

Abstract
NSD2 is a histone methyltransferase that specifically dimethylates histone H3 lysine 36 (H3K36me2), a modification associated with gene activation. Dramatic overexpression of NSD2 in t(4;14) multiple myeloma (MM) and an activating mutation of NSD2 discovered in acute lymphoblastic leukemia are significantly associated with altered gene activation, transcription, and DNA damage repair. The partner proteins through which NSD2 may influence critical cellular processes remain poorly defined. In this study, we utilized proximity-based labeling (BioID) combined with label-free quantitative MS to identify high confidence NSD2 interacting partners in MM cells. The top 24 proteins identified were involved in maintaining chromatin structure, transcriptional regulation, RNA pre-spliceosome assembly, and DNA damage. Among these, an important DNA damage regulator, poly(ADP-ribose) polymerase 1 (PARP1), was discovered. PARP1 and NSD2 have been found to be recruited to DNA double strand breaks upon damage and H3K36me2 marks are enriched at damage sites. We demonstrate that PARP1 regulates NSD2 via PARylation upon oxidative stress. In vitro assays suggest the PARylation significantly reduces NSD2 histone methyltransferase activity. Furthermore, PARylation of NSD2 inhibits its ability to bind to nucleosomes and further get recruited at NSD2-regulated genes, suggesting PARP1 regulates NSD2 localization and H3K36me2 balance. This work provides clear evidence of cross-talk between PARylation and histone methylation and offers new directions to characterize NSD2 function in DNA damage response, transcriptional regulation, and other pathways.
Keywords: histone methylation, DNA damage, protein complex, proteomics, epigenetics, NSD2, PARP1, PARylation
Introduction
NSD2 (MMSET/WHSC1) is a member of the nuclear receptor-binding SET domain (NSD)3 family, functioning as a histone methyltransferase (HMT) that catalyzes dimethylation of histone H3 lysine 36 specifically (1, 2). NSD2 was identified to be the key gene disrupted and overexpressed by fusion to the immunoglobulin heavy-chain promoter/enhancer in the t(4;14) translocation, found in almost 15–20% of multiple myeloma (MM) cases (3, 4). NSD2 overexpression in t(4;14)+ MM cells leads to increased cell proliferation and altered downstream gene expression due to a genome wide increase in H3K36 dimethylation and a decrease in gene repression-associated H3K27 trimethylation (H3K27me3) (5–7). Recently, a gain-of-function point mutation of NSD2 (E1099K) has been discovered in multiple leukemia cell lines and patient samples that similarly causes elevated H3K36me2 and altered gene activation (8–12).
The H3K36me2 mark serves important roles in DNA damage repair (13–15). Upon radiation, H3K36me2 marks are enriched at double strand breaks (DSB) to recruit DNA repair components, including NBS1 and Ku70, initiating nonhomologous end joining (NHEJ) (16, 17). This modification has also been found to promote the resolution of RAD51 dsDNA filaments during late stage homologous recombination (HR) (18). NSD2 was identified as a member of the DNA damage response pathway through genetic screening and is present at sites of DNA damage upon UV laser microirradiation (19, 20). It serves an important role to facilitate the recruitment of 53BP1, a DNA damage response regulator, to DSBs (20). NSD2 is also involved in initiation of class switch recombination during B-cell development. Deregulation of NSD2 causes decreased activation-induced cytidine deaminase-mediated DNA breaks, which further leads to defects of class switch recombination (21, 22). Our lab previously discovered that NSD2 promotes both NHEJ and HR in U2OS cells. Furthermore, MM cells overexpressing NSD2 display an enhanced rate of DNA damage repair that allows cells to survive upon chemotherapy treatment (23).
In addition to its histone methyltransferase function, NSD2 has been found to play important roles in multiple molecular pathways. For instance, NSD2 has been reported to interact with the bromodomain protein 4 (BRD4) and the positive transcription elongation factor β (P-TEFβ) to facilitate transcriptional elongation of rapidly induced genes (24). NSD2 also associates with HIRA, the histone H3.3-specific histone chaperone, to promote histone H3.3 deposition along gene bodies of rapidly activated genes, suggesting a role for NSD2 in chromatin assembly (24). Moreover, NSD2 is involved in repression of microRNA miR-126 in MM through an interaction with the transcriptional corepressor TRIM28 and the histone deacetylases HDAC1/2 that results in maintaining a high protein level of the oncoprotein c-MYC (25).
An initial screen for NSD2 partner proteins identified a preliminary set of 19 interaction partners (25). However, this work was carried out in nonphysiologically relevant 293 human embryonic kidney cells and required the use of relatively harsh conditions to extract NSD2 from cells, which are tightly bound to chromatin. Thus, we have now utilized BioID proximity-based labeling (26, 27), which does not require the use of harsh conditions that may disrupt weaker but biologically relevant complexes, to obtain a more complete understanding of the biological functions and mechanisms of NSD2. This label-free quantitative study minimizes nonspecific contamination and helps define the NSD2 interaction network. Using this method, we have identified with high confidence 24 nuclear interacting partners of NSD2 involved in multiple molecular pathways and biological processes including chromatin remodeling, gene expression regulation, DNA damage repair, and RNA splicing. The mapped protein network provides novel insights into the functions and mechanisms of NSD2 operative in multiple myeloma.
Results
Study 1: identification of NSD2 interacting partners by BioID (proximity labeling and IP-MS)
NSD2 protein binds tightly to the chromatin scaffold and remains insoluble in the nuclear pellet after standard nuclear extraction. Accordingly, an enzyme shearing mixture is required to digest chromatin and release NSD2 into the soluble nuclear fraction, which might disrupt any physiological NSD2 complex. Because of this, we adopted BioID, a method in which a promiscuous biotin ligase moiety is engineered into the protein of interest, to biotinylate proteins based on proximity and screen for NSD2 interacting proteins. To identify physiologically relevant interacting proteins of NSD2, we chose the TKO NSD2-low cell line (5), which was derived from the t(4;14)+ KMS11 MM cell line in which the rearranged and overexpressed NSD2 allele was disrupted and only the WT NSD2 expresses. These cells were transduced with retrovirus encoding a BirA-tagged version of NSD2. The resulting TKO cell line stably expressed NSD2-BirA and mimicked high expression of NSD2 in t(4;14)+ MM. These TKO + NSD2-BirA cells were cultured in medium supplemented with biotin for the BirA domain to biotinylate proteins that are in close proximity to NSD2. NSD2 and its biotinylated partners were extracted in SDS-containing lysis buffer, captured, and subsequently analyzed by nanocapillary LC-MS (Fig. 1A). TKO cells expressing NSD2-BirA had an elevated histone H3K36me2 level, suggesting fusion of the BirA tag did not affect NSD2 histone methyltransferase activity (Fig. 1B). Six biological replicates of NSD2-BirA BioID IP-MS were analyzed. A total of 94 proteins identified in at least five of the six biological replicates were considered high confident targets (Fig. 1C). In our study design, the same workflow was performed on TKO cells in the absence of BirA-NSD2 as a negative control for endogenous biotinylated proteins and nonspecific binders to streptavidin beads. Of the 55 proteins identified at least twice in three biological replicates of the TKO control, 31 were in common with NSD2-BirA (Fig. 1C). As a result, 63 unique NSD2 high confident interacting proteins were identified (Table S1). Gene set enrichment analysis of the 63 proteins using EnrichR (28) suggested that NSD2 partner proteins are highly associated with chromatin remodeling and chromatin modification according to the GO biological process 2018. This validated our process and NSD2 functions as a chromatin regulator (Fig. 1D).
Figure 1.
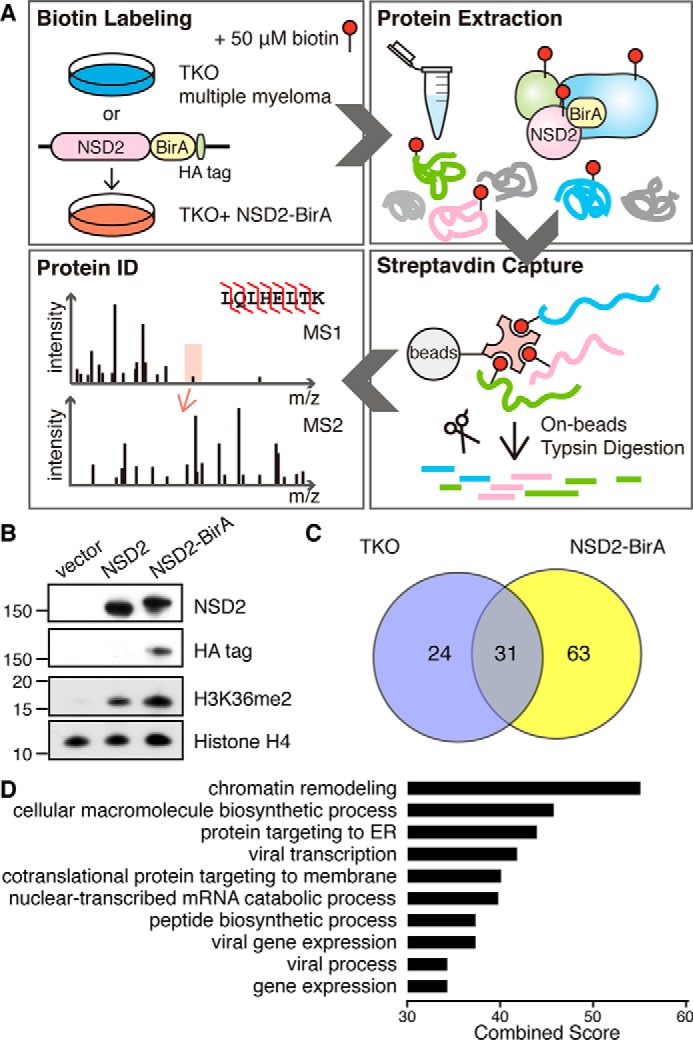
NSD2 interacting partners identified by BioID. A, study design: TKO control cells or TKO MM cells stably expressing NSD2-BirA were cultured in media supplemented with 50 μm biotin for 48 h and then lysed. Biotinylated proteins were captured by streptavidin beads. On-bead trypsin digestion was performed and the resulting peptides were desalted and identified by nanocapillary LC-MS. B, immunoblot of TKO cells stably expressing empty vector, NSD2, or NSD2-BirA. Because the NSD2-BirA construct contains a C-terminal HA tag, the HA tag blot confirmed expression of the NSD2-BirA construct in TKO cells. C, a Venn diagram depicting unique and common interacting proteins identified in NSD2-BirA cells and TKO control cells. D, gene set enrichment analysis of NSD2-binding partners organized according to GO biological process using EnrichR (28).
Study 2: label-free quantitative MS to capture NSD2 interaction partners
To further identify highly confident NSD2 interaction partners and reduce noise from BirA random labeling, we developed a BirA control to eliminate nonspecific proteins from the screen. A BirA ligase along with a C-terminal nuclear localization signal peptide was cloned into a retrovirus vector and stably transduced into the TKO cell line. The control cell line was then used to perform comparative quantification of biotinylated proteins in the NSD2-BirA experiment versus BirA alone. Three biological replicates of TKO expressing NSD2-BirA or BirA control were cultured in medium supplemented with biotin (Fig. 2A). The same numbers of cells were harvested and lysed. The same amount of total protein was used for affinity capture to ensure highly comparable conditions. Each sample was analyzed in technical triplicate for protein identification and quantification. A statistical platform based on a hierarchical linear model was utilized to search for enrichment of proteins from cells expressing NSD2-BirA. Due to affinity enrichment, the data are presented as an asymmetrical volcano plot where the x axis represents the relative abundance difference between NSD2-BirA versus BirA control and the y axis is the instantaneous q-value, a measure of how confident the abundance difference is statistically (Fig. 2B). A threshold of 2-fold difference and 5% false discovery rate was set for quantification, which responds to a -log10 (instantaneous q-value) of 1.3. In total, 42 proteins were identified with statistically significant enrichment in the NSD2-BirA state, and among those, 24 were nuclear proteins (colored red in the plot). Because NSD2 localizes in the nucleus physiologically, we considered these 24 nuclear hits as confident interacting partners (Table S2). Variation in signal intensity for each quantified peptide was assigned as a percent of the total for each different source (Fig. 2C). Small variations in biological and technical replicates and large variations in NSD2-BirA versus BirA control suggested our quantification study was highly reliable.
Figure 2.

NSD2-binding partners identified by label-free quantitative MS. A, study design: three biological replicates of TKO cells stably expressing NSD2-BirA or nuclear-localized BirA control were cultured with biotin, lysed, and quantified. Equal amounts of total protein from each sample was used for streptavidin capture and trypsin digestion. The resulting peptides were purified and analyzed by nanocapillary LC-MS in triplicate. High resolution MS1 was used for relative peptide quantification and MS2 was used for protein identification. B, volcano plot of quantified proteins is shown. The x axis is the fold-change in protein abundance of NSD2-BirA relative to BirA control. The y axis (instantaneous q-value) measures the statistical confidence of the variation between NSD2-BirA and BirA control. The vertical dotted lines represent a 2-fold change threshold. The horizontal dotted line indicates a false discovery rate of 5%. Twenty-four nuclear targets above threshold are colored red. C, box and whisker plot indicating variations in signal intensity of each quantified protein. Variation attributed to the treatment (NSD2-BirA versus BirA control), biological replicates, technical replicates, or residuals (variation not explained by any of the previously described sources) are indicated. D, gene ontology KEGG pathway analysis of the 24 high confidence NSD2 partners. E, Venn diagram of overlapping hits from the BioID IP-MS qualitative Study 1 (see Fig. 1) and label-free quantitative Study 2.
GO analysis of the 24 interacting targets using EnrichR through KEGG pathway 2016 suggested NSD2-binding proteins are highly associated with the base excision repair pathway (Fig. 2D), which is responsible for removing base lesions from the genome throughout the cell cycle. The spliceosome is the second top pathway on the graph, suggesting that NSD2 might function beyond transcription elongation to facilitate spliceosome assembly or function.
The 63 hits identified in the BioID IP-MS qualitative Study 1 and 24 label-free quantitative hits in Study 2 have 16 overlapping targets (Fig. 2E). As described above, BRD4 was previously identified as a NSD2-interacting protein implicated in transcription elongation. In Study 1, BRD4 is one of the 63 high confident hits. Meanwhile, in the label-free quantitative study BRD4 was enriched 6-fold in NSD2-BirA cells, suggesting that proximity-based methods for identifying NSD2-associated proteins are mechanistically valid.
Poly(ADP-ribose) polymerase 1 (PARP1) is a NSD2-binding protein
PARP1 protein level increased 5-fold in the NSD2-BirA sample compared with the BirA control (Fig. 2B). It was also identified in all 6 biological replicates of BioID IP-MS analysis suggesting it is a true binding protein of NSD2. Because NSD2 may have a role in DNA repair, we chose to further investigate this interaction. The MM cell line KMS-11 and as well as KMS-11-derived NTKO cells (5) that both are t(4;14)+ and express high levels of NSD2 were used to capture endogenous interaction of NSD2 and PARP1. Reciprocal co-immunoprecipitation results confirm that PARP1 and NSD2 physically interact under physiological conditions in MM cells (Fig. 3A).
Figure 3.
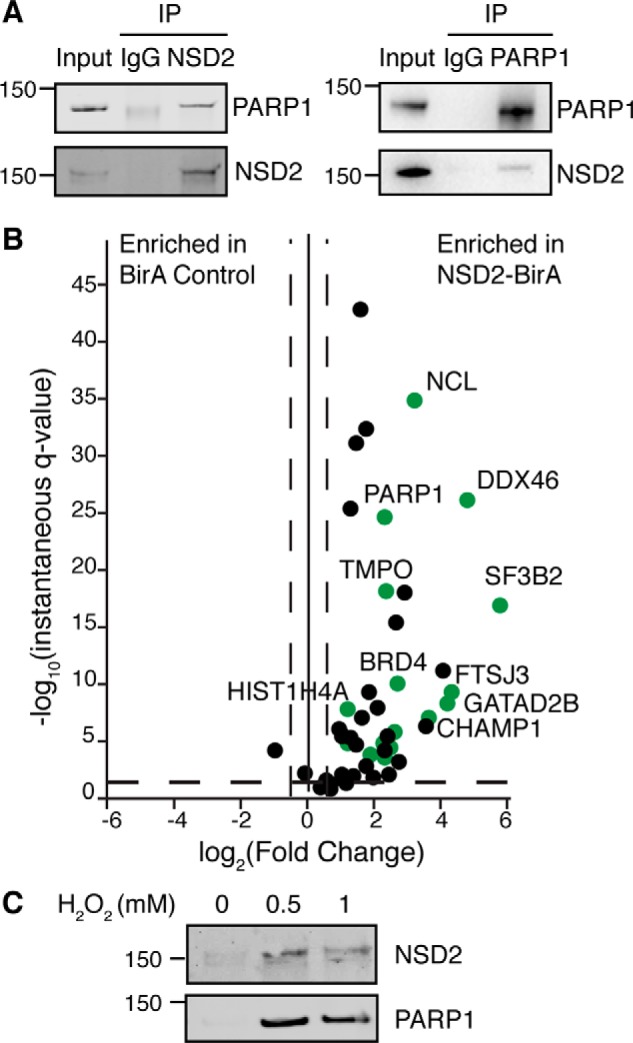
PARP1 is a NSD2-binding partner. A, reciprocal co-immunoprecipitation of endogenous PARP1 and NSD2 from NTKO and KMS11 cells. B, volcano plot of NSD2-binding partners with PARP1 substrates colored in green. PARP1 substrate information was adopted from Refs. 33 and 34. C, NTKO cells were treated with 0.5 and 1 mm hydrogen peroxide for 15 min and harvested to extract nuclear proteins. PARylated proteins were then affinity captured with a poly(ADP-ribose)-binding macrodomain resin and immunoblotted for PARP1 and NSD2 as indicated.
PARP1 is a nuclear DNA-dependent ADP-ribosyltransferase that catalyzes poly(ADP-ribosyl)ation (PAR) of nuclear proteins (29). It serves critical roles in different molecular pathways by modifying itself and other proteins. The PARylation activity of PARP1 is necessary for mediating DNA damage repair. Previous proteomics-based approaches have been widely used to identify PARP1 PARylation targets (30–34). Here we compared NSD2 interacting partners and PARP1 targets identified in multiple previously published papers (33, 34). PARP1 targets were plotted in green on the volcano plot (Fig. 3B) and indicated in Table S2. Interestingly, 17 of the 24 identified NSD2 interacting partners can be PARylated by PARP1. Furthermore, NSD2 itself was identified as a PARylation target in other screening experiments (33, 34). Because it has been reported that PARP1 may be activated upon DNA damage and oxidative stress, we induced oxidative stress in NTKO cells by hydrogen peroxide treatment and captured PARylated proteins using an Archaeoglobus fulgidus macrodomain (35). Results indicate that PARP1 becomes auto-PARylated upon oxidative stress and enrichment of NSD2 after PARylated protein precipitation was observed in stressed cells but not in the control cells, suggesting that NSD2 can be PARylated upon DNA damaging stimuli in vivo (Fig. 3C).
NSD2 is a PARP1 substrate
Because NSD2 interacts with PARP1 under physiological conditions and was PARylated upon oxidative stress, we further tested if PARP1 was responsible for PARylating NSD2 in vitro. Recombinant NSD2 protein was incubated with PARP1, with and without NAD+ and DNA cofactors. When incubating with PARP1 enzyme, NAD+ and DNA, a ∼50 kDa mass shift of NSD2 was observed indicating poly(ADP-ribosyl)ation (Fig. 4A, Fig. S1A, lane 5). The molecular weight shift corresponded to about 90 PAR units. In the same reaction, auto-PARylation of PARP1 was observed as an indistinct broad band in the PAR blot that blocked recognition with the PARP1 antibody (Fig. 4A, Fig. S1A, lane 7). PARylation required cofactor NAD+. Upon titration of NAD+, NSD2 PARylation plateaued when PARylated NSD2 reached about 200 kDa, suggesting the PAR modification is highly regulated or only a finite amount of PAR can be added to NSD2 (Fig. 4B, Fig. S1B). As controls, the absence of DNA in the reaction as well as the presence of PARP1 inhibitor Olaparib significantly diminished PARP1 enzymatic activity (Fig. 4A, Fig. S1A, lanes 3 and 6).
Figure 4.

NSD2 is a PARP1 substrate. A, recombinant NSD2 protein was incubated with or without PARP1 enzyme and cofactors indicated. Reactions were incubated at room temperature for 10 min and then analyzed by immunoblot. B, effect of NAD+ titration on the in vitro PARylation reaction. 0.25 μm NSD2 and 0.25 μm PARP1 were incubated with NAD+ concentrations of 1 μm, 5 μm, 10 μm, 50 μm, 100 μm, 500 μm, and 1 mm at room temperature for 10 min. The reactions were analyzed by immunoblot with the indicated antibodies. C, an in vitro histone methyltransferase assay was performed using radioactive [3H]SAM, recombinant NSD2 enzyme. No radioactivity incorporation was observed when using recombinant PARP1 as substrate. Purified nucleosomes were used as a positive control. Incorporated 3H signal was normalized to the positive control. Two independent experiments are shown.
Because several nonhistone proteins have been reported to be HMT substrates (36) and AURKA was recently identified as a novel nonhistone target of NSD2 that regulates p53 stability (37), we performed a NSD2 methyltransferase assay using PARP1 as the substrate. Significantly, no PARP1 methylation was detected in vitro suggesting that PARP1 was not a substrate for NSD2 (Fig. 4C). Taken together, our data show that NSD2 is a substrate of PARP1 but not the other way around.
PARylation of NSD2 by PARP1 decreases NSD2 histone methyltransferase activity
Previous studies showed that PARylation can regulate enzyme activity (38, 39). Therefore, we tested whether PAR modification of NSD2 affected its histone methyltransferase activity. To test this, NSD2 and PARP1 were incubated with and without the NAD+ cofactor required for PARylation prior to HMT assay. Olaparib was added into the reaction to terminate PARP1 enzyme activity (Fig. 5A). After incubation, an aliquot of the reaction was analyzed by immunoblot to measure PARylation of NSD2 (Fig. 5B, Fig. S2, lane 8). Significantly, the histone methyltransferase assay revealed that PARylated NSD2 has a 2-fold decrease of enzyme activity compared to unmodified enzyme (Fig. 5C, lane 8 versus 9). Controls of incubation of NSD2 with NAD+, activated DNA, or Olaparib alone did not affect NSD2 activity (lanes 4–6). Interestingly, the interaction of PARP1 and NSD2 alone did not have a significant effect on NSD2 histone methyltransferase activity (lane 7). Thus, our results suggest that PARylation of NSD2 may regulate its activity and the PAR modification, but not the interaction of PARP1, and NSD2 inhibits NSD2 HMT function.
Figure 5.

PARylation of NSD2 inhibits its enzyme activity. A, schematic of the experimental strategy. NSD2 was incubated with individual cofactors or together with PARP1 for PARylation reaction as outlined in Step 1. Subsequently the reactions were terminated by adding Olaparib and incubated on ice for 30 min as in Step 2. B, one-third of the PARylation reaction was removed and analyzed by immunoblot to assay for PARylation. C, 3H-labeled SAM and purified HeLa oligonucleosomes were added for the NSD2 histone methyltransferase reaction as outlined in Step 3 and methylation activity was analyzed by a filter-binding assay that measures incorporation of 3H-labeled methyl groups. Two independent experiments are shown.
PARylation of NSD2 disrupts its association with chromatin
Next, we assessed whether the decrease of PARylated NSD2 HMT activity was due to loss of enzyme activity or inhibition of chromatin binding. To test this, we performed the PARylation reaction first, then provided SAM and biotinylated oligonucleosomes for the histone methyltransferase assay. Biotinylated nucleosomes were then captured and analyzed along with any associated NSD2 (Fig. 6A). Because the nucleosomes used were isolated from HeLa cells, basal H3K36me2 was observed (Fig. 6, B and C, Fig. S3, lane 2). Increased H3K36me2 was detected when unmodified NSD2 protein was in the reaction (lanes 3 and 5). However, when NSD2 was PARylated, the H3K36me2 level remained the same as without addition of NSD2 (lane 4), further supporting that PARylation of NSD2 inhibits its enzymatic function. Significantly, whereas unmodified NSD2 proteins were co-purified with biotinylated nucleosomes (Fig. 6B, Fig. S3, lanes 3 and 5), association of PARylated NSD2 with nucleosome was greatly reduced (lane 4 versus lane 3), suggesting that PARylation blocks the NSD2-chromatin interaction. NSD2 and PARP1 interaction without NAD+ had no effect on NSD2 chromatin binding and concomitantly NSD2 could catalyze increased H3K36me2 levels (Fig. 6, B and C, Fig. S3, lane 5). In summary, our data suggest the PARylation of NSD2 by PARP1 prevents it from binding to chromatin, resulting in decreased histone methyltransferase activity.
Figure 6.

PARylation of NSD2 disrupts its association with chromatin in vitro. A, schematic of the experimental strategy. After the PARylation reaction, SAM and biotinylated HeLa oligonucleosomes were allowed to bind to NSD2. Streptavidin magnetic beads were added into the reaction and incubated for 1 h at room temperature. B, biotinylated nucleosomes along with bound NSD2 were captured and analyzed by immunoblot against NSD2, histone H3K36me2, and total histone H4. C, histone H3K36me2 band relative intensities were quantified using ImageJ. Two independent experiments are shown.
PARP1 PARylates NSD2 and reduces its recruitment to target genes
We further evaluated whether NSD2 can be PARylated by PARP1 in the cell. To test endogenous NSD2 PARylation by PARP1, we pre-treated NTKO cells with or without Olaparib and precipitated all PARylated proteins upon oxidative stress. As shown in Fig. 7A (left panel), NSD2 was PARylated and captured upon H2O2 treatment. However, cells pre-treated with Olaparib to inhibit PARP1 activity had much less PARylated NSD2, suggesting PARP1 PARylates NSD2 in the cell upon oxidative stress.
Figure 7.

PARylation of NSD2 disrupts its association with chromatin in the cell. A, NTKO cells pretreated with 5 μm Olaparib for 24 h were treated with 0.5 mm hydrogen peroxide for 15 min and harvested to extract nuclear proteins. PARylated proteins were then affinity captured with a poly(ADP-ribose)-binding macrodomain resin and immunoblotted for PARP1 and NSD2 as indicated. B, nuclear fractionation assay of KMS11 cells. Soluble and insoluble nuclear fractions were analyzed by immunoblot for NSD2. HDAC2 was used as soluble loading control and histone H3 was used as insoluble loading control. Relative soluble and insoluble NSD2 was quantified using ImageJ. C, ChIP-qPCR against NSD2 on the NSD2-regulated genes JAM2 and CLS2 promoters and gene bodies using NTKO cells with or without treatment of hydrogen peroxide. IgG was used as negative control. Enrichment was calculated as a percentage of total input DNA. Three independent biological replicates are shown.
Next, we investigated if PARylation affects NSD2 binding to chromatin in the cell. We performed a nuclear fractionation assay that separates soluble nuclear proteins from chromatin-bound nuclear proteins. When treated with H2O2, soluble NSD2 increased and insoluble NSD2 decreased compared with nuclear loading controls, suggesting PARylation of NSD2 released it from chromatin binding (Fig. 7B, Fig. S4). The modest localization change of NSD2 might be due to only partial PARylation of NSD2 in the cell in response to H2O2. Alternatively, FRAP studies by our group and others showed that NSD2 is tightly bound to chromatin and exchanges with free molecules slowly (3, 12). If for example, only free NSD2 were readily PARylated, a limited H2O2 treatment would only be expected to yield a partial increase in net soluble NSD2 levels, given the slow release of bound NSD2 from chromatin.
Furthermore, we investigated how NSD2 PARylation affects individual gene loci. JAM2 and GLS2 were previously shown as NSD2-regulated genes and down-regulated when the NSD2 level was depleted (6). We performed ChIP-qPCR using primers that target both promoters and gene body regions of JAM2 and GLS2. NSD2 binding was significantly decreased at both gene loci when NTKO cells were treated with H2O2 (Fig. 7C). Taken together, our results suggest PARP1 PARylates NSD2 upon oxidative stress to inhibit its ability to bind chromatin and recruitment to target genes in vivo.
Discussion
In this study, we mapped the interaction network of NSD2 in a multiple myeloma model by both qualitative and quantitative high precision MS. Under physiological conditions, NSD2 remains bound to the chromatin. The BioID method (∼10 nm labeling radius) (40) enabled us to better capture the real neighboring proteins of NSD2. Twenty-four high confidence interacting proteins were identified including the previously described NSD2 interacting partner BRD4.
PARP1 has drawn a large amount of attention in recent years due to the success of its inhibitors (Olaparib, Niraparib, and Rucaparib) in clinical trials (41). In our study, PARP1 was identified as a novel interacting partner of NSD2. NSD2 was previously identified in proteomics-based studies as a PARP1 substrate (33, 34). We successfully validated the physical interaction and demonstrated that in MM cells NSD2 can be PARylated in the context of a DNA damaging stimulus. Our results further suggest that PARP1 PARylates NSD2 to dissociate it from chromatin binding and reduces its recruitment to target genes in the cell. Our findings are evidence of a novel mechanism by which PARP1 may regulate NSD2 localization.
A growing number of studies have revealed that PARP1 regulates gene expression through PARylation of histone methyltransferases and demethylases (42). On one hand, at the transcription start site of PARP1-regulated genes, PARP1 colocalizes with H3K4me3, a histone mark enriched at the promoter of activated genes, and protects this mark by inhibiting the corresponding histone demethylase KDM5B, consequently promoting gene expression (43). On the other hand, PARP1 facilitates repression of retinoic acid-dependent genes by PARylating and inhibiting H3K9 demethylase KDM4D to maintain H3K9me2, a repressive histone mark, at the promoter regions of these genes (44). Moreover, PARP1 directly PARylates EZH2, the catalytic subunit of the PRC2 complex that specifically catalyzes histone H3K27me3, a transcriptional repressive mark that antagonizes H3K36me2 (38, 39). Our data show that PARP1 may also inhibit H3K36me2, which localizes to promoter regions and transcription start sites of actively transcribed genes through NSD2 activity. One conceivable functional role for PARP1 action on NSD2 may be that upon sensing DNA damage, PARP1-dependent PARylation of NSD2 may reduce the level of H3K36me2 at actively transcribed genes to inhibit gene expression.
H3K36me2 is enriched at DSBs upon DNA damage. However, the balance of methylation and demethylation is critical for the repair. Metnase is another enzyme responsible for writing H3K36me2 marks at DSBs to recruit DNA repair machinery proteins including NBS1 and Ku70. The loss of Metnase causes decreased H3K36me2 and deficient DNA repair (14). Similarly, reduced H3K36me2 due to loss of NSD2 in U2OS cells was associated with both decreased HR and NHEJ repair (23). However, a study in Caenorhabditis elegans suggests when JMJD-5, the H3K36me2 demethylase, is deleted or catalytic activity disabled, the animals are hypersensitive to IR and cannot resolve RAD51 foci during HR (18). Collectively, the information suggests that the level of H3K36me2 at the DSB sites might be crucial for proper DNA damage repair by both NHEJ and HR. As an important early stage sensor and regulator of DNA damage repair, PARP1 may regulate the H3K36me2 level by PARylating NSD2 and potentially other epigenetic regulators to maintain the histone mark balance.
NSD2 serves as an epigenetic writer that dimethylates histone H3 at lysine 36 to activate gene transcription. ChIP studies suggest that NSD2 is bound to chromatin globally across the genome in t(4;14)+ MM cells (45). However, under physiological conditions, how NSD2 regulates the activity of specific genes remains unclear. The identification of several transcription factors as binding partners with NSD2 in our study may provide hints at how NSD2 specificity is achieved. ZBTB33 (Kaiso) and GATAD2B are both transcriptional repressors and bind to the DNA in a sequence-specific fashion (46–49). Further characterization of interactions of NSD2 to ZBTB33 and GATAD2B may show novel mechanisms of how NSD2 regulates downstream gene expression. Furthermore, the identification of DDX46 and SF3B2 as NSD2 interacting partners suggest a novel function of NSD2 in RNA splicing. Both DDX46 and SF3B2 are involved in pre-spliceosome assembly (50, 51). Interestingly, SF3BS is a nonhistone target of PRMT9, an arginine methyltransferase (52). Because little is known about nonhistone targets of NSD2 so far, the interaction of NSD2 with SF3B2 might provide a new idea of NSD2 regulating nonhistone proteins through its methyltransferase activity. Furthermore, NSD2 has a critical role in facilitating transcription elongation (24), and the identification of DDX46 and SF3B2 as binding partners might suggest a new mechanism of functional interactions between elongation and splicing.
In summary, this study identifies new interacting partners of the epigenetic regulator NSD2 and reveals a potential novel function in DNA damage repair. These results illustrate the importance for the balance and cross-talk between PARylation and histone modification in regulating DNA repair. Further studies of this regulation may provide new insights of therapeutic possibilities in multiple myeloma.
Experimental procedures
Cloning and cell culture
KMS11-derived TKO and NTKO MM cell lines were cultured in RPMI 1640 media (Life Technologies) supplemented with 10% fetal bovine serum. Phoenix-AMPHO packaging cells were cultured in Dulbecco's modified Eagle's medium 1640 (Life Technologies) supplemented with 10% fetal bovine serum. The NSD2 cDNA was cloned into the retroviral vector pRetroX-IRES-DsRedExpress (Clontech) using NotI and ClaI restriction sites. pcDNA3.1 MCS-BirA(R118G)-HA was a gift from Kyle Roux (Addgene plasmid number 36047) (26). The BirA cDNA was PCR amplified from the pcDNA3.1 vector and inserted in-frame to the C terminus of NSD2 using ClaI and BamHI restriction sites. For the control, BirA with a C-terminal nuclear localization signal was cloned into the retroviral vector. Retroviruses to express NSD2-BirA and BirA control were generated by transfection of packaging Phoenix-AMPHO cells with the plasmids described above using Lipofectamine 3000 reagent (Invitrogen). One million TKO cells were resuspended in 2 ml of virus media along with 6 μg/ml of Polybrene (Millipore) and then followed by spinoculation for 90 min at 2000 rpm. DsRed-positive cells were sorted (BD FACS Aria II) 72 h post-transduction and expanded.
BioID
BioID was performed as described in Ref. 27. For each experimental replicate, one 15-cm dish of TKO or TKO-NSD2-BirA cells was cultured and supplemented in the presence of 50 μm biotin for 48 h, washed 3 times in PBS, lysed in BioID lysis buffer (50 mm Tris-HCl, pH 7.4, 500 mm NaCl, 0.2% SDS, 1 mm DTT, 1× Halt protease inhibitor (Thermo Fisher Scientific)), and quenched. Supernatants were saved after centrifuging at 14,000 × g for 10 min at 4 °C. Total proteins were quantified by BCA assay (Pierce) and the same amount to total proteins in each replicate was used for streptavidin capture. PierceTM Streptavidin Magnetic Beads (60 μl, Thermo Fisher Scientific) were washed 3 times with lysis buffer and incubated with protein lysate overnight at 4 °C on a tube rotator. Protein-bound magnetic beads were washed with three different wash buffers (27) and were then ready for digestion.
For on-bead digestion, magnetic beads were first washed twice in 100 mm ammonium bicarbonate, and bound proteins were reduced by addition of DTT to a final concentration of 10 mm and incubation at 50 °C for 30 min. Alkylation was then done by incubation in 100 mm iodoacetamide (Sigma) at room temperature for 30 min in the dark. Bound proteins were then washed with 100 mm ammonium bicarbonate and digested with 1 μg of trypsin (Promega) overnight at 37 °C. The reaction was stopped by adding 2 μl of formic acid. ZipTip (Millipore) cleanup was performed to desalt the samples. The resulting peptides were further dried in a SpeedVac centrifuge.
Mass spectrometry data acquisition
Nanocapillary LC-MS/MS analyses were performed with a 75-μm × 10.5-cm PicoChip column (New Objective, MA) packed with 1.9-μm Reprosil C18 beads. A 150 μm × 3-cm trap packed with 3-μm beads was installed in-line. Solvent A consisted of 0.1% formic acid in water and solvent B was 0.1% formic acid in acetonitrile. Peptides were resuspended in 30 μl of solvent A for LC-MS, and 6 μl were injected. For the label-free quantitative study, each sample was injected in technical triplicate. A total of 18 LC-MS/MS runs (3 biological replicates × 3 technical replicates × 2 states) were injected in random order for the data presented in Fig. 2. Peptides were trapped at 5 μl/min for 5 min with 100% Solvent A, then separated at a flow rate of 300 nl/min with a gradient from 5 to 40% B in 35 min. After a 4-min ramp to 60% B, the column was washed at 95% B for 3 min and re-equilibrated to 5% B for 10 min with a total analysis time of 60 min.
The LC was coupled by electrospray to an LTQ Velos Orbitrap mass spectrometer operating in data-dependent MS/MS mode with a top 10 method. The capillary temperature was 275 °C. MS1 scans were collected from 400 to 2000 m/z with resolving power equal to 60,000 at 400 m/z. The MS1 AGC was set to 1 × 106. Precursors were isolated with a 1.5 m/z isolation width, and the CID normalized collision energy was set to 35%. Dynamic exclusion was set to 60 s and charge 1+ ions were excluded as well. The ion trap AGC target was set to 3 × 105.
Protein identification
Mascot searching was performed for BioID protein identification. Raw files were converted to .mgf as input. Human SwissProt AC 2018_02 database (20,317 sequences) was used for the search. Trypsin was used as the proteolytic enzyme with up to two missed cleavages. Oxidation on methionine and deamination on asparagine and glutamine were chosen as variable modifications. Carbamidomethyl on cysteine was set as static modification. Peptide mass tolerance was 10 ppm; MS/MS tolerance was 0.6 Da.
Bioinformatic and statistical analysis
For the label-free quantitative study, three biological replicates were created using a different batch of cells on different days, and each biological replicate was injected in technical triplicate. One technical injection of TKO-BirA control biological replicate 1 failed due to a LC problem. This resulted in 17 LC-MS/MS runs used for this experiment. Proteome Discoverer (version 2.1; Thermo Fisher) was used for identification and peptide quantification. A spectrum selector was used to import spectrum from .raw files. The Sequest HT search engine was used to cross-reference identified peptides against human Uniprot fasta database. The mass tolerance was set to be 10 ppm for precursors and 0.6 Da for fragment ions. Oxidation on methionine and deamination on asparagine and glutamine were chosen as variable modifications. Carbamidomethyl on cysteine was set as static modification. Trypsin was set as the proteolytic enzyme with up to two missed cleavages. The precursor ion area detector in Proteome Discoverer v2.1 was used for quantification.
In total, 2955 peptides were identified. Each protein was considered identified when at least two unique peptides were identified with at least two independent peptide spectral matches observed from the biological replicates. In total, 52 proteins were identified by 270 peptides. For differential expression analysis, all peptide intensities were standardized across all measures of the peptide. For each protein, each peptide species was treated as an independent set of observations on the protein. Standardized peptide intensities were tested for differential expression using a hierarchical linear model with technical replicates nested within biological replicates with a modified form of the technique described elsewhere (53, 54). The resulting F test probabilities were then corrected for multiple testing (55) with a false discovery rate of 5%. All statistical analyses were done using a custom script for SAS 9.4 (Cary, NC). The SAS script used for differential expression calculations is included in supporting data Xiaoxiao_20170201.sas.zip.
Co-immunoprecipitation
Nuclear extracts were prepared using the Nuclear Complex Co-IP Kit (Active Motif, CA) according to the manufacturer's protocol and quantified by BCA assay (Pierce). Nuclear proteins (200 μg) were incubated with 2 μg of PARP1 antibody (39559, Active Motif), rabbit IgG (2729S, CST), NSD2 antibody (ab75359, Abcam), or mouse IgG2b (ab91366, Abcam) at 4 °C overnight on a tube rotator. Next, 40 μl of Protein G Mag-Sepharose beads (Sigma) were added into the mixture and incubated for 1 h. The protein complex bound beads were washed 3 times with PBS + 0.5% Nonidet P-40 and 3 times with PBS. Bound proteins were then eluted by boiling for 10 min in 20 μl of 1 × SDS loading buffer and analyzed by Western blotting with antibodies against NSD2 and PARP1.
PARylated protein precipitation
Cells were washed with PBS and incubated in PBS with 0.5 or 1 mm hydrogen peroxide (Sigma) at 37 °C for 15 min. Nuclear proteins were extracted and 200 μg of were diluted in 500 μl of PAR lysis buffer (50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 1 mm EDTA, 1% Triton X-100, 10% glycerol, 0.5% deoxycholate, 1 mm DTT, 1× Halt Protease Inhibitor). In total, 20 μl of poly(ADP-ribose) binding macrodomain resin (Tulip Biolabs, PA) was pre-washed with PAR lysis buffer and added into the nuclear protein mixture to incubate overnight at 4 °C on a tube rotator. The beads were washed 3 times with PAR lysis buffer. Bound PARylated proteins were eluted by boiling for 10 min in 20 μl of 1× SDS loading buffer and analyzed by immunoblot with antibodies against NSD2 and PARP1 (ab194586, Abcam).
In vitro PARylation assay
Recombinant NSD2 methyltransferase (0.25 μm, Reaction Biology, PA) was incubated with 0.25 μm PARP1 enzyme (Tulip Biolabs, PA) in 10 μl of PARylation buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm MgCl2, 1 mm DTT). Other components including 10 μg/ml of calf thymus DNA (Sigma), 50 μm NAD+, and 500 nm Olaparib (Selleckchem) were added to the reaction as indicated. The reactions were incubated at room temperature for 10 min and inactivated by adding 10 μl of 2× SDS loading buffer, and boiling for 2 min. The elution was resolved by SDS-PAGE and immunoblotted with antibodies against NSD2, PARP1, and poly(ADP-ribose) polymer (ab14459, Abcam).
In vitro histone methyltransferase assay
PARylation reaction containing 0.25 μm recombinant NSD2, 0.25 μm PARP1 enzyme, 10 μg/ml of DNA, and 50 μm NAD+ in 15 μl of PARylation buffer was set up to PARylate NSD2. After a 10-min incubation at room temperature, 500 nm Olaparib or ddH2O was added to stop the reaction. One-third (5 μl) of the reaction was removed to confirm PARylation by immunoblot. The remaining 10-μl reaction was diluted in 40 μl of HMT buffer (50 mm Tris-HCl, pH 8.5, 50 mm NaCl, 10 mm MgCl2, 2 mm tris(2-carboxyethyl)phosphine) with 1 μm HeLa oligonucleosomes (Reaction Biology, PA) and 2 μm [3H]SAM (PerkinElmer Life Sciences) and incubated for 1 h in a 30 °C water bath. The HMT reaction was terminated by adding trichloroacetic acid (TCA) to a final concentration of 10%. Precipitated proteins were spotted onto glass microfiber filters (GE Healthcare) and washed with ice-cold 10% TCA and 100% ethanol. The filters were air dried in the chemical hood and measured by liquid scintillation counting for 3H incorporation.
NSD2-nucleosome association assay
A PARylation reaction of 15 μl was set up as described above. After terminating with Olaparib, the reaction was diluted in HMT buffer with 1 μm biotinylated HeLa oligonucleosomes (HMT-35-160, Reaction Biology, PA) and 2 μm SAM (New England Biolabs) to a final volume of 60 μl and incubated for 30 min at 30 °C. Following incubation, 20 μl of the reaction was kept as the input fraction. The remaining 40 μl of the reaction was diluted in 460 μl of wash buffer (50 mm Tris-HCl, pH 8.5, 100 mm NaCl, 5 mm MgCl2, 2 mm tris(2-carboxyethyl)phosphine, 0.5% Nonidet P-40, Halt protease inhibitor) with 50 μl of pre-washed streptavidin magnetic beads (88817, Pierce) and incubated at room temperature for 1 h. Nucleosome-bound complexes were washed three times in wash buffer and eluted by boiling in 30 μl of 1× SDS loading buffer. Input and elution were resolved by SDS-PAGE and blotted with antibodies against NSD2, PARP1, histone H4 (ab7311, Abcam), and H3K36me2 (2901S, CST).
Nuclear fractionation assay
Nuclear fractionation assay was performed according to the manufacturer's protocol (Subcellular Protein Fractionation Kit for Cultured Cells, Thermo). Soluble and insoluble nuclear protein fractions were saved and quantified by BCA assay (Pierce). The same amount of soluble and insoluble proteins from hydrogen peroxide-treated and untreated samples were resolved by SDS-PAGE and blotted with antibodies against NSD2, HDAC2 (05-814, Millipore), and histone H3 (9715S, CST).
ChIP-qPCR
Cells (107) treated with or without 0.5 mm hydrogen peroxide were cross-linked with 0.8% formaldehyde for 7 min at room temperature. Glycine was added to 250 mm and incubated for 5 min. Cells were then spun down and resuspended in 1 ml of lysis buffer (5 mm HEPES, pH 7.5, 14 mm NaCl, 0.1 mm EDTA, 10% glycerol, 1% Triton X-100) and incubated for 10 min on ice to isolate nuclei. The extracted nuclei were spun down and washed in 1 ml of wash buffer (2 mm Tris, pH 7.6, 20 mm NaCl, 0.1 mm EDTA) for 10 min on ice. The resulting nuclei were then resuspended in 950 μl of shearing buffer (10 mm Tris, pH 7.6, 1 mm EDTA) and transferred to 1 ml of milliTUBE (Covaris, MA) for sonication with E220 Focused-ultrasonicators (Covaris, MA) at a duty factor of 10%, peak incidence 140, and cycle/burst 200 for 35 min. Sonicated chromatin samples were diluted with 1 ml of ChIP buffer twice (30 mm Tris, pH 8.0, 300 mm NaCl, 2 mm EDTA, 2% Triton X-100) and incubated with 10 μg of NSD2 antibody (6) or IgG2b antibody for immunoprecipitation at 4 °C overnight. Input (1%) was saved until reversing cross-link. Pre-blocked 25 μl of Dynabeads protein A/G (Thermo) were added into the samples and incubated for 2 h at 4 °C with rotation. The protein-bound magnetic beads were separated and washed twice with the following buffers: ChIP buffer (15 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100), high salt buffer (20 mm Tris, pH 8.0, 500 mm NaCl, 2 mm EDTA, 0.1% SDS, 1% Triton X-100), LiCl buffer (10 mm Tris, pH 8.0, 1 mm EDTA, 0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholate) and 10 mm Tris, pH 8.0, buffer, then eluted and reverse cross-linked with 100 μl of elution buffer (1% SDS, 200 mm NaCl, 10 mm EDTA, 50 mm Tris, pH 8.0) at 65 °C for 4 h. RNase A (3 μg, 20 mg/ml, Thermo) and proteinase K were added and incubated at 45 °C for 1 h. DNA samples were purified using Qiagen PCR purification kit. Primers for JAM2 and CLS2 were previously described (6). qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Enrichment was calculated as a percentage of total input DNA.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (56) partner repository with dataset identifier PXD013759.
Author contributions
X. H., N. L. K., and J. D. L. conceptualization; X. H., L. F., A. J. S., and R. L. B. data curation; X. H. investigation; X. H. methodology; X. H. writing-original draft; R. D. L. software; R. D. L. formal analysis; R. L. B., N. L. K., and J. D. L. supervision; R. L. B., N. L. K., and J. D. L. writing-review and editing; N. L. K. and J. D. L. funding acquisition; J. D. L. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grant R01 CA195732 (to J. D. L.), a Leukemia and Lymphoma Society Specialized Center of Excellence grant (to J. D. L.), and National Institutes of Health Grant P41 GM108569 (to N. L. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2, Figs. S1–S4, and data.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD013759.
- NSD
- nuclear receptor-binding SET domain
- HMT
- histone methyltransferase
- MM
- multiple myeloma
- DSB
- double strand break
- NHEJ
- nonhomologous end joining
- HR
- homologous recombination
- BRD4
- bromodomain protein 4
- PARP1
- poly(ADP-ribose) polymerase 1
- IP
- immunoprecipitation
- SAM
- S-adenosylmethionine
- HA
- hemagglutinin
- qPCR
- quantitative PCR.
References
- 1. Bennett R. L., Swaroop A., Troche C., and Licht J. D. (2017) The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harb. Perspect. Med. 7, a026708 10.1101/cshperspect.a026708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuo A. J., Cheung P., Chen K., Zee B. M., Kioi M., Lauring J., Xi Y., Park B. H., Shi X., Garcia B. A., Li W., and Gozani O. (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44, 609–620 10.1016/j.molcel.2011.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keats J. J., Maxwell C. A., Taylor B. J., Hendzel M. J., Chesi M., Bergsagel P. L., Larratt L. M., Mant M. J., Reiman T., Belch A. R., and Pilarski L. M. (2005) Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 105, 4060–4069 10.1182/blood-2004-09-3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mirabella F., Wu P., Wardell C. P., Kaiser M. F., Walker B. A., Johnson D. C., and Morgan G. J. (2013) MMSET is the key molecular target in t(4;14) myeloma. Blood Cancer J. 3, e114 10.1038/bcj.2013.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lauring J., Abukhdeir A. M., Konishi H., Garay J. P., Gustin J. P., Wang Q., Arceci R. J., Matsui W., and Park B. H. (2008) The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111, 856–864 10.1182/blood-2007-05-088674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martinez-Garcia E., Popovic R., Min D. J., Sweet S. M., Thomas P. M., Zamdborg L., Heffner A., Will C., Lamy L., Staudt L. M., Levens D. L., Kelleher N. L., and Licht J. D. (2011) The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117, 211–220 10.1182/blood-2010-07-298349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng Y., Sweet S. M., Popovic R., Martinez-Garcia E., Tipton J. D., Thomas P. M., Licht J. D., and Kelleher N. L. (2012) Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. U.S.A. 109, 13549–13554 10.1073/pnas.1205707109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oyer J. A., Huang X., Zheng Y., Shim J., Ezponda T., Carpenter Z., Allegretta M., Okot-Kotber C. I., Patel J. P., Melnick A., Levine R. L., Ferrando A., Mackerell A. D. Jr., Kelleher N. L., Licht J. D., and Popovic R. (2014) Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 28, 198–201 10.1038/leu.2013.204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jaffe J. D., Wang Y., Chan H. M., Zhang J., Huether R., Kryukov G. V., Bhang H. E., Taylor J. E., Hu M., Englund N. P., Yan F., Wang Z., McDonald R. E. 3rd, Wei L., Ma J., et al. (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 45, 1386–1391 10.1038/ng.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding L. W., Sun Q. Y., Tan K. T., Chien W., Mayakonda A., Yeoh A. E. J., Kawamata N., Nagata Y., Xiao J. F., Loh X. Y., Lin D. C., Garg M., Jiang Y. Y., Xu L., Lim S. L., et al. (2017) Mutational landscape of pediatric acute lymphoblastic leukemia. Cancer Res. 77, 390–400 10.1158/0008-5472.CAN-16-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J. H., Loh M. L., Ma X. T., Rusch M., Wu G., Harvey R. C., Wheeler D. A., Hampton O. A., Carroll W. L., Chen I. M., Gerhard D. S., Gastier-Foster J. M., Relling M. V., Smith M. A., Devidas M., et al. (2013) Comparison of mutational profiles of diagnosis and relapsed pediatric B-acute lymphoblastic leukemia: a report from the COG ALL target project. Blood 122 [Google Scholar]
- 12. Swaroop A., Oyer J. A., Will C. M., Huang X., Yu W., Troche C., Bulic M., Durham B. H., Wen Q. J., Crispino J. D., MacKerell A. D. Jr, Bennett R. L., Kelleher N. L., and Licht J. D. (2019) An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 38, 671–686 10.1038/s41388-018-0474-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jha D. K., and Strahl B. D. (2014) An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun. 5, 3965 10.1038/ncomms4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fnu S., Williamson E. A., De Haro L. P., Brenneman M., Wray J., Shaheen M., Radhakrishnan K., Lee S. H., Nickoloff J. A., and Hromas R. (2011) Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. U.S.A. 108, 540–545 10.1073/pnas.1013571108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pai C. C., Deegan R. S., Subramanian L., Gal C., Sarkar S., Blaikley E. J., Walker C., Hulme L., Bernhard E., Codlin S., Bähler J., Allshire R., Whitehall S., and Humphrey T. C. (2014) A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun 5, 4091 10.1038/ncomms5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Liu L., Yang S., Song N., Zhou X., Gao J., Yu N., Shan L., Wang Q., Liang J., Xuan C., Wang Y., Shang Y., and Shi L. (2014) Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. U.S.A. 111, 7096–7101 10.1073/pnas.1324036111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao L. L., Wei F., Du Y., Song B., Wang D., Shen C., Lu X., Cao Z., Yang Q., Gao Y., Wang L., Zhao Y., Wang H., Yang Y., and Zhu W. G. (2016) ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene 35, 402 10.1038/onc.2015.311 [DOI] [PubMed] [Google Scholar]
- 18. Amendola P. G., Zaghet N., Ramalho J. J., Vilstrup Johansen J., Boxem M., and Salcini A. E. (2017) JMJD-5/KDM8 regulates H3K36me2 and is required for late steps of homologous recombination and genome integrity. PLoS Genet. 13, e1006632 10.1371/journal.pgen.1006632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hajdu I., Ciccia A., Lewis S. M., and Elledge S. J. (2011) Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 108, 13130–13134 10.1073/pnas.1110081108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pei H., Zhang L., Luo K., Qin Y., Chesi M., Fei F., Bergsagel P. L., Wang L., You Z., and Lou Z. (2011) MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128 10.1038/nature09658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nguyen H. V., Dong J., Panchakshari R. A., Kumar V., Alt F. W., and Bories J. C. (2017) Histone methyltransferase MMSET promotes AID-mediated DNA breaks at the donor switch region during class switch recombination. Proc. Natl. Acad. Sci. U.S.A. 114, E10560–E10567 10.1073/pnas.1701366114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pei H., Wu X., Liu T., Yu K., Jelinek D. F., and Lou Z. (2013) The histone methyltransferase MMSET regulates class switch recombination. J. Immunol. 190, 756–763 10.4049/jimmunol.1201811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shah M. Y., Martinez-Garcia E., Phillip J. M., Chambliss A. B., Popovic R., Ezponda T., Small E. C., Will C., Phillip M. P., Neri P., Bahlis N. J., Wirtz D., and Licht J. D. (2016) MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 35, 5905–5915 10.1038/onc.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarai N., Nimura K., Tamura T., Kanno T., Patel M. C., Heightman T. D., Ura K., and Ozato K. (2013) WHSC1 links transcription elongation to HIRA-mediated histone H3.3 deposition. EMBO J. 32, 2392–2406 10.1038/emboj.2013.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Min D. J., Ezponda T., Kim M. K., Will C. M., Martinez-Garcia E., Popovic R., Basrur V., Elenitoba-Johnson K. S., and Licht J. D. (2013) MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia 27, 686–694 10.1038/leu.2012.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roux K. J., Kim D. I., and Burke B. (2013) BioID: a screen for protein-protein interactions. Curr. Protoc. Protein Sci. 74, Unit 19.23 [DOI] [PubMed] [Google Scholar]
- 28. Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., and Ma'ayan A. (2016) EnrichR: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gibson B. A., and Kraus W. L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 10.1038/nrm3376 [DOI] [PubMed] [Google Scholar]
- 30. Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M. O., and Nielsen M. L. (2013) Proteome-wide identification of poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285 10.1016/j.molcel.2013.08.026 [DOI] [PubMed] [Google Scholar]
- 31. Zhen Y., and Yu Y. (2018) Proteomic analysis of the downstream signaling network of PARP1. Biochemistry 57, 429–440 10.1021/acs.biochem.7b01022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gagné J. P., Pic E., Isabelle M., Krietsch J., Ethier C., Paquet E., Kelly I., Boutin M., Moon K. M., Foster L. J., and Poirier G. G. (2012) Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 40, 7788–7805 10.1093/nar/gks486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gibson B. A., Zhang Y., Jiang H., Hussey K. M., Shrimp J. H., Lin H., Schwede F., Yu Y., and Kraus W. L. (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353, 45–50 10.1126/science.aaf7865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S. C., Young C., Hottiger M. O., and Nielsen M. L. (2016) Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 7, 12917 10.1038/ncomms12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karras G. I., Kustatscher G., Buhecha H. R., Allen M. D., Pugieux C., Sait F., Bycroft M., and Ladurner A. G. (2005) The macro domain is an ADP-ribose binding module. EMBO J. 24, 1911–1920 10.1038/sj.emboj.7600664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang X., Wen H., and Shi X. (2012) Lysine methylation: beyond histones. Acta Biochim. Biophys. Sin. (Shanghai) 44, 14–27 [DOI] [PubMed] [Google Scholar]
- 37. Park J. W., Chae Y. C., Kim J. Y., Oh H., and Seo S. B. (2018) Methylation of Aurora kinase A by MMSET reduces p53 stability and regulates cell proliferation and apoptosis. Oncogene 37, 6212–6224 10.1038/s41388-018-0393-y [DOI] [PubMed] [Google Scholar]
- 38. Caruso L. B., Martin K. A., Lauretti E., Hulse M., Siciliano M., Lupey-Green L. N., Abraham A., Skorski T., and Tempera I. (2018) Poly(ADP-ribose) polymerase 1, PARP1, modifies EZH2 and inhibits EZH2 histone methyltransferase activity after DNA damage. Oncotarget 9, 10585–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamaguchi H., Du Y., Nakai K., Ding M., Chang S. S., Hsu J. L., Yao J., Wei Y., Nie L., Jiao S., Chang W. C., Chen C. H., Yu Y., Hortobagyi G. N., and Hung M. C. (2018) EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene 37, 208–217 10.1038/onc.2017.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim D. I., Birendra K. C., Zhu W., Motamedchaboki K., Doye V., and Roux K. J. (2014) Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. U.S.A. 111, E2453–2461 10.1073/pnas.1406459111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown J. S., Kaye S. B., and Yap T. A. (2016) PARP inhibitors: the race is on. Br. J. Cancer 114, 713–715 10.1038/bjc.2016.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ciccarone F., Zampieri M., and Caiafa P. (2017) PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 63, 123–134 10.1016/j.semcdb.2016.11.010 [DOI] [PubMed] [Google Scholar]
- 43. Krishnakumar R., and Kraus W. L. (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol. Cell 39, 736–749 10.1016/j.molcel.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Le May N., Iltis I., Amé J. C., Zhovmer A., Biard D., Egly J. M., Schreiber V., and Coin F. (2012) Poly(ADP-ribose) glycohydrolase regulates retinoic acid receptor-mediated gene expression. Mol. Cell 48, 785–798 10.1016/j.molcel.2012.09.021 [DOI] [PubMed] [Google Scholar]
- 45. Popovic R., Martinez-Garcia E., Giannopoulou E. G., Zhang Q., Zhang Q., Ezponda T., Shah M. Y., Zheng Y., Will C. M., Small E. C., Hua Y., Bulic M., Jiang Y., Carrara M., Calogero R. A., et al. (2014) Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 10, e1004566 10.1371/journal.pgen.1004566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prokhortchouk A., Hendrich B., Jørgensen H., Ruzov A., Wilm M., Georgiev G., Bird A., and Prokhortchouk E. (2001) The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Gene Dev. 15, 1613–1618 10.1101/gad.198501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brackertz M., Boeke J., Zhang R., and Renkawitz R. (2002) Two highly related p66 proteins comprise a new family of potent transcriptional repressors interacting with MBD2 and MBD3. J. Biol. Chem. 277, 40958–40966 10.1074/jbc.M207467200 [DOI] [PubMed] [Google Scholar]
- 48. Brackertz M., Gong Z., Leers J., and Renkawitz R. (2006) p66α and p66β of the Mi-2/NuRD complex mediate MBD2 and histone interaction. Nucleic Acids Res. 34, 397–406 10.1093/nar/gkj437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koh D. I., Han D. Y., Ryu H., Choi W. I., Jeon B. N., Kim M. K., Kim Y., Kim J. Y., Parry L., Clarke A. R., Reynolds A. B., and Hur M. W. (2014) KAISO, a critical regulator of p53-mediated transcription of CDKN1A and apoptotic genes. Proc. Natl. Acad. Sci. U.S.A. 111, 15078–15083 10.1073/pnas.1318780111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Will C. L., Urlaub H., Achsel T., Gentzel M., Wilm M., and Lührmann R. (2002) Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J. 21, 4978–4988 10.1093/emboj/cdf480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cretu C., Schmitzová J., Ponce-Salvatierra A., Dybkov O., De Laurentiis E. I., Sharma K., Will C. L., Urlaub H., Lührmann R., and Pena V. (2016) Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol. Cell 64, 307–319 10.1016/j.molcel.2016.08.036 [DOI] [PubMed] [Google Scholar]
- 52. Hadjikyriacou A., Yang Y., Espejo A., Bedford M. T., and Clarke S. G. (2015) Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate RNA splicing factor SF3B2. J. Biol. Chem. 290, 16723–16743 10.1074/jbc.M115.659433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wildburger N. C., Ali S. R., Hsu W. C., Shavkunov A. S., Nenov M. N., Lichti C. F., LeDuc R. D., Mostovenko E., Panova-Elektronova N. I., Emmett M. R., Nilsson C. L., and Laezza F. (2015) Quantitative proteomics reveals protein-protein interactions with fibroblast growth factor 12 as a component of the voltage-gated sodium channel 1.2 (Nav1.2) macromolecular complex in mammalian brain. Mol. Cell. Proteomics 14, 1288–1300 10.1074/mcp.M114.040055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wildburger N. C., Lichti C. F., LeDuc R. D., Schmidt M., Kroes R. A., Moskal J. R., and Nilsson C. L. (2015) Quantitative proteomics and transcriptomics reveals metabolic differences in attracting and non-attracting human-in-mouse glioma stem cell xenografts and stromal cells. EuPA Open Proteomics 8, 94–103 10.1016/j.euprot.2015.06.006 [DOI] [Google Scholar]
- 55. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 2346101 [Google Scholar]
- 56. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (56) partner repository with dataset identifier PXD013759.