where noted. The control bath solution contained 150 NaCl, 4 NaOH, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH 7.4). Ten minutes after seal formation, the bath solution was switched to a divalent-free salt solution containing 150 Na+ or 150 NMDG+, 154 Cl−, 10 glucose, 10 HEPES, and an appropriate amount of NaOH or HCl to establish a pH of 7.4. Salts and drugs were obtained from Sigma. ATP dissolved in the appropriate extracellular solution (containing predominately NaCl or NMDG-Cl) was applied using either a Rapid Solution Changer RSC-200 (Bio-Logic Science Instruments, Seyssinet-Pariset, France) or a Perfusion Fast-Step SF-77B (Warner Instruments, Hamden, CT).
We waited 10 min for complete exchange of the intracellular contents with pipette solution (48). Then, a two-ramp voltage protocol was applied twice to each cell, once in the presence of extracellular Na+, and once in the presence of extracellular NMDG+. In each case, a control voltage ramp (−80 to 60 mV, 0.7 mV/ms) was applied in the absence of ATP to measure the leak current (Ifirst ramp) and then reapplied 500 ms after the start of a 3-s application of ATP to measure the total current containing an agonist-gated component (Isecond ramp). ATP-gated current was extracted by subtracting the leak current from the total current (IATP = Isecond ramp − Ifirst ramp). IATP versus applied voltage ramp was plotted, and Erev was measured at the x-intercept. Then, the relative permeability of NMDG+ (PNMDG/PNa) was calculated as shown in Equation 1,
| (Eq. 1) |
where ΔErev equaled Erev, NMDG − Erev, Na, and R, T, and F have their usual values (51).
In silico modeling
Data were simulated using Apple computers, MatLab 2019a (MathWorks, Natick, MA), and scripts written by Gil Toombes (Molecular Physiology and Biophysics Section, NINDS, National Institutes of Health, Bethesda, MD).
Data analysis
Igor Pro (Wavemetrics, Grants Pass, OR) and MatLab were used for off-line data analysis and construction of figures. In the text, data are described as the mean ± S.E. for the number of experiments stated. In the figures, data are presented as box-and-whisker diagrams with median values, interquartile ranges, and whiskers equal to standard deviations. Significant differences among groups were determined using Prism (GraphPad, San Diego) by one-way ANOVA with Tukey's post hoc or Student's t test where appropriate. We considered data sets in which p < 0.01 was significantly different from control.
Author contributions
X. L., D. S. K. S., and T. M. E. conceptualization; X. L., D. S. K. S., J. C., and T. M. E. data curation; X. L., D. S. K. S., and T. M. E. formal analysis; X. L. and T. M. E. validation; X. L., D. S. K. S., and T. M. E. investigation; X. L., D. S. K. S., J. C., and T. M. E. methodology; X. L., D. S. K. S., and J. C. writing-review and editing; T. M. E. resources; T. M. E. software; T. M. E. supervision; T. M. E. funding acquisition; T. M. E. visualization; T. M. E. writing-original draft; T. M. E. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Annette Nicke for the gift of the rat and mouse P2X7 slice variants; Stanko Stojilkovic for the truncated and Δ18P2X7aR mutants; and Laura Janks, Stephanie Michalski, and Manju Tewari for comments on the manuscript.
This work was supported by National Institutes of Health Grant 1R01GM112188. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- P2XR
- P2X receptor
- rP2X7aR and rP2X7kR
- rat P2X7 receptor splice variants
- BU
- bead unit
- [Ca2+]i
- intracellular Ca2+
- NMDG
- N-methyl-d-glucamine
- TM1 and TM2
- first and second transmembrane domains
- Erev
- reversal potential of agonist-gated currents
- Pf%
- fractional Ca2+ current
- ΔF380
- change in fluorescence (380 nm excitation, 510 emission)
- PNMDG/PNa
- NMDG permeability relative to Na+
- HEK293T cells
- human embryonic kidney cells expressing the SV40 T-antigen
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate
- eGFP
- enhanced GFP
- r
- rat
- MΩ
- megohm
- nS
- nanosiemens
- YFP
- yellow fluorescent protein
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum.
References
- 1. Nicke A., Grutter T., and Egan T. M. (2018) in The Oxford Handbook of Neuronal Ion Channels (Bhattacharjee A., ed) pp. 1–32, Oxford University Press, Oxford, UK [Google Scholar]
- 2. Schmid R., and Evans R. J. (2019) ATP-Gated P2X receptor channels: molecular insights into functional roles. Annu. Rev. Physiol. 81, 43–62 10.1146/annurev-physiol-020518-114259 [DOI] [PubMed] [Google Scholar]
- 3. Skaper S. D., Debetto P., and Giusti P. (2010) The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J. 24, 337–345 10.1096/fj.09-138883 [DOI] [PubMed] [Google Scholar]
- 4. Wiley J. S., Sluyter R., Gu B. J., Stokes L., and Fuller S. J. (2011) The human P2X7 receptor and its role in innate immunity. Tissue Antigens 78, 321–332 10.1111/j.1399-0039.2011.01780.x [DOI] [PubMed] [Google Scholar]
- 5. Di Virgilio F. (2015) P2X receptors and inflammation. Curr. Med. Chem. 22, 866–877 10.2174/0929867322666141210155311 [DOI] [PubMed] [Google Scholar]
- 6. Sperlágh B., and Illes P. (2014) P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol. Sci. 35, 537–547 10.1016/j.tips.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 7. Savio L. E. B., de Andrade Mello P., da Silva C. G., and Coutinho-Silva R. (2018) The P2X7 receptor in inflammatory diseases: angel or demon? Front. Pharmacol. 9, 52 10.3389/fphar.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adinolfi E., Giuliani A. L., De Marchi E., Pegoraro A., Orioli E., and Di Virgilio F. (2018) The P2X7 receptor: a main player in inflammation. Biochem. Pharmacol. 151, 234–244 10.1016/j.bcp.2017.12.021 [DOI] [PubMed] [Google Scholar]
- 9. Di Virgilio F., Dal Ben D., Sarti A. C., Giuliani A. L., and Falzoni S. (2017) The P2X7 receptor in infection and inflammation. Immunity 47, 15–31 10.1016/j.immuni.2017.06.020 [DOI] [PubMed] [Google Scholar]
- 10. Di Virgilio F., Sarti A. C., and Grassi F. (2018) Modulation of innate and adaptive immunity by P2X ion channels. Curr. Opin. Immunol. 52, 51–59 10.1016/j.coi.2018.03.026 [DOI] [PubMed] [Google Scholar]
- 11. Janks L., Sprague R. S., and Egan T. M. (2019) ATP-Gated P2X7 receptors require chloride channels to promote inflammation in human macrophages. J. Immunol. 202, 883–898 10.4049/jimmunol.1801101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yip L., Woehrle T., Corriden R., Hirsh M., Chen Y., Inoue Y., Ferrari V., Insel P. A., and Junger W. G. (2009) Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J. 23, 1685–1693 10.1096/fj.08-126458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taylor S. R., Gonzalez-Begne M., Dewhurst S., Chimini G., Higgins C. F., Melvin J. E., and Elliott J. I. (2008) Sequential shrinkage and swelling underlie P2X7-stimulated lymphocyte phosphatidylserine exposure and death. J. Immunol. 180, 300–308 10.4049/jimmunol.180.1.300 [DOI] [PubMed] [Google Scholar]
- 14. Sluyter R. (2017) The P2X7 receptor. Adv. Exp. Med. Biol. 1051, 17–53 10.1007/5584_2017_59 [DOI] [PubMed] [Google Scholar]
- 15. Jimenez-Mateos E. M., Smith J., Nicke A., and Engel T. (2018) Regulation of the P2X7 receptor expression and function in the brain. Brain Res. Bull. 10.1016/j.brainresbull.2018.12.008 [DOI] [PubMed] [Google Scholar]
- 16. Kaczmarek-Hájek K., Lörinczi E., Hausmann R., and Nicke A. (2012) Molecular and functional properties of P2X receptors–recent progress and persisting challenges. Purinergic Signal. 8, 375–417 10.1007/s11302-012-9314-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bartlett R., Stokes L., and Sluyter R. (2014) The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675 10.1124/pr.113.008003 [DOI] [PubMed] [Google Scholar]
- 18. North R. A., and Surprenant A. (2000) Pharmacology of cloned P2X receptors. Annu. Rev. Pharmacol. Toxicol. 40, 563–580 10.1146/annurev.pharmtox.40.1.563 [DOI] [PubMed] [Google Scholar]
- 19. Liang X., Samways D. S., Wolf K., Bowles E. A., Richards J. P., Bruno J., Dutertre S., DiPaolo R. J., and Egan T. M. (2015) Quantifying Ca2+ current and permeability in ATP-gated P2X7 receptors. J. Biol. Chem. 290, 7930–7942 10.1074/jbc.M114.627810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicke A., Kuan Y. H., Masin M., Rettinger J., Marquez-Klaka B., Bender O., Górecki D. C., Murrell-Lagnado R. D., and Soto F. (2009) A functional P2X7 splice variant with an alternative transmembrane domain 1 escapes gene inactivation in P2X7 knock-out mice. J. Biol. Chem. 284, 25813–25822 10.1074/jbc.M109.033134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz N., Drouot L., Nicke A., Fliegert R., Boyer O., Guse A. H., Haag F., Adriouch S., and Koch-Nolte F. (2012) Alternative splicing of the N-terminal cytosolic and transmembrane domains of P2X7 controls gating of the ion channel by ADP-ribosylation. PLoS ONE 7, e41269 10.1371/journal.pone.0041269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He M. L., Zemkova H., Koshimizu T. A., Tomić M., and Stojilkovic S. S. (2003) Intracellular calcium measurements as a method in studies on activity of purinergic P2X receptor-channels. Am. J. Physiol. Cell Physiol. 285, C467–C479 10.1152/ajpcell.00042.2003 [DOI] [PubMed] [Google Scholar]
- 23. Egan T. M., and Khakh B. S. (2004) Contribution of calcium ions to P2X channel responses. J. Neurosci. 24, 3413–3420 10.1523/JNEUROSCI.5429-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Samways D. S., Li Z., and Egan T. M. (2014) Principles and properties of ion flow in P2X receptors. Front. Cell. Neurosci. 8, 6 10.3389/fncel.2014.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M., Toombes G. E., Silberberg S. D., and Swartz K. J. (2015) Physical basis of apparent pore dilation of ATP-activated P2X receptor channels. Nat. Neurosci. 18, 1577–1583 10.1038/nn.4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Samways D. S., Migita K., Li Z., and Egan T. M. (2008) On the role of the first transmembrane domain in cation permeability and flux of the ATP-gated P2X2 receptor. J. Biol. Chem. 283, 5110–5117 10.1074/jbc.M708713200 [DOI] [PubMed] [Google Scholar]
- 27. Boué-Grabot E., Archambault V., and Séguéla P. (2000) A protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X(2) ATP-gated channels. J. Biol. Chem. 275, 10190–10195 10.1074/jbc.275.14.10190 [DOI] [PubMed] [Google Scholar]
- 28. Allsopp R. C., and Evans R. J. (2015) Contribution of the juxta-transmembrane intracellular regions to the time-course and permeation of ATP-gated P2X7 receptor ion channels. J. Biol. Chem. 290, 14556–14566 10.1074/jbc.M115.642033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mansoor S. E., Lü W., Oosterheert W., Shekhar M., Tajkhorshid E., and Gouaux E. (2016) X-ray structures define human P2X(3) receptor gating cycle and antagonist action. Nature 538, 66–71 10.1038/nature19367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fryatt A. G., Dayl S., Stavrou A., Schmid R., and Evans R. J. (2019) Organization of ATP-gated P2X1 receptor intracellular termini in apo and desensitized states. J. Gen. Physiol. 151, 146–155 10.1085/jgp.201812108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pippel A., Stolz M., Woltersdorf R., Kless A., Schmalzing G., and Markwardt F. (2017) Localization of the gate and selectivity filter of the full-length P2X7 receptor. Proc. Natl. Acad. Sci. U.S.A. 114, E2156–E2165 10.1073/pnas.1610414114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kracun S., Chaptal V., Abramson J., and Khakh B. S. (2010) Gated access to the pore of a P2X receptor: structural implications for closed-open transitions. J. Biol. Chem. 285, 10110–10121 10.1074/jbc.M109.089185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Migita K., Haines W. R., Voigt M. M., and Egan T. M. (2001) Polar residues of the second transmembrane domain influence cation permeability of the ATP-gated P2X(2) receptor. J. Biol. Chem. 276, 30934–30941 10.1074/jbc.M103366200 [DOI] [PubMed] [Google Scholar]
- 34. Yan Z., Li S., Liang Z., Tomić M., and Stojilkovic S. S. (2008) The P2X7 receptor channel pore dilates under physiological ion conditions. J. Gen. Physiol. 132, 563–573 10.1085/jgp.200810059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smart M. L., Gu B., Panchal R. G., Wiley J., Cromer B., Williams D. A., and Petrou S. (2003) P2X7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J. Biol. Chem. 278, 8853–8860 10.1074/jbc.M211094200 [DOI] [PubMed] [Google Scholar]
- 36. Stojilkovic S. S., Tomic M., He M. L., Yan Z., Koshimizu T. A., and Zemkova H. (2005) Molecular dissection of purinergic P2X receptor channels. Ann. N.Y. Acad. Sci. 1048, 116–130 10.1196/annals.1342.011 [DOI] [PubMed] [Google Scholar]
- 37. Allsopp R. C., Farmer L. K., Fryatt A. G., and Evans R. J. (2013) P2X receptor chimeras highlight roles of the amino terminus to partial agonist efficacy, the carboxyl terminus to recovery from desensitization, and independent regulation of channel transitions. J. Biol. Chem. 288, 21412–21421 10.1074/jbc.M113.464651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Allsopp R. C., and Evans R. J. (2011) The intracellular amino terminus plays a dominant role in desensitization of ATP-gated P2X receptor ion channels. J. Biol. Chem. 286, 44691–44701 10.1074/jbc.M111.303917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allsopp R. C., Lalo U., and Evans R. J. (2010) Lipid raft association and cholesterol sensitivity of P2X1–4 receptors for ATP: chimeras and point mutants identify intracellular amino-terminal residues involved in lipid regulation of P2X1 receptors. J. Biol. Chem. 285, 32770–32777 10.1074/jbc.M110.148940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lalo U., Roberts J. A., and Evans R. J. (2011) Identification of human P2X1 receptor-interacting proteins reveals a role of the cytoskeleton in receptor regulation. J. Biol. Chem. 286, 30591–30599 10.1074/jbc.M111.253153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wen H., and Evans R. J. (2009) Regions of the amino terminus of the P2X receptor required for modification by phorbol ester and mGluR1α receptors. J. Neurochem. 108, 331–340 10.1111/j.1471-4159.2008.05761.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ormond S. J., Barrera N. P., Qureshi O. S., Henderson R. M., Edwardson J. M., and Murrell-Lagnado R. D. (2006) An uncharged region within the N terminus of the P2X6 receptor inhibits its assembly and exit from the endoplasmic reticulum. Mol. Pharmacol. 69, 1692–1700 10.1124/mol.105.020404 [DOI] [PubMed] [Google Scholar]
- 43. North R. A. (2002) Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067 10.1152/physrev.00015.2002 [DOI] [PubMed] [Google Scholar]
- 44. Yan Z., Khadra A., Li S., Tomic M., Sherman A., and Stojilkovic S. S. (2010) Experimental characterization and mathematical modeling of P2X7 receptor channel gating. J. Neurosci. 30, 14213–14224 10.1523/JNEUROSCI.2390-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bässler E. L., Ngo-Anh T. J., Geisler H. S., Ruppersberg J. P., and Gründer S. (2001) Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J. Biol. Chem. 276, 33782–33787 10.1074/jbc.M104030200 [DOI] [PubMed] [Google Scholar]
- 46. Heckman K. L., and Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 10.1038/nprot.2007.132 [DOI] [PubMed] [Google Scholar]
- 47. Surprenant A., Rassendren F., Kawashima E., North R. A., and Buell G. (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738 10.1126/science.272.5262.735 [DOI] [PubMed] [Google Scholar]
- 48. Samways D. S., and Egan T. M. (2007) Acidic amino acids impart enhanced Ca2+ permeability and flux in two members of the ATP-gated P2X receptor family. J. Gen. Physiol. 129, 245–256 10.1085/jgp.200609677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Virginio C., Church D., North R. A., and Surprenant A. (1997) Effects of divalent cations, protons and calmidazolium at the rat P2X7 receptor. Neuropharmacology 36, 1285–1294 10.1016/S0028-3908(97)00141-X [DOI] [PubMed] [Google Scholar]
- 50. Evans R. J., Lewis C., Virginio C., Lundstrom K., Buell G., Surprenant A., and North R. A. (1996) Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J. Physiol. 497, 413–422 10.1113/jphysiol.1996.sp021777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hille B. (2001) Ion Channels of Excitable Membranes. 3rd Ed., pp. 13–17, Sinauer Associates Inc., Sunderland, MA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.