

Abstract
A new family of cyclometalated ruthenium(II) complexes [Ru(N^N)2 (C^N)]+ derived from the π-extended benzo[h]imidazo[4,5-f]quinolone ligand appended with thienyl groups (n=1–4, compounds 1–4) was prepared and characterized for their chemical, photophysical, and photobiological properties. The lipophilicities of 1–4, determined as octanol-water partition coefficients (log Po/w), were positive and increased with the number of thienyl units. The absorption and emission bands of the C^N compounds were red-shifted by up to 200 nm relative to the analogous Ru(II) diimine systems. All of the complexes exhibited dual emission, with the intraligand fluorescence (1IL, C^N-based) shifting to lower energies with increasing n and the metal-to-ligand charge transfer phosphorescence (3MLCT, N^N-based) remaining unchanged. Compounds 1–3 exhibited excited state absorption (ESA) profiles consistent with lowest-lying 3MLCT states when probed by nanosecond transient absorption (TA) spectroscopy with 532-nm excitation, and had contributions from 1IL(C^N) states with 355-nm excitation. These assignments were supported by the lifetimes observed (<10 ns for the 1IL states, and around 20 ns for the 3MLCT states) as well as a noticeable ESA for 3 with 355-nm excitation that did not occur with 532-nm excitation. Compound 4 was the only member of the family with two 3MLCT emissive lifetimes (15, 110 ns), and the TA spectra collected with both 355- and 532-nm excitation was assigned to the 3IL state, which was corroborated by its 4–6 µs lifetime. The ESA for 4 had a rise time of approximately 10 ns and an initial decay of 110 ns, which suggests a possible 3MLCT-3IL excited state equilibrium that results in delayed emission from the 3MLCT state. Compound 4 was nontoxic toward human skin melanoma cells (SKMEL28) in the dark (EC50 = >300 µM); 1–3 were cytotoxic and yielded EC50 values between 1–20 µM. The photocytotoxicites with visible light ranged from 87 nM with a phototherapeutic index (PI) of 13 for 1 to approximately 1 µM (PI = >267) for 4. With red light, EC50 values varied from 270 nM (PI=21) for 3 to 12 µM for 4 (PI = >25). The larger PIs for 4, especially with visible light, were attributed to the much lower dark cytotoxicity for this compound. Since the dark cytotoxicity contributes substantially to the observed photocytotoxicity for 1–3, it was not possible to assess whether the 3IL state of 4 led to a much more potent phototoxic mechanism in the absence of dark toxicity. There was no stark contrast in cellular uptake and accumulation by laser scanning confocal and differential interference contrast (DIC) microscopy to explain the large differences in dark toxicities between 1–3 and 4. Nevertheless, the study highlights a new family of Ru(II) C^N complexes where π-conjugation beyond a certain point results in low dark cytotoxicity with high photocytotoxicity, opposing the notion that cyclometalated Ru(II) systems are too toxic to be phototherapeutic agents.
Graphical Abstract
SYNOPSIS TOC.
A Ru(II) complex bearing the cyclometalating IBQ-4T ligand has been prepared, which is one of only a few examples of a Ru(II) C^N complex with a large phototherapeutic index (PI). The new compound has a low-energy intraligand triplet excited state that leads to photocytotoxicity toward melanoma cancer cells.
INTRODUCTION
There has been a longstanding interest in Ru(II) coordination complexes derived from diimine ligands (N^N) for use as photosensitizers in a variety of applications ranging from phototherapy to solar energy conversion. This enthusiasm for [Ru(bpy)3]2+ (bpy=2,2′-bipyridine) and its many derivatives over the years stems from the interesting photochemical and photophysical properties of the parent compound, notably, strong absorption and emission of visible light due to relatively long-lived (τ≈1 µs) metal-to-ligand charge transfer (MLCT) excited states.1–3 In our own group, we have been heavily involved in the exploration of Ru(II)-based compounds for photodynamic therapy (PDT), with one photosensitizer (TLD1433) having successfully completed a human clinical trial (ClinicalTrials.gov identifier: ) for treating bladder cancer with PDT.4
Relative to the well-known Ru(II) N^N complexes, the analogous complexes derived from cyclometalating ligands (C^N) have been understudied. As of 2015 the Cambridge Structural Database (CSD) contained fewer than 20 [Ru(N^N)2(C^N)]+ complexes,5,6 and these were narrow in scope and based only on the deprotonated phenylpyridine C^N ligand (phpy−). The lag with research related to this compound class has been attributed to modest metal-based oxidation potentials, short excited state lifetimes, and weak photoluminescence related, in part, to the much lower excited state energies of the C^N systems.7 However, their lower energy MLCT states also make these systems attractive in terms of longer wavelength absorption and enhanced thermal and photochemical stability, features that the dye-sensitized solar cells (DSSCs) community have exploited.8–11
The larger 3MLCT-3MC energy gaps associated with Ru(II) C^N complexes have prevented their exploitation as photochemotherapy (PCT) agents, where the phototoxic mechanism is based on photoinduced ligand loss from dissociative 3MC excited states.12 Likewise, the smaller energy gaps between the 3MLCT states and the ground states results in reduced singlet oxygen (1O2) quantum yields,13,14 making many Ru(II) C^N compounds poor PDT agents. In addition, many Ru(II) C^N systems, with their reduced charges and higher lipophilicities compared to their Ru(II) N^N cousins, are cytotoxic in the dark, resulting in very marginal phototherapeutic indices (PI). Such systems have little utility in phototherapy applications that require an otherwise nontoxic prodrug.12,14–16
This dark cytotoxicity has been demonstrated for complexes of the type [Ru(bpy)(phpy−)(N^N)]Cl (where N^N=1,10-phenanthroline (phen), dipyrido[3,2-f:2′,3′-h]quinoxaline (dpq), dipyrido[3,2-a:2′,3′-c]phenazine (dppz), or benzo[i]dipyrido[3,2-a;2′,3′-c]phenazine (dppn)).15,16 We also found this to be the case for [Ru(bpy)2 (C^N)]Cl complexes where C^N is deprotonated benzo[h]quinoline (bhq−), 4,9,12-triazadibenzo[a,c]naphthalene (pbpq−), or 4,9,14-triazadibenzo[a,c]anthracene (pbpz−);13 and [Ru(4,4′-dmb)2 (C^N)]Cl systems where 4,4′-dmb=4,4′-dimethyl-2,2′-bipyridine and C^N is deprotonated 2-(2-thiophene)-1H-benzo[h]imidazo[4,5-f]quinoline (IBQ-1T), 2-(2,2′-bithiophene)-1H-benzo[h]imidazo[4,5-f]quinoline (IBQ-2T), and 2-(2,2′:5′,2′′-terthiophene)-1H-benzo[h]imidazo[4,5-f]quinoline (IBQ-3T).14
In our studies, we systematically extended the π-conjugation on the C^N ligand of the cyclometalated Ru(II) compounds instead of the N^N ligand (the latter had been done by others). Surprisingly, we found that there was an abrupt reduction in the dark cytotoxicity on going from the [Ru(bpy)2(pbpz−)]Cl complex to the more π-expansive [Ru(bpy)2(pbpn−)]Cl complex (pbpn−=4,9,16-triazadibenzo[a,c]naphthacene),13 and on going from the [Ru(4,4′-dmb)2 (IBQ-3T)]Cl complex to the more π-expansive [Ru(4,4′-dmb)2(IBQ-4T)]Cl complex (IBQ-4T=2-(2,2′:5′,2′′:5′′,2′′′-tetrathiophene)-1H-benzo[h]imidazo[4,5-f]quinoline.14 From these two studies, we established that (i) not all Ru(II) C^N complexes are inherently cytotoxic, and (ii) cytotoxicity does not always positively correlate with lipophilicity in this compound class. We further wondered whether the abrupt elimination of cytotoxicity we observed at a critical degree of π-conjugation (in families where π-conjugation was varied systematically) was a general trend.
To address this question, we prepared complexes 1–4 shown in Chart 1. These Ru(II) C^N complexes are derived from the IBQ-nT C^N ligand combined with bpy N^N ligands, and are thus close relatives of the previously published IBQ-nT series with 4,4′-dmb as the N^N coligands. The hypothesis for this investigation was that an abrupt change in dark cytotoxicity would occur between 3 and 4, further supporting our assertion that π-extension of the C^N ligand in Ru(II) cyclometalated complexes provides an opportunity to create potent photobiological agents with low cytotoxicity.
Chart 1.

Molecular Structures of Ru(II) C^N Complexes 1–4.
EXPERIMENTAL PROCEDURES
Materials.
Benzo[h]quinoline and iodopentoxide, were purchased from Sigma-Aldrich. The formylthiophenes 2-thiophenecarboxaldehyde, 2,2′-bithiophene-5-carboxaldehyde, and 5-[5-(2-thienyl)-2-thienyl]thiophene-2-carbaldehyde were purchased from Maybridge. Ethylene diamine was purchased from Alfa Aesar, and benzaldehyde was purchased from ACP. All of these chemicals were used without further purification. [Ru(bpy)2Cl2]·2H2O was prepared as previously described.17 The cyclometalating ligands IBQ-nT, where n=1–4, were synthesized from benzo[h]quinoline-5,6-dione18 and the corresponding formylthiophenes by a method previously reported by us.14 Formyltetrathiophene was synthesized following a published method,19,20 with full characterization details reported previously by us.14 We are not aware of any unexpected, new, and/or significant hazards or risks associated with the reported work. However, utmost precaution should be taken when working with novel compounds of unknown risk.
Instrumentation.
Microwave synthesis was carried out using a CEM Discover microwave reactor. NMR spectra were collected at the Dalhousie University Nuclear Magnetic Resonance Research Resource at either 500 MHz using a Bruker AVANCE 500 spectrometer or 300 MHz using a Bruker AVANCE 300 spectrometer. 13C NMR spectra were collected at the Joint School for Nanoscience and Nanotechnology at 176 MHz using an Agilent 700 spectrometer with a cryoprobe. ESI mass spectra were obtained using a Bruker microTOF focus mass spectrometer at the Dalhousie University Mass Spectrometry Laboratory. An Agilent/Hewlett–Packard 1100 series instrument (ChemStation Rev. A. 10.02 software) was used for HPLC analyses using a Hypersil GOLD C18 reversed-phase column with an A-B gradient (98% → 40% A; A = 0.1% formic acid in H2O, B = 0.1% formic acid in MeOH). Ligands and complexes were analyzed by HPLC at 300 μM in DMSO and 100 μM in MeCN, respectively. Retention times are estimated with ± 0.1 min error.
Synthesis.
Cyclometalated Ru(II) complexes were synthesized by an adapted literature procedure,21 which we have previously reported.13 These complexes as their PF6 − salts were characterized by 1H NMR, 1H-1H COSY NMR, and mass spectrometry. Rf values and HPLC retention times were also determined for each compound. For biological experiments, the PF6− salts were converted to their corresponding Cl− salts via anion metathesis on Amberlite IRA-410 with MeOH.
[Ru(bpy)2 (IBQ-1T)]PF6 (1).
Ru(bpy)2 Cl2 ·2H2O (135 mg, 0.26 mmol) and IBQ-1T (60 mg, 0.2 mmol) were added to a microwave vessel with triethylamine (1 mL) and ethylene glycol (3 mL) and subjected to microwave irradiation at 120 °C for 1 h. The purple mixture was transferred to ~30 mL of a saturated KPF6 solution with stirring. The precipitate that formed was isolated by vacuum filtration with a fine sintered-glass frit. The crude product was purified by silica column chromatography with 1% MeOH in DCM. Purple solid (36 mg, 21%): Rf = 0.67 (10% H2O + 2.5% sat’d KNO3 in MeCN); 1H NMR (500 MHz, MeCN-d3) δ = 8.66 (d, J = 7.9 Hz, 1H, c), 8.49 (d, J = 8.1 Hz, 1H, 3A), 8.39 – 8.34 (m, 2H, 3B, 3C), 8.30 (d, J = 8.2 Hz, 1H, 3D), 8.03 (td, J = 7.9, 1.6 Hz, 1H, 4A), 7.98 (d, J = 5.2 Hz, 1H, 6A), 7.95 (d, J = 5.7 Hz, 1H, 6C), 7.89 – 7.84 (m, 3H, 4B, 6B, f), 7.81 (dd, J = 5.3, 1.4 Hz, 1H, a), 7.77 – 7.70 (m, 3H, 4C, 4D, g), 7.66 (d, J = 5.9 Hz, 1H, 6D), 7.55 (dd, J = 5.1, 1.1 Hz, 1H, i), 7.47 (ddd, J = 7.6, 5.3, 1.2 Hz, 1H, 5A), 7.35 (dd, J = 8.0, 5.2 Hz, 1H, b), 7.30 – 7.24 (m, 2H, 5B, e), 7.23 (dd, J = 5.1, 3.6 Hz, 1H, h), 7.07 – 7.00 (m, 2H, 5C, 5D), 6.65 (d, J = 7.1 Hz, 1H, d); 13C NMR (176 MHz, MeCN-d3) δ = 157.69, 157.07, 156.68, 155.41, 155.32, 154.25, 150.38, 150.29, 149.39, 147.69, 145.62, 139.91, 136.37, 135.50, 134.90, 133.93, 133.88, 133.60, 131.83, 129.72, 128.27, 128.19, 128.01, 127.54, 126.99, 126.33, 126.15, 125.97, 125.64, 123.35, 123.19, 122.94, 122.87, 122.85, 121.99, 121.71, 113.01, 112.64; MS (ESI+) m/z: 714.3 [M-PF6]+; HRMS (ESI+) m/z for C38 H26 N7 RuS: cald 714.1008, found 714.1003; HPLC retention time: 25.27 min.
[Ru(bpy)2 (IBQ-2T)]PF6 (2).
Ru(bpy)2 Cl2 ·2H2O (135 mg, 0.26 mmol) and IBQ-2T (77 mg, 0.2 mmol) were added to a microwave vessel with triethylamine (1 mL) and ethylene glycol (3 mL) and subjected to microwave irradiation at 120 °C for 1 h. The purple mixture was transferred to ~30 mL of saturated KPF6 solution with stirring. The precipitate that formed was isolated by vacuum filtration with a fine sintered-glass frit. The crude product was purified by silica column chromatography with 1% MeOH in DCM. Purple solid (15 mg, 8.0%): Rf = 0.72 (10% H2O + 2.5% sat’d KNO3 in MeCN); 1H NMR (500 MHz, MeCN-d3) δ 11.82 (s, 1H, NH) 8.65 (dd, J = 8.3, 1.1 Hz, 1H, c), 8.49 (d, J = 8.2 Hz, 1H, 3A), 8.40 – 8.34 (m, 2H, 3B, 3C), 8.30 (d, J = 8.1 Hz, 1H, 3D), 8.02 (t, J = 7.9 Hz, 1H, 4A), 7.99 – 7.95 (m, 2H, 6A, 6C), 7.93 – 7.83 (m, 3H, 4B, 6B, f), 7.81 (dd, J = 5.3, 1.4 Hz, 1H, a), 7.77 – 7.69 (m, 3H, 4C, 4D, g), 7.66 (d, J = 5.8 Hz, 1H, 6D), 7.50 – 7.43 (m, 1H, 5A), 7.42 (dd, J = 5.1, 1.1 Hz, 1H, k), 7.38 (dd, J = 3.6, 1.1 Hz, 1H, i), 7.36 – 7.30 (m, 2H, 5B, b), 7.29 – 7.24 (m, 2H, e, h), 7.11 (dd, J = 5.1, 3.6 Hz, 1H, j), 7.04 (t, J = 6.6 Hz, 2H, 5C, 5D), 6.65 (d, J = 7.1 Hz, 1H, d); 13C NMR (176 MHz, MeCN-d3) δ = 157.68, 157.06, 156.67, 155.44, 155.32, 154.28, 150.38, 150.30, 149.38, 147.71, 145.19, 139.96, 138.51, 136.42, 136.37, 134.90, 133.88, 133.60, 132.48, 131.83, 129.80, 128.34, 128.07, 127.27, 126.99, 126.34, 126.32, 126.15, 125.98, 125.73, 124.67, 123.34, 123.18, 122.95, 122.87, 121.66, 113.57, 112.77; MS (ESI+) m/z: 796.33 [M-PF6]+; HRMS (ESI+) m/z for C42H28N7RuS2 : cald 796.0886, found 796.0861; HPLC retention time: 28.00 min.
[Ru(bpy)2 (IBQ-3T)]PF6 (3).
Ru(bpy)2 Cl2 ·2H2O (135 mg, 0.26 mmol) and IBQ-3T (93 mg, 0.2 mmol) were added to a microwave vessel with triethylamine (1 mL) and ethylene glycol (3 mL) and subjected to microwave irradiation at 120 °C for 1 h. The purple mixture was transferred to ~30 mL of saturated KPF6 solution with stirring. The precipitate that formed was isolated by vacuum filtration with a fine sintered-glass frit. The crude product was purified by silica column chromatography with 1% MeOH in DCM. Purple solid (8 mg, 8.0%): Rf = 0.76 (10% H2O + 2.5% sat’d KNO3 in MeCN); 1H NMR (500 MHz, MeCN-d3) δ 8.62 (s, 1H, c), 8.49 (d, J = 8.2 Hz, 1H, 3A), 8.37 (t, J = 8.3 Hz, 2H, 3B, 3C), 8.30 (d, J = 8.1 Hz, 1H, 3D), 8.06 – 8.00 (m, 1H, 4A), 8.00 – 7.93 (m, 2H, 6A, 6C), 7.89 – 7.83 (m, 2H, 4B, f), 7.81 (dd, J = 5.4, 1.3 Hz, 1H, a), 7.77 – 7.68 (m, 4H, 4C, 4D, 6B, g), 7.66 (d, J = 5.8 Hz, 1H, 6D), 7.46 (dd, J = 7.4, 5.6 Hz, 1H, 5A), 7.38 (dd, J = 5.1, 1.1 Hz, 1H, m), 7.34 (dd, J = 8.1, 5.3 Hz, 1H, b), 7.31 – 7.24 (m, 5H, 5B, e, h, i, j), 7.21 (d, J = 3.8 Hz, 1H, k), 7.09 (dd, J = 5.1, 3.6 Hz, 1H, l), 7.07 – 7.00 (m, 2H, 5C, 5D), 6.65 (d, J = 7.1 Hz, 1H, d); 13C NMR (176 MHz, MeCN-d3) δ = 157.68, 157.06, 156.67, 155.45, 155.32, 154.29, 150.38, 150.37, 150.28, 149.37, 147.69, 145.13, 139.96, 137.88, 136.68, 136.37, 136.36, 135.24, 134.90, 133.87, 133.59, 132.66, 131.83, 128.31, 128.12, 128.06, 127.27, 126.99, 126.39, 126.33, 126.15, 125.96, 125.43, 125.39, 124.78, 124.73, 124.27, 123.34, 123.17, 122.94, 122.88, 121.64, 113.56, 112.79; MS (ESI+) m/z: 878.47 [M-PF6]+; HRMS (ESI+) m/z for C46H30N7RuS3: cald 878.0763, found 878.0785; HPLC retention time: 29.99
[Ru(bpy)2 (IBQ-4T)]PF6 (4).
Ru(bpy)2 Cl2 ·2H2O (151 mg, 0.29 mmol) and IBQ-4T (120 mg, 0.22 mmol) were added to a microwave vessel with triethylamine (1 mL) and ethylene glycol (3 mL) and subjected to microwave irradiation at 120 °C for 1 h. The purple mixture was transferred to ~30 mL of saturated KPF6 solution with stirring. The precipitate that formed was isolated by vacuum filtration with a fine sintered-glass frit and washed with 50 mL DI H2O and 200 mL diethyl ether. The crude product was purified by silica column chromatography with a gradient of 0% → 1% MeOH in DCM. Purple solid (26 mg, 11%): Rf = 0.72 (10% H2O + 2.5% sat’d KNO3 in MeCN); 1H NMR (500 MHz, MeCN-d3) δ 8.62 (s, 1H, c), 8.48 (d, J = 8.3 Hz, 1H, 3A), 8.40 – 8.33 (m, 2H, 3B, 3C), 8.29 (d, J = 8.2 Hz, 1H, 3D), 8.02 (dd, J = 8.7, 7.2 Hz, 1H, 4A), 8.00 – 7.91 (m, 2H, 6A, 6C), 7.89 – 7.83 (m, 2H, 4B, f), 7.81 (d, J = 5.3 Hz, 1H, a), 7.79 – 7.69 (m, 4H, 4C, 4D, 6B, g), 7.66 (d, J = 5.8 Hz, 1H, 6D), 7.46 (dd, J = 7.4, 5.5 Hz, 1H, 5A), 7.37 (dd, J = 5.1, 1.1 Hz, 1H, o), 7.34 (t, J = 6.8 Hz, 1H, b), 7.32 – 7.24 (m, 5H, 5B, e, h, i, j), 7.24 – 7.16 (m, 3H, k, l, m), 7.08 (dd, J = 5.1, 3.6 Hz, 1H, n), 7.07 – 7.00 (m, 2H, 5C, 5D), 6.65 (d, J = 7.1 Hz, 1H, d); 13C NMR (176 MHz, MeCN-d3) δ = 157.69, 157.07, 156.68, 155.50, 155.32, 154.27, 150.39, 150.29, 149.38, 147.77, 145.00, 139.98, 137.84, 136.47, 136.37, 136.17, 135.70, 135.45, 135.17, 134.90, 133.90, 133.63, 132.73, 131.96, 131.83, 130.02, 128.31, 128.23, 128.03, 126.99, 126.39, 126.33, 126.17, 125.99, 125.53, 125.40, 125.05, 124.95, 124.95, 124.90, 124.78, 124.24, 123.36, 123.19, 122.95, 122.88, 122.76, 121.91, 121.75, 112.68; MS (ESI+) m/z: 960.20 [M–PF6]+; HRMS (ESI+) m/z for C50H32N7S4Ru: cald 960.0640, found 960.0641; HPLC retention time: 31.31 min.
Lipophilicity.
The lipophilicities of the complexes were determined using a modified “shake flask” method. A solution of 1-octanol saturated with water was prepared by combining 16 mL of 1-octanol (99.9%) with 4 mL of deionized water (MilliQ). A solution of water saturated with 1-octanol was prepared from 16 mL of deionized water (MilliQ) and 4 mL of 1-octanol (99.9%). The two saturated solutions were shaken at room temperature for 24 h (New Brunswick Classic C25KC Incubator Shaker set at 230 rpm). The C^N complexes were prepared at 50 µM in 500 µL of saturated 1-octanol in microfuge tubes, and then an equal volume of saturated water was added (total volume = 1 mL). The mixtures were shaken by hand exactly 200 times, centrifuged at 11,000 rpm for 2 minutes (BioRad Model 16K Microcentrifuge), and then separated and analyzed by absorption spectroscopy using a microplate reader (Molecular Devices SpectraMax M2e). The concentrations of the C^N complexes in both layers were calculated from absorption standard curves constructed from known concentrations of the compounds. If no absorption was detected in a layer, the assumption that <0.1% of the initial concentration ended up in that layer was used for the log P calculation. Formally, log Po/w is the log of the ratio of concentrations in the two layers.
Spectroscopy.
The photophysical measurements were performed on dilute solutions (5–20 μM) of the PF6 − salts of the metal complexes in spectroscopic grade MeCN. Molar extinction coefficients (ε) were determined at the local absorption maxima from the slope of absorption vs. concentration plots for five concentrations (20 μM and four serial dilutions of 25%), and measured in duplicate. Emission quantum yields (Φem) and singlet oxygen quantum yields (ΦΔ) were determined using [Ru(bpy)3](PF6)2 as the standard according to Eqn 1, where I, A, and η are the integrated emission intensity, the absorbance at the excitation wavelength, and the refractive index of the solvent, respectively, and the subscript s denotes the standard. Reference quantum yields used for [Ru(bpy)3](PF6)2 are: Φem = 0.012 at 298 K in aerated MeCN,22 Φem = 0.38 at 77 K in frozen 4:1 v/v EtOH:MeOH,1 and ΦΔ = 0.56 in aerated MeCN.23
| (Eqn 1) |
Oxygen was removed from the solutions by argon purging in a long-stemmed cuvette (Luzchem QSC10S) for emission experiments, or by freeze-pump-thaw (5x) using custom Schlenk-style cuvettes for transient absorption measurements. For the 77 K measurements, samples were dissolved in 4:1 EtOH:MeOH in a 5 mm i.d. NMR tube, and then frozen in liquid nitrogen in a quartz-tipped cold finger Dewar (Wilmad Labglass).
UV-vis absorption spectra were recorded on a Jasco V-730 spectrometer. Steady-state luminescence spectra were measured on a PTI Quantamaster equipped with a K170B PMT for measuring ultraviolet to visible emission, and a Hamamatsu R5509–42 NIR PMT for measuring NIR emission (<1400 nm) and quantifying 1O2 emission (centered around 1276 nm). Emission and excitation spectra were corrected for the wavelength dependences of lamp output and detector response.
Transient absorption (TA) spectra and lifetimes were measured on an Edinburgh Instruments LP-980 spectrometer equipped with a PMT-LP detector and a Continuum Minilite Nd:YAG laser emitting at 355 nm or 532 nm (7–9 mJ pulse−1). This instrumentation was also used in its fluorescence mode to measure emission lifetimes.
Cytotoxicity and photocytotoxicity.
Cell viability experiments were carried out in triplicate in 96-well TC-treated microtiter plates (Corning Costar, Acton, MA). DPBS (200 μL) was added to the peripheral outer wells (to minimize evaporation from the inner sample wells) and warm culture medium (25 μL) was added to the inner sample wells. SKMEL28 cells (∼550,000−600,000 cells mL−1) were transferred in 50 μL aliquots to the inner wells, and then the plates were incubated at 37 °C, 5% CO2 in a water-jacketed incubator (Thermo Electron Corp., FormaSeries II, model 3110, HEPA Class 100) for 3 h.
The Ru(II) C^N complexes were serially diluted with DPBS and prewarmed at 37 °C. Aliquots of 25 μL of the appropriate dilutions were added to cells, and the plates were incubated at 37 °C under 5% CO2 for 16 h. Dark (control) microplates were kept in the incubator, and light-treated microplates were irradiated under one of the following conditions: visible light (400−700 nm, 34.7 mW cm−2) using a 190 W BenQ MS 510 overhead projector or red light (625 nm, 27.8 mW cm−2) from an LED array (PhotoDynamic Inc., Halifax, NS). The light-treated plates were irradiated for approximately48 and 60 min, respectively, to give a fluence of 100 J cm−2.
Following the dark or light treatment, the microplates were incubated for 48 h followed by the addition of 10 µL of prewarmed alamarBlue reagent (Life Technologies DAL 1025) to all sample wells. The plates were subsequently incubated for another 15−16 h. Cell viability was determined by fluorescence using a Cytofluor 4000 fluorescence microplate reader (excitation = 530 ± 25 nm and emission = 620 ± 40 nm.
Sigmoidal fits of the dose−response curves (Graph Pad Prism 6.0.) for the dark and light conditions were carried out to obtain EC50 values, corresponding to the effective concentrations required to reduce cell viability by 50%, according to eq 1 (below), where yi and yf are the initial and final fluorescence signal intensities. EC50 values were generally reproducible to within ±25% in the sub-micromolar regime, ±10% below 10 μM, and ±5% above 10 μM. Phototherapeutic indices (PIs) are defined as the dark EC50 values divided by the light EC50 values.
| (Eqn 2) |
Cellular Imaging.
Sterile glass-bottom Petri dishes (MatTek) were coated with 200 μL poly-L-lysine (Ted Pella) and incubated at 37 °C, 5% CO2 for 1 h. The dishes were then washed three times with DPBS and left uncovered to dry at room temperature for 15 min. SKMEL28 cells (approximately 100,000 cells delivered in 500 µL) were added to the poly-L-lysine-coated glass bottom Petri dishes and allowed to adhere for 2 h in a humidified incubator at 37 °C, 5% CO2. Metal compounds (500 μL of a 50 μM solution in PBS prewarmed to 37 °C) were added and the sample dishes were incubated for an additional 15 min prior to receiving a dark treatment or a visible light treatment (24 min from a 190 W BenQ MS 510 overhead projector with 400−700 nm output, power density = 34.7 mW cm−2, total light dose ≈ 50 J cm−2). Dark samples were protected from light and held at room temperature for the same amount of time. Images were collected at 15 min after treatment with a Carl Zeiss LSM 510 laser scanning confocal microscope equipped with a 40x oil objective lens. An argon−krypton laser (λex=458/488 nm) was used for excitation, and the emission was captured through a 505 nm long-pass filter. All images were collected with a pinhole diameter of 100 μm and processed using the Zeiss LSM Image Browser Version 4.2.0.121 software (Carl Zeiss Inc.).
RESULTS AND DISCUSSION
Synthesis and Characterization.
Since it was discovered that phpy− can be substituted with electron withdrawing groups to produce cyclometalated Ru(II) complexes with conversion efficiencies equal to those of the isothiocyanate-based champion dyes,24 there has been a rapid increase in the exploration of functionalized C^N ligands for DSSCs.9,11 However, examples of functionalized C^N ligands have been largely limited to phpy− substituted with electron withdrawing atoms, groups, or rings, and functionalized C^N ligand based on bhq− are even more scarce. To fill this void and increase the structural landscape of cyclometalating ligands, our group is investigating π-extended C^N ligands (and their Ru(II) and Ir(III) complexes).13,14,25–27 Our studies have focused on these systems for photobiological applications, but the properties that make good sensitizers for this purpose are often the attributes that are important for other light-based technologies, including DSSCs.
Herein we report four new Ru(II) cyclometalated complexes derived from deprotonated thienyl-appended IBQ ligands. The IBQ-nT ligands were prepared as described previously,14 and then complexed to Ru(bpy)2 Cl2 using an adapted method for preparing [Ru(bpy)2 (N′-methyl-2,3′-bipyridinium)](PF6)2.21 The reported reflux reaction with triethylamine in ethanol was adapted for microwave synthesis with ethylene glycol as the solvent to reduce reaction times to 1 h and eliminate the need for the addition of silver(I) salts. Compounds 1–4 were isolated as their hexafluorophosphate salts and purified by silica gel chromatography in yields that ranged from 8–21%. These unoptimized yields are similar to those reported for the related [Ru(4,4′-dmb)2 (IBQ-nT)]Cl2 complexes.14 Structural and photophysical characterization was performed on the hexafluorophosphate salts of the complexes in MeCN to facilitate comparison to other Ru(II) N^N and C^N studies in the literature that are often carried out using this salt and solvent. For biological experiments, the (PF6)2 salts were converted to their corresponding Cl2 salts using anion-exchange chromatography. The Cl2 salt form is commonly employed for biological studies of Ru(II) complexes.
ESI (+ve) mass spectrometry confirmed the target [Ru(bpy)2 (IBQ-nT)]+ molecular ion peak for each compound, and the structures of the final complexes 1–4 were confirmed by a detailed analysis of their 1D 1H and 2D 1H-1H COSY as well as 13C NMR spectra (Figures S1–S8) based on assignments previously reported for their [Ru(bpy)2 (IBQ-nT)]PF6 counterparts.14 Crystals suitable for X-ray crystallographic analysis were not obtained, but the previously reported X-ray crystallographic structure of the [Ru(bpy)2 (bhq)]+ complex5 proved useful in assigning the ligand 1H NMR signals. The most informative features from the published solid-state structure of that simpler system are that (i) the Ru-N bond distances for rings A and B (Figures S1, S3, S5, S7) and the cyclometalating ligand are elongated, and (ii) the Ru-N bond distances in rings C and D and the Ru-C bond distance for the cyclometalating ligand are shortened. The hydrogens of 1–4 exhibited 1H NMR chemical shifts consistent with these observations. The ring hydrogens of the N^N ligands were shifted downfield in the order A > B > C > D; hydrogen c on the cyclometalating ligand was the most deshielded aromatic hydrogen in the spectra; and hydrogen d was the most shielded. The coligand hydrogens were assigned following the trends established for [Ru(bpy)2 (N^N)]2+ polypyridyl complexes in general, with H3 > H4 > H6 > H5. The remaining C^N assignments were made through close scrutiny of the 1H-1H COSY spectra and the previously assigned C^N ligand spectra. The purity of each complex was established to be >95% by reverse-phase HPLC (Figures S9–S12), where retention times for the complexes increased with n as would be expected for increasing lipophilicity.
Lipophilicity.
The octanol-water partition coefficients (log Po/w) for C^N complexes 1–4 were determined in order to compare their lipophilicities. This parameter is sometimes used in medicinal chemistry to predict cell membrane permeability (and, in turn, cytotoxicity), whereby positive and negative values for log Po/w signify preferences of the compound for octanol and water, respectively. Generally, Ru(II) C^N complexes are considered more lipophilic than their Ru(II) N^N counterparts, with the cyclometalated compounds characterized by more positive log Po/w values,16 and this appeared to be the case for the present series as well. All four C^N complexes had positive log Po/w values (Figure 1), whereas the parent diimine complex [Ru(bpy)3]Cl2 was assigned a log Po/w value of <−3. No [Ru(bpy)3]Cl2 was detected in the octanol phase and thus it was assumed that >99.9% of the compound was in the aqueous phase.
Figure 1.
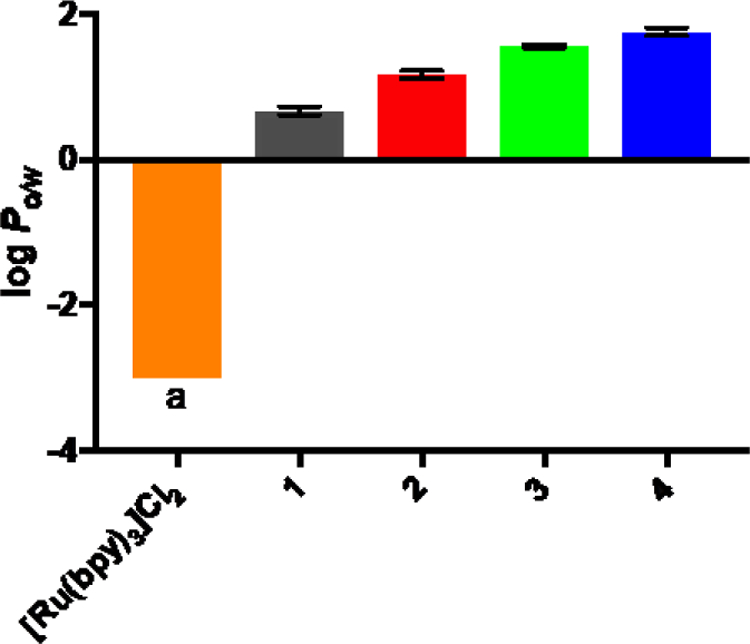
Octanol-water partition coefficients (log Po/w) for complexes 1–4 as their chloride salts with [Ru(bpy)3]Cl2 included for comparison. aThe concentration in octanol was too low to determine and was assumed to be <0.1%.
For the present Ru(II) C^N series, the log Po/w value was positively correlated to the number of thiophene rings in the IBQ-nT ligand. Compound 4 was the most lipophilic and 1 was the least. Compared to the analogous [Ru(4,4′-dmb)2 (IBQ-nT)]Cl complexes,14 the absence of methyl substituents in the bpy coligands of the present [Ru(bpy)2 (IBQ-nT)]Cl series resulted in slightly reduced lipophilicity for a given complex but with the same overall trend: positive log Po/w values that increased with the number of thienyl groups. If cellular uptake increases with lipophilicity and lipophilicity parallels cytotoxicity as observed for some cyclometalated Ru(II) compounds,16,28,29 compound 4 would be predicted to possess the greatest dark cytotoxicity of the series. However, we previously observed that the [Ru(4,4′-dmb)2 (IBQ-4T)]Cl cousin of 4 was the least dark cytotoxic of its series, underscoring that cytotoxicity does not positively correlate with lipophilicity across all Ru(II) C^N compounds. In other words, cellular uptake may not positively correlate with lipophilicity (or cellular uptake may not positively correlate with cytotoxicity).
Photophysical Properties.
Absorption.
Four main electronic transitions appeared in the UV-vis spectra of the four complexes. Shown in Figure 2 and tabulated in Table 1, these transitions can be assigned in accordance with similar complexes.8,13 The intense sharp peaks at ≤290 nm were due to intraligand (IL) ππ* transitions on the bpy ligands, and the broader bands occurring in the 330–450 nm range were assigned to IL ππ* transitions on the IBQ-nT ligands. These bands increased in intensity and shifted to lower energy as the number of thiophene rings increased, both being consistent with the expanding conjugation in the ligands; the spectra in Figure 2have been normalized to the bpy ππ* signal to emphasize these differences.
Figure 2.
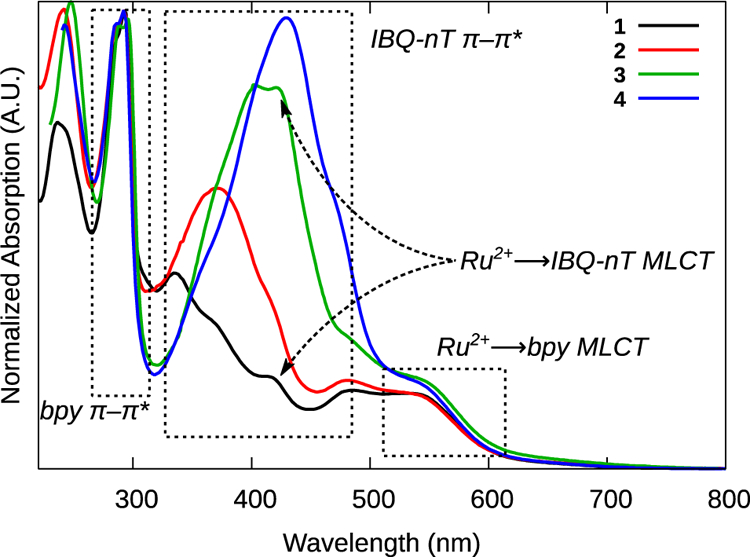
Normalized ground state absorption spectra of complexes 1–4 as PF6− salts, in MeCN at room temperature.
Table 1:
Molar Extinction Coefficients at Selected Wavelengths for Complexes 1–4
| Cmpd | Absorption wavelength/nm (log ε) |
|---|---|
| 1 | 296 (4.73), 341 (4.39) 420 (4.07), 540 (3.99) |
| 2 | 296 (4.73), 376 (4.57), 420 (4.32), 540 (4.00) |
| 3 | 296 (4.87), 401 (4.80), 420 (4.80), 540 (4.20) |
| 4 | 296(4.86), 420 (4.90), 433 (4.91), 544 (4.19) |
Broad MLCT transitions occurred between 350 and 650 nm, likely due to the decreased symmetry (C1) of these complexes. The low intensity bands near 540 nm were assigned to MLCT transitions between the Ru2+ center and the N^N bpy ligands. The analogous transition between Ru2+ and the C^N IBQ-nT ligands appeared as a small peak near 420 nm in all four spectra, but was most evident for complexes 1 and 3. This more energetic transition is consistent with the elevated C^N π* orbital, which has been previously determined electrochemically for similar complexes.8,30–32 The similarities in the energies of the Ru2+(dπ)→IBQ-nT(π*) MLCT transitions suggest that the π* acceptor orbital is proximal to the metal center (localized to the IBQ portion of IBQ-nT) and does not extend onto the thienyl rings in these complexes. An alternate explanation is that bpy is the π* acceptor orbital for all of the MLCT bands, with varying contributions from the highest occupied molecular orbital (HOMO) of π-symmetry at the C^N phenyl ring.24,33 Our assignment is based on that of Turro and coworkers for Ru(II) C^N systems containing the π-expansive dppn ligand.32
Emission.
The excitation by light of an appropriate wavelength produced dual emission in all four C^N complexes at room temperature and at 77K: blue-green fluorescence from the IBQ-nT ligand-centered 1IL state and NIR phosphorescence from the 3MLCT state (Figure 3, Table 2 and Table S1). This dual emission is a common phenomenon in decoupled bichromophoric systems and thus suggests that the two chromophores in this family are not electronically coupled. Either transition could be emphasized by selection with the appropriate excitation wavelength. This behavior was observed previously for a related family of Ru(II) C^N complexes and carefully confirmed by comparing excitation spectra collected at different emission wavelengths with those of the free ligands.14 With as low as 1% contamination of the metal complex with the free ligand, the excitation spectra of the complexes are superimposable with the spectra of their free ligands. The fact that unique excitation signatures were obtained for the complexes suggests that the blue-green emission observed is from metal-bound ligand rather than free ligand.
Figure 3.
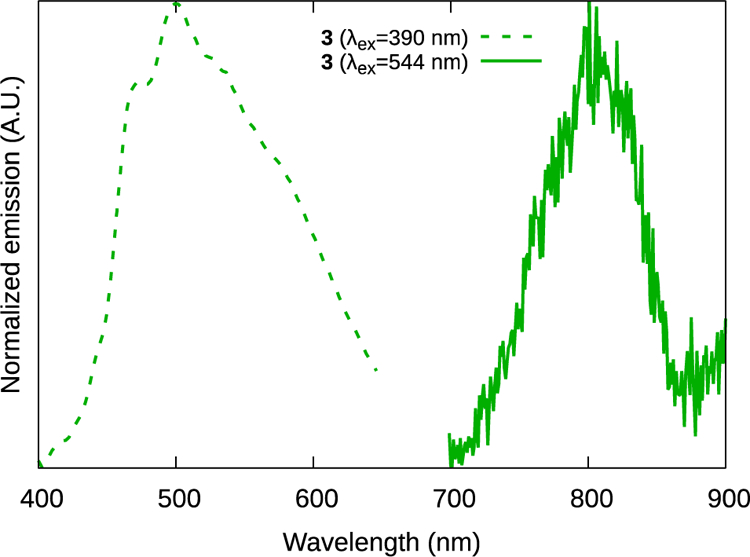
1IL and 3MLCT emission from complex 3.
Table 2:
Spectroscopic Data for Complexes 1–4 in MeCN at 298 Ka
| IL fluorescence | MLCT phosphorescence | 1O2 phosphorescence | ||
|---|---|---|---|---|
| Cmpd | λem, nm (Ar); τem, ns | λem, nm (Ar); τem, ns | Φp (Ar) | Φ∆ (air) |
| 1 | 400 (340); 7 | 807 (538); 21 | 2.3 × 10−4 (538) | 5.2 × 10−2 (538) |
| 2 | 453 (368); 5 | 808 (538); 22 | 1.6 × 10−4 (544) | 5.6 × 10−2 (538) |
| 3 | 500 (390); 5 | 805 (544); 20 | 1.1 × 10−4 (544) | 5.2 × 10−2 (544) |
| 4 | 527 (410); 6 | 805 (540); 15, 110 | 1.3 × 10−4 (540) | 1.4 × 10−1 (540) |
Excitation wavelengths (nm) are indicated in parentheses.
The 1IL fluorescence energy decreased from around 3.1 eV to around 2.4 eV as the π network expanded through the series by the sequential addition of a thiophene ring (Table 2). Likewise, the optimal excitation energy decreased. By contrast, the 3MLCT excitation and emission energies did not correlate with the number of thiophene rings in the C^N ligand. Complexes 1–4 yielded weak (Φp≈10−4, Figure 3) unstructured 3MLCT phosphorescence centered near 810 nm in Ar-sparged MeCN at RT. The 3MLCT emission did not vary significantly between the complexes, indicating that the π* acceptor orbital of the 3MLCT was similar for each complex. This information permits the assignment of the orbital parentage of the two transitions to IBQ-nT ππ* and Ru2+(dπ)→bpy(π*) for 1IL and 3MLCT, respectively. The assignment of the 3MLCT state as Ru2+(dπ)→bpy(π*) rather than Ru2+(dπ)→IBQ-nT(π*) is based on the bpy π* orbital having been established as the lowest-lying π*-acceptor ligand in a variety of [Ru(N^N)2 (C^N)]+ complexes.8
At 77 K (in 4:1 MeOH:EtOH) the 3MLCT phosphorescence became stronger and was blue-shifted by approximately 75–80 nm (Table S1). The low-temperature phosphorescence also displayed characteristic 3MLCT vibronic structure with spacings of around 1210 cm−1, which is indicative of diimine involvement in the emissive excited state.34 Values of ΔEs ≈1238–1382 cm−1 are comparable to that for [Ru(bpy)3]2+ (ΔEs =1127 cm−1) under the same conditions, which would be expected for a polar 3MLCT excited state.
Emissive lifetimes for 1–4 (Table 2) were determined with 355-nm laser excitation. The room temperature 3MLCT emission was very short-lived, around 20 ns, and comparable to what we have previously observed in similar C^N complexes.13,14 These short lifetimes are not surprising, and are consistent with the energy gap law; [Ru(bpy)3]2+ has a 3MLCT emission lifetime of approximately 1 μs1 while its cyclometalated cousin [Ru(bpy)3 (C^N)]+ has a lifetime of only 12 ns.8 The 3MLCT lifetimes did not change as the thiophene chain lengthened, further supporting the position that the π*-acceptor orbital of the 3MLCT state was centered on the bpy ligand. Only complex 4 had a biexponential emissive decay, exhibiting a 110 ns decay that was not present in the other complexes. This may suggest that the emissive 3MLCT state can be populated from an additional pathway in 4 only, resulting in delayed emission from the 3MLCT state.
Transient Absorption Spectroscopy.
The triplet excited states of complexes 1–4 were explored with nanosecond transient absorption (TA) spectroscopy. Their spectroscopic characteristics were analyzed in the context of what is known for the TA profiles of archetypal Ru(II) N^N complexes35, related Ru(II) C^N complexes,8,31,35,36 plus other Ru(II) dyads based around pyrenyl36 and oligothienyl37–40 chromophores. The TA signatures of these types of compounds generally consist of three key features: a net positive signal in the UV, a strong mid-visible bleach, and a broad but very weak red-NIR absorption.35 For Ru(II) N^N complexes, the mid-visible bleach results from the loss of the 1A1 → MLCT absorption in the ground state. The two new absorptions arise from the ESA of the diimine ligand radical anion formed as the 3MLCT state, and the red-NIR signal includes ligand-to-metal charge transfer (LMCT) transitions involving the ancillary N^N ligands. The latter tends to be weak, broad and featureless, and partly overlapping the bleach.
The nanosecond TA spectra of 1–4 were measured in degassed (5× freeze-pump-thaw) MeCN with excitation at 355 and 535 nm. Lifetimes were not deconvoluted from the excitation pulse, which was on the order of 3–4 ns. The time-sliced spectra for 1–4 over the first 80 ns are shown in Figure 4, and the lifetimes are tabulated in Table 3. Additional TA spectra for each compound at short and longer time delays, exciting at both 355 and 532 nm, are shown in the Supporting Information (Figures S13–S18). The emissive and TA lifetimes in Tables 2 and 3, respectively, were used to construct the proposed photophysical model in Scheme 1.
Figure 4.

Transient absorption spectra of 1–4 as excited by 355-nm (left column) or 532-nm (right column) light. The spectra collected with 355-nm excitation for 1–3 have 1IL emission superimposed on the signal that has not been subtracted.
Table 3.
Transient Absorption Lifetimes at the Indicated Wavelengths Using λex=355 or 532 nm
| Cmpd | τTA, ns [λ, nm]; λex=355 nm | τTA, ns [λ, nm]; λex =532 nm |
|---|---|---|
| 1 | 6 [420], 20 [360, 420, 540] | 20 [357, 547, 670] |
| 2 | 5–8 [430, 540], 21 [540] | 20 [367, 537, 700] |
| 3 | 6 [640], 23 [530, 640] | 21 [357, 480, 640] |
| 4 | 10 [660]a, 110–120 [440, 660], 4000–5000 [440, 660] | 10 [357, 437, 657]a, 110–130 [357, 437, 657], 4500–6300 [437, 657] |
These lifetimes were associated with signal growth rather than decay.
Scheme 1.

Simplified Jablonksi diagram illustrating the photophysical pathways in the series. For clarity, only compounds 3 and 4 are shown, and the relative energy levels are not to scale.
aLifetime observed as a growth in the TA spectra corresponding to the formation of the 3IL state from the 3MLCT state.
bLifetime observed as a decay in the TA and emissive spectra, possibly corresponding to repopulation of the 3MLCT state from the 3IL state leading to delayed 3MLCT emission.
The TA profiles of 1–3 excited with 355 nm laser pulses exhibited two lifetimes, one around 7 ns and the other around 21 ns. These values compare well with the emissive lifetimes collected with the same excitation wavelength (Table 2), which were assigned to accessible C^N 1IL and N^N 3MLCT states, respectively (Emission section). At t=20 ns, these spectra had negligible contributions from the faster decaying, superimposed 1IL fluorescence and reflected the signature of the 3MLCT ground-state bleach. As expected, the spectra collected for 1–3 with 532-nm excitation, where the C^N 1IL state is inaccessible via direct population (or via indirect population from N^N 1MLCT states), resembled the t=20 ns spectra with 355-nm excitation and lacked the shorter lifetime component (Figure 4).
Unlike 1–3, the ESA spectra of 4 exhibited a much longer-lived (τTA ≈5 μs) component (Figures 5 and S16), indicating the availability of an excited state(s) exclusive to this complex. The shape of the ESA spectrum of 4 at t=20 ns resembled that of the IBQ-4T ligand alone (in 1M KI in DMSO, with lifetime τ≈20 μs, Figure S18). The bleach near 440 nm corresponds to the loss of the C^N ligand-based 1ππ* ground state absorption, and the broad ESA centered around 660 nm is qualitatively similar to the triplet-triplet ESA assigned to the 3ππ* state in the TA spectrum of tetrathiophene.41 These results suggest that the long-lived excited state discerned for 4 is the C^N-based 3IL state, residing primarily on the oligothienyl unit.
Figure 5.
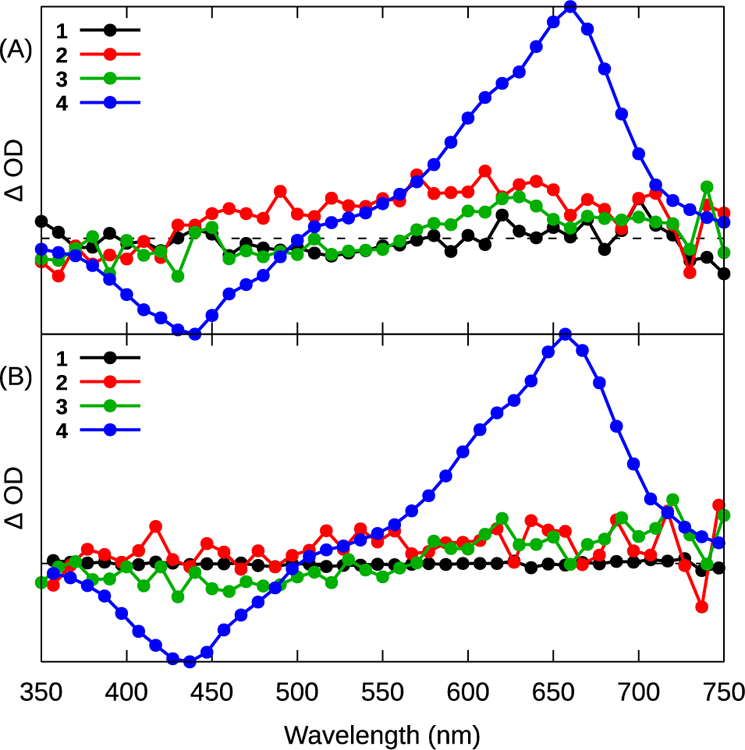
Transient absorption spectra collected for complexes 1–4 with (A) λpump =355 nm or (B) λpump =532 nm at a delay time of 90 ns with 10 ns integration.
The population of the long-lived C^N 3IL state was observed by the growth of its TA spectrum (Figure 6A), τ≈10 ns, concomitant with the bleach of the N^N MLCT bleach at ≈560 nm. The C^N 3IL absorption subsequently decayed with two kinetically separate modes, with lifetimes of ≈110 ns and ≈5 μs (Figure 6B), with the shorter component being 30–40× more intense, based on the pre-exponential factors of the fit functions.
Figure 6.
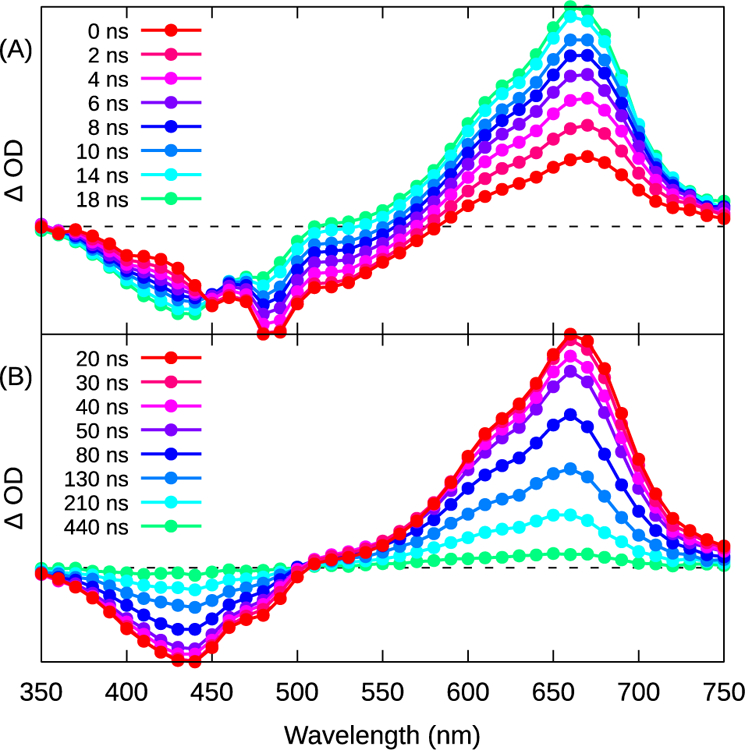
Transient absorption spectra of 4 at (A) shorter (2 ns integration) and (B) longer (10 ns integration) periods after excitation. λex=355 nm. The spectra with λex=532 nm were similar (see Figure S17).
Only complex 4 exhibited delayed emission from the N^N 3MLCT state (Table 2). The lifetime of this delayed emission matches the shorter 110 ns component of the C^N 3IL decay in the TA experiments, pointing to a re-population of the N^N 3MLCT state from the C^N 3IL, shown schematically by the Jablonski diagram in Scheme 1. This model proposes an equilibrium between the N^N 3MLCT state (1.54 eV, by emission) and the C^N 3IL state (1.81 eV for discrete tetrathiophene,42 but likely somewhat lower for the present ligand owing to more extensive π-conjugation) in 4. The close energy match between these levels is important for establishing the equilibrium,36 and similar triplet reservoirs are known in tethered pyrene-type structures.36,43 The higher energy of the C^N 3IL state in 1, 2, and 3 does not permit this population, and consequently neither a long-lived µs TA transient nor a delayed emission from the 3MLCT was observed in these compounds.
The measurable growth of the C^N 3IL TA absorption in 4, the connection between this growth and the decay of the N^N 3MLCT transient spectrum, the similarity of the 335 nm and the 532 nm-excited ESA spectra after around 20 ns, and the failure to observe any C^N 3IL signal in 1–3 all suggest that there was no direct population of C^N 3IL from the C^N 1IL state in any of the complexes.
Singlet Oxygen Sensitization.
The quantum yields for 1O2 generation (Φ∆) were determined for compounds 1–4 as their hexafluorophosphate salts in air-saturated MeCN from the phosphorescence of sensitized 1O2 centered near 1268 nm (Table 2). The standard employed was [Ru(bpy)3](PF6)2 in MeCN (Φ∆=0.56),23 which is considered to be an effective 1O2 sensitizer. The hexafluorophosphate salts of the complexes in MeCN were used for the Φ∆ measurements because the phosphorescence from 1O2 is quenched in water,44 and the hexafluorophosphate salts are completely soluble in MeCN whereas the chloride salts are not. In addition, pure water is not representative of a complex biological environment, where it is expected that the metal complexes may associate with biological macromolecules and possibly experience a more hydrophobic environment. The use of the hexafluorophosphate salts in MeCN is also advantageous for comparing Φ∆ for this series with values for other metal complexes, often reported for this salt and solvent. While the calculated values for Φ∆ do not represent true values for in vitro or in vivo conditions, they do highlight the relative potentials of the compounds in this series to generate 1O2.
All four Ru(II) C^N compounds were less effective than the [Ru(bpy)3](PF6)2 standard at 1O2 sensitization. The longest wavelength maxima in the excitation spectra for sensitized 1O2 emission by these complexes ranged from 538 to 540 nm, corresponding to the N^N-based 1MLCT absorption for 1–4. Excitation of the complexes at their respective maxima gave 1O2 quantum yields of 5–6% for 1–3 and 14% for 4. These values are much smaller than those determined for the corresponding thienyl-based Ru(II) diimine complexes,45 but agree well with their cyclometalated [Ru(4,4′-dmb)2 (IBQ-nT)]+ counter-parts,14 and related cyclometalated systems derived from pbpn/pbpz/pbpq/bhq C^N ligands.13 The much lower values of Φ∆ for Ru(II) C^N complexes has been attributed to their shorter intrinsic 3MLCT lifetimes that result from the lower-energy 3MLCT states in these systems, where nonradiative decay competes more effectively with 1O2 sensitization and phosphorescence. The differences in 1O2 yields between 1–3 and 4 in this series (and in the corresponding [Ru(4,4′-dmb)2 (IBQ-nT)]+ family14) most likely stems from the contribution of low-lying, thienyl-based 3IL states to the excited state dynamics in 4, which results in a prolonged triplet state lifetime (τTA ≈5 μs) that increases the 1O2 quantum yield. The modest Φ∆ value of 14% for 4 relative to its Ru(II) N^N cousin, where Φ∆ approaches unity efficiency, could be explained by the proposed excited state 3MLCT-3IL interaction, which shortens the 3IL state lifetime substantially. Nevertheless, the ability to populate 3IL states in 4 exclusively with both 355- and 532-nm excitation was anticipated to not only impact Φ∆ but potentially influence photobiological activity.
Photobiological Properties.
Cytotoxicity and photocytotoxicity.
To explore potential photobiological activities within the series, the cytotoxicities of compounds 1–4 were determined toward SKMEL28 cells in the dark (sham treatment) or with a light treatment. The light treatments were 100 J cm−2 of broadband visible or monochromatic red (625 nm) delivered at a fluence rate of approximately 28 mW cm−2. Briefly, cells were dosed with the metal complexes in the range of 1 nM to 300 µM, incubated for 16 h followed by either a dark or a light treatment, and then assessed for viability by addition of the resazurin cell viability indicator46 48 h later. Cytotoxicity and photocytotoxicity were quantified by nonlinear fitting of the dose-response curves to extract the effective concentration required to reduce cell viability by half (EC50) relative to non-treated control cells. The phototherapeutic index (PI) was calculated as the ratio of the dark to light EC50 values, and is a measure of the amplification of cytotoxic activity by the light treatment. The results are shown in Table 4 and Figure 7.
Table 4.
(Photo)cytotoxicities and Phototherapeutic Indices of Complexes 1–4 toward SKMEL28 Cells
| Cmpd | Dark EC50 (μM) | Visa EC50 (μM) | Vis PI | Redb EC50 (μM) | Red PI |
|---|---|---|---|---|---|
| 1 | 1.12 ± 0.05 | 0.087 ± 0.006 | 13 | 0.590 ± 0.070 | 2 |
| 2 | 19.5 ± 1.30 | 0.108 ± 0.002 | 181 | 10.5 ± 0.2 | 2 |
| 3 | 5.53 ± 0.18 | 0.114 ± 0.019 | 49 | 0.270 ± 0.020 | 21 |
| 4 | >300 | 1.13 ± 0.07 | >267 | 12.0 ± 0.4 | >25 |
EC50 values reported ± SEM (μM).
400–700 nm, 28 mW cm−2, 100 J cm−2
633 nm 28 mW cm−2, 100 J cm−2
Figure 7.

(Photo)cytotoxic (a) and phototherapeutic index (b) activity plots for compounds 1–4 against SKMEL28 melanoma cells with the following treatment conditions: dark (black circles), visible light (blue circles), or red light (red circles). Representative dose-response plot for SKMEL28 cells treated with compound 4 (c) and subjected to a dark (black curve), visible light (blue curve), or red light (red curve) treatment.
Ru(II) C^N complexes 1–3 were cytotoxic to cells in the absence of a light, with the EC50 value for 1 being on the order of those for cytotoxic chemotherapy drugs (near 1 µM). The cytotoxicities of 2 and 3 were slightly less (EC50 =5–20 µM), and 4 was completely nontoxic toward SKMEL28 cells in the dark (EC50 =>300 µM). The trend, which admittedly is not well understood at present, appears to be an abrupt decrease in cytotoxicity at some critical degree of π-conjugation (in this case on going from n=1 to 4) rather than a systematic decrease in cytotoxicity with increasing π-extension. This trend was observed previously in the [Ru(4,4′-dmb)2 (IBQ-nT)]Cl series, where there was an abrupt change in the dark cytotoxicity on going from n=3 to n=4 in terms of the number of thienyl units. It that series, the intracellular luminescence from the complexes viewed with laser scanning confocal microscopy suggested that the absence of cytotoxicity with n=4 could be due to reduced cellular uptake (and possibly reduced nuclear accumulation as a consequence). Confocal imaging also showed a significant reduction in cellular uptake for 4 relative to 1–3 (vide infra). As discussed with regard to the lipophilicity study, decreased cellular uptake with increased lipophilicity contradicts the existing dogma. However, if the tetrathiophene group induces extracellular aggregation, then reduced cellular uptake in the absence of a light trigger is not unexpected.
All four of the complexes exhibited amplified cytotoxicity when activated with visible or red light. Visible light yielded PIs ranging from 13 in the case of 1 to more than 267 for 4. The enhancement for 1 and 2 with red light, on the other hand, was only about two-fold, whereas for 3 and 4 it was 21 and >25, respectively. The elevated dark cytotoxicity for compounds 1–3 certainly attributed to attenuated PIs relative to their Ru(II) diimine counterparts in general. However, the PIs for 2 and 4 with visible light were significant nonetheless (Figure 7b). The visible EC50 values for 1–3 were submicromolar and ranged from 87 to 144 nM, while the red EC50 values were submicromolar for 1 and 3 only and approximately 10 µM for 2. However, the marginal PIs for 1 and 3 with either light condition reflected the baseline toxicity that led to the smaller EC50 values obtained for photocytotoxicity. Despite EC50 values for photocytotoxicity that were larger than for the other compounds, 4 was the most phototoxic compound of the series because dark toxicity did not contribute significantly to the observed photocytotoxicity (Figure 7c). Visible and red EC50 values were approximately 1 and 12 µM, respectively. The photocytotoxicity with red light for 4 was similar to what was measured previously for its [Ru(4,4′-dmb)2 (IBQ-4T)]Cl relative,14 but its potency with visible light was reduced by more than four-fold. This difference highlights that subtle changes to the ancillary N^N ligands in these Ru(II) C^N complexes can fine tune the photobiological activity in some cases.
The present study shows that not only do ancillary N^N ligands affect photobiological activity but that the thienyl units in the π-extended C^N also have an impact, with a critical change in the photophysical and photobiological properties on going from n=3 to n=4. This marked switch from cytotoxic chemotherapeutic to a nontoxic compound but potent photosensitizer with one additional thiophene ring has now been corroborated in two Ru(II) C^N families (using bpy and 4,4′-dmb ancillary N^N ligands) and is reminiscent of an earlier discovery we made when studying the cyclometalated Ru(II) complex containing the π-extended pbpn C^N ligand and its less π-expansive relatives:13 there is a critical degree of π-conjugation required for reducing the cytotoxicity of cyclometalated Ru(II) complexes. Past this point, compounds that were once thought to be too cytotoxic to act as photobiological agents may now be considered for applications such as PDT. Further, the π-expanded Ru(II) C^N complexes provide important exceptions to two existing dogmas: (i) cytotoxicity increases with lipophilicity, and (ii) cyclometalated Ru(II) compounds are too toxic to act as PDT agents.
The dark cytotoxicity associated with compounds 1–3 in the present series and their close analogs in the [Ru(4,4′-dmb)2 (IBQ-nT)]Cl family complicates the task of relating the photophysical properties to their photobiological activities. In the analogous Ru(II) diimine families, there is a clear trend between elevated photocytotoxicity and accessibility of low-lying 3IL states, which are highly-photosensitizing with much larger 1O2 quantum yields.4,45 Substantial dark cytotoxicity precludes making similar correlations for the Ru(II) C^N complexes of this study. The absence of dark cytotoxicity for 4 and its much lower photocytotoxicity relative to its Ru(II) diimine counterpart (approximately an order of magnitude lower under the same conditions) appear to implicate 1O2 as the mediator of the photobiological activity for this compound. This phototoxic mechanism is consistent with the much smaller value of Φ∆ measured for 4 (approximately ten-fold less) relative to [Ru(bpy)2 (IP-4T)]Cl2 if both systems function by 1O2 sensitization.
Cellular Imaging.
SKMEL28 cells treated with 1–4 were further analyzed by taking advantage of the inherent 1IL emission (Figure 3) from the complexes. The effects of complexes 1–4 (25 µM) on SKMEL28 cells were documented by differential interference contrast (DIC) and laser-scanning confocal microscopy (Figure 8). The cells were incubated with the compound for 15 min in the dark and then kept in the dark (sham) or irradiated with visible light (50 J cm−2, 35 mW cm−2) followed by imaging within 15 min after the treatment (total time from dosing to imaging was approximately 1 h). The conditions were different than those used in the dose-response cytotoxicity assays to ensure that enough cells remained viable at the imaging time to assess any qualitative differences among the four compounds and two treatment conditions. Overlays of the DIC and emission images were constructed to highlight cellular morphology and localization/uptake of the compounds at t=1 h.
Figure 8.

Laser-scanning confocal microscopy and DIC images of SKMEL28 human melanoma cells dosed with compounds 1–4 (25 µM) and kept in the dark (a) or exposed to a visible light treatment of 50 J cm−2 delivered at a rate of 35 mW cm−2 (b). Scale bar = 20 µm.
The representative images in Figure 8 demonstrate that all four Ru(II) C^N complexes were taken up by cells in the dark. This observation is based on the assumption that the emission signal from the metal complex reflects its location and that a certain degree of cellular integrity must be maintained to observe a signal. Qualitatively, there did not appear to be a difference in the cellular uptake between the dark and the light condition for any compound.
A substantial amount of cellular debris was evident for SKMEL28 cells treated with 1–3 in the dark, which agreed with the dark cytotoxicity measured for these compounds in the dose-response cytotoxicity assays. The extent of cell damage in the dark elicited by 1–3 made it difficult to ascertain any light amplification of this cytotoxicity. While the use of confocal microscopy is not an accurate tool for assessing intracellular distribution, the Ru(II) C^N compounds appeared to accumulate in the nucleus (for cells that were healthy enough for localization to be discerned). This was also true for cells treated with lower concentrations of metal complex in the dark at shorter incubation times, where the cells appeared healthy.
Cells treated with 4 in the dark luminesced brightly, suggesting effective cellular uptake. Light activation of 4 resulted in a significant amount of cellular debris, which corroborated the dose-response (photo)cytotoxicity findings whereby 4 gave the largest PI. With lower concentrations and shorter incubation times, 4 still luminesced in cells. This was somewhat surprising given that its relative [Ru(4,4′-dmb)2 (IP-4T)]Cl showed minimal intracellular emission in the same cell line (and presumably low uptake) even with higher concentrations and longer incubation times.14 In fact, reduced cellular uptake (and thus reduced nuclear accumulation) was used to explain the absence of dark cytotoxicity for that metal complex. In the case of 4, reduced uptake or differential localization cannot explain the stark differences in dark cytotoxicity within the series. Similarly, photoactivated uptake cannot explain the large PI of 4.
While the imaging study did not inform on mechanism of action for the dark cytotoxicity of 1–3 or explain the absence of cytotoxicity for 4 (efforts to understand these properties are ongoing), the study was critical for demonstrating that effective nuclear uptake does not necessarily lead to dark cytotoxicity. It also highlights the fact that two similar Ru(II) C^N complexes, differing only by two methyl groups on the bpy coligands ([Ru(bpy)2 (IBQ-4T)]Cl in the present study versus [Ru(4,4′-dmb)2 (IBQ-4T)]Cl in our previous study14), can have nearly identical (photo)cytotoxicity profiles yet drastically different intracellular luminescence properties which may reflect differences in uptake and localization. Thus, care must be taken in using one to explain the other and vice versa.
SUMMARY AND CONCLUSIONS
Four new cyclometalated Ru(II) complexes were prepared from two bpy N^N coligands combined with a IBQ-nT C^N ligand of varying thienyl chain length (n=1–4). The structural rationale behind the series was to systematically alter the π-conjugation on the C^N ligand, and to our knowledge this is only the third study of tris-bidentate Ru(II) systems that explore π-expansion through the C^N component. The log Po/w values measured for complexes 1–4 were all positive, with lipophilicities increasing with the number of thienyl rings in the cyclometalating ligand. The compounds exhibited emission from both the 1IL or 3MLCT states that could be selectively induced by choosing the excitation wavelength. The energy of the 1IL fluorescence decreased as the thiophene chain lengthened, but the energy of the 3MLCT did not, permitting the assignments of the orbital contributions to these transitions as the C^N-based π→π* fluorescence and dπ(Ru2+)→π*(N^N) phosphorescence, respectively. Complex 4 exhibited an additional long-lived (τTA≈5 μs) dark state by TA spectroscopy that was not observed in the emission measurements and appeared to be in equilibrium with the N^N-based 3MLCT, leading to delayed phosphorescence. Based on its lifetime and TA spectral signature, this state was assigned as the C^N-localized 3IL state. All four compounds were weak 1O2 sensitizers, but the longest-lived 4 was three times more potent than the others, presumably due to the contribution of the 3IL state to the excited state dynamics for this compound.
Complexes 1–3 were cytotoxic to SKMEL28 cells in the order 1 > 3 > 2, with EC50 values ranging from 1 µM for 1 to 20 µM for 2. Compound 4 was nontoxic in the dark, demonstrating that dark cytotoxicity does not parallel lipophilicity within this series. The PIs with visible light were as low as 13 for 1 owing to its high dark cytotoxicity, and >267 for 4 due to the absence of dark cytotoxicity. Red light was much less effective at amplifying the cytotoxic effects of these compounds, with the maximum PI being >25 for the best photobiological agent of the family and only around 2 for the least photoactive members. The relative cytotoxicities of the compounds could not be attributed to differences in cellular uptake according to assessment by laser scanning confocal microscopy using the inherent 1IL emission from the compounds. Compound 4 appeared to be taken up as effectively as the other compounds. In addition, the uptake between dark- and light-treated SKMEL28 cells dosed with compounds 1–4 appeared very similar, so the absence of dark cytotoxicity for only 4 could not be attributed to photoactivated uptake. The photophysical and photobiological trends within the series agreed well with what was previously observed in the analogous [Ru(4,4′-dmb)2 (IBQ-nT)]Cl series, where there was a clear difference in cellular uptake and nuclear accumulation for n=4 with a relatively large PI that could be explained by photoactivated uptake and corroborated by confocal microscopy. Thus, it was surprising that the confocal images of cells treated with 4 showed significant cellular uptake and nuclear accumulation in the dark that looked very similar for the cells exposed to visible light.
The underlying differences in the dark cytotoxicities between 1–3 and 4 in the present series yet striking similarities in the imaging experiments (and by extension similarities in their cellular uptake and nuclear accumulation profiles) is an area under active investigation but beyond the scope of this study. Nevertheless, this study serves as the third example of a cyclometalated Ru(II) series based on π-expanded C^N ligands, whereby there is a critical π-conjugation length that leads to an abrupt change in dark cytotoxicity. The implication is that cyclometalated Ru(II) compounds that were once thought to be too toxic can now serve as potent photobiological agents (low dark cytotoxicity with a large phototherapeutic margin) provided the C^N ligand is of the critical π-conjugation length. This number has been established as n=4 in two related series: [Ru(4,4′-dmb)2 (IBQ-nT)]Cl and now [Ru(bpy)2 (IBQ-nT)]Cl. The phenomenon could be a general one given that it has also been demonstrated in a series based on [Ru(bpy)2 (pbpn)]Cl (and its less-conjugated relatives) as well. Finally, this study highlights that subtle changes to the auxiliary N^N ligands in the [Ru(N^N)2 (IBQ-4T)]Cl construct can be used to fine-tune the photobiological activity given that 4,4’-methyl substitution of bpy leads to more potent light cytotoxicity with a larger PI. The inherent modularity of these organometallic systems provides the opportunity to explore a vast structure-activity landscape for optimizing their chemical, photophysical, and photobiological properties for phototherapy applications.
Supplementary Material
ACKNOWLEDGMENT
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA222227. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge financial support from the University of North Carolina at Greensboro, the Natural Sciences and Engineering Council of Canada, the Canadian Institutes of Health Research, the Canadian Foundation for Innovation, the Nova Scotia Research and Innovation Trust, and Acadia University.
Footnotes
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
1D 1H and 2D 1H–1H COSY and 13C NMR spectra, HPLC traces, and transient absorption decay spectra for the Ru(II) C^N complexes (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Juris A; Balzani V; Barigelletti F; Campagna S; Belser P; von Zelewsky A Ru(II) Polypyridine Complexes: Photophysics, Photochemistry, Eletrochemistry, and Chemiluminescence. Coord. Chem. Rev 1988, 84, 85–277. [Google Scholar]
- (2).Balzani V; Juris A Photochemistry and Photophysics of Ru(II) polypyridine Complexes in the Bologna Group. From Early Studies to Recent Developments. Coord. Chem. Rev 2001, 211, 97–115. [Google Scholar]
- (3).Kalyanasundaram K Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: London, 1992. [Google Scholar]
- (4).Monro S; Colón KL; Yin H; Roque J; Konda P; Gujar S; Thummel RP; Lilge L; Cameron CG; McFarland SA Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 2019, 119, 797–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ertl CD; Ris DP; Meier SC; Constable EC; Housecroft CE; Neuburger M; Zampese JA Sticking and Patching: Tuning and Anchoring Cyclometallated Ruthenium(II) Complexes. Dalton. Trans 2015, 44, 1557–1570. [DOI] [PubMed] [Google Scholar]
- (6).Allen FH The Cambridge Structural Database: A Quarter of a Million Crystal Structures and Rising. Acta Crystallogr. B 2002, 58, 380–388. [DOI] [PubMed] [Google Scholar]
- (7).Caspar JV; Meyer TJ Application of the Energy Gap Law to Nonradiative, Excited-State Decay. J. Phys. Chem 1983, 87, 952–957. [Google Scholar]
- (8).Muro-Small ML; Yarnell JE; McCusker CE; Castellano FN Spectroscopy and Photophysics in Cyclometalated RuII–Bis(Bipyridyl) Complexes. Eur. J. Inorg. Chem 2012, 2012, 4004–4011. [Google Scholar]
- (9).Bomben P; Robson K; Koivisto B; Berlinguette C Cyclometalated Ruthenium Chromophores for the Dye-Sensitized Solar Cell. Coord. Chem. Rev 2012, 256, 1438–1450. [Google Scholar]
- (10).Bomben PG; Gordon TJ; Schott E; Berlinguette CP A Trisheteroleptic Cyclometalated RuII Sensitizer That Enables High Power Output in a Dye-Sensitized Solar Cell. Angew. Chem. Int. Ed 2011, 50, 10682–10685. [DOI] [PubMed] [Google Scholar]
- (11).Robson KCD; Koivisto BD; Yella A; Sporinova B; Nazeeruddin MK; Baumgartner T; Grätzel M; Berlinguette CP Design and Development of Functionalized Cyclometalated Ruthenium Chromophores for Light-Harvesting Applications. Inorg. Chem 2011, 50, 5494–5508. [DOI] [PubMed] [Google Scholar]
- (12).Albani BA; Peña B; Dunbar KR; Turro C New Cyclometallated Ru(II) Complex for Potential Application in Photochemotherapy? Photochem. Photobiol. Sci 2014, 13, 272–280. [DOI] [PubMed] [Google Scholar]
- (13).Sainuddin T; McCain J; Pinto M; Yin H; Gibson J; Hetu M; McFarland SA Organometallic Ru(II) Photosensitizers Derived from π-Expansive Cyclometalating Ligands: Surprising Theranostic PDT Effects. Inorg. Chem 2016, 55, 83–95. [DOI] [PubMed] [Google Scholar]
- (14).Ghosh G; Colón KL; Fuller A; Sainuddin T; Bradner E; McCain J; Monro SMA; Yin H; Hetu MW; Cameron CG; et al. Cyclometalated Ruthenium(II) Complexes Derived from α-Oligothiophenes as Highly Selective Cytotoxic or Photocytotoxic Agents. Inorg. Chem 2018, 57, 7694–7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Peña B; David A; Pavani C; Baptista MS; Pellois J-P; Turro C; Dunbar KR Cytotoxicity Studies of Cyclometallated Ruthenium(II) Compounds: New Applications for Ruthenium Dyes. Organometallics 2014, 33, 1100–1103. [Google Scholar]
- (16).Huang H; Zhang P; Chen H; Ji L; Chao H Comparison between Polypyridyl and Cyclometalated Ruthenium(II) Complexes: Anticancer Activities against 2D and 3D Cancer Models. Chem. Eur. J 2015, 21, 715–725. [DOI] [PubMed] [Google Scholar]
- (17).Sullivan BP; Salmon DJ; Meyer TJ Mixed Phosphine 2, 2’-Bipyridine Complexes of Ruthenium. Inorg. Chem 1978, 17, 3334–3341. [Google Scholar]
- (18).Kosuge T; Senoo A; Ohrui H; Muratsubaki M Azafluorene Derivative and Organic Light-Emitting Device Using the Derivative US 8,110,685 B2, February 7, 2012.
- (19).Krishnaswamy NR; Kumar C Absorption, Emission and Mass Spectral Characteristics of Oligothiophenes and Their Derivatives. Indian J. Chem. Sect. B Org. Chem. Med. Chem 1993, 32B, 766–771. [Google Scholar]
- (20).McFarland SA Metal-Based Thiophene Photodynamic Compounds and Their Use US20130331367 A1, December 12, 2013.
- (21).Koizumi T; Tomon T; Tanaka K Synthesis and Electrochemical Properties of Bis(Bipyridine)Ruthenium(II) Complexes Bearing Pyridinyl- and Pyridinylidene Ligands Induced by Cyclometalation of N′-Methylated Bipyridinium Analogs. J. Organomet. Chem 2005, 690, 1258–1264. [Google Scholar]
- (22).Foxon SP; Metcalfe C; Adams H; Webb M; Thomas JA Electrochemical and Photophysical Properties of DNA Metallo-Intercalators Containing the Ruthenium(II) Tris(1-Pyrazolyl)Methane Unit. Inorg. Chem 2007, 46, 409–416. [DOI] [PubMed] [Google Scholar]
- (23).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord Chem. Rev 2002, 233/234, 351–371. [Google Scholar]
- (24).Bessho T; Yoneda E; Yum J-H; Guglielmi M; Tavernelli I; Imai H; Rothlisberger U; Nazeeruddin MK; Grätzel M New Paradigm in Molecular Engineering of Sensitizers for Solar Cell Applications. J. Am. Chem. Soc 2009, 131, 5930–5934. [DOI] [PubMed] [Google Scholar]
- (25).Wang L; Yin H; Cui P; Hetu M; Wang C; Monro S; Schaller RD; Cameron CG; Liu B; Kilina S; et al. Near-Infrared-Emitting Heteroleptic Cationic Iridium Complexes Derived from 2,3-Diphenylbenzo[g]Quinoxaline as in Vitro Theranostic Photodynamic Therapy Agents. Dalton Trans 2017, 46, 8091–8103. [DOI] [PubMed] [Google Scholar]
- (26).Wang C; Lystrom L; Yin H; Hetu M; Kilina S; McFarland SA; Sun W Increasing the Triplet Lifetime and Extending the Ground-State Absorption of Biscyclometalated Ir(III) Complexes for Reverse Saturable Absorption and Photodynamic Therapy Applications. Dalton Trans 2016, 45, 16366–16378. [DOI] [PubMed] [Google Scholar]
- (27).Liu B; Monro S; Lystrom L; Cameron CG; Colón K; Yin H; Kilina S; McFarland SA; Sun W Photophysical and Photobiological Properties of Dinuclear Iridium(III) Bis-Tridentate Complexes. Inorg. Chem 2018, 57, 9859–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fetzer L; Boff B; Ali M; Xiangjun M; Collin J-P; Sirlin C; Gaiddon C; Pfeffer M Library of Second-Generation Cycloruthenated Compounds and Evaluation of Their Biological Properties as Potential Anticancer Drugs: Passing the Nanomolar Barrier. Dalton Trans 2011, 40, 8869–8878. [DOI] [PubMed] [Google Scholar]
- (29).Puckett CA; Barton JK Methods to Explore Cellular Uptake of Ruthenium Complexes. J. Am. Chem. Soc 2007, 129, 46–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bomben PG; Robson KCD; Sedach PA; Berlinguette CP On the Viability of Cyclometalated Ru(II) Complexes for Light-Harvesting Applications. Inorg. Chem 2009, 48, 9631–9643. [DOI] [PubMed] [Google Scholar]
- (31).Motley TC; Troian-Gautier L; Brennaman MK; Meyer GJ Excited-State Decay Pathways of Tris(Bidentate) Cyclometalated Ruthenium(II) Compounds. Inorg. Chem 2017, 56, 13579–13592. [DOI] [PubMed] [Google Scholar]
- (32).Peña B; Leed NA; Dunbar KR; Turro C Excited State Dynamics of Two New Ru(II) Cyclometallated Dyes: Relation to Cells for Solar Energy Conversion and Comparison to Conventional Systems. J. Phys. Chem. C 2012, 116, 22186–22195. [Google Scholar]
- (33).Kreitner C; Heinze K Excited State Decay of Cyclometalated Polypyridine Ruthenium Complexes: Insight from Theory and Experiment. Dalton Trans 2016, 45, 13631–13647. [DOI] [PubMed] [Google Scholar]
- (34).Hissler M; Connick WB; Geiger DK; McGarrah JE; Lipa D; Lachicotte RJ; Eisenberg R Platinum Diimine Bis(Acetylide) Complexes: Synthesis, Characterization, and Luminescence Properties. Inorg. Chem 2000, 39, 447–457. [DOI] [PubMed] [Google Scholar]
- (35).McCusker CE; McCusker JK Synthesis and Spectroscopic Characterization of CN-Substituted Bipyridyl Complexes of Ru(II). Inorg. Chem 2011, 50, 1656–1669. [DOI] [PubMed] [Google Scholar]
- (36).McClenaghan ND; Leydet Y; Maubert B; Indelli MT; Campagna S Excited-State Equilibration: A Process Leading to Long-Lived Metal-to-Ligand Charge Transfer Luminescence in Supramolecular Systems. Coord. Chem. Rev 2005, 249, 1336–1350. [Google Scholar]
- (37).Majewski MB; de Tacconi NR; MacDonnell FM; Wolf MO Ligand-Triplet-Fueled Long-Lived Charge Separation in Ruthenium(II) Complexes with Bithienyl-Functionalized Ligands. Inorg. Chem 2011, 50, 9939–9941. [DOI] [PubMed] [Google Scholar]
- (38).Barbieri A; Ventura B; Flamigni L; Barigelletti F; Fuhrmann G; Bäuerle P; Goeb S; Ziessel R Binuclear Wirelike Dimers Based on Ruthenium(II)−Bipyridine Units Linked by Ethynylene−Oligothiophene−Ethynylene Bridges. Inorg. Chem 2005, 44, 8033–8043. [DOI] [PubMed] [Google Scholar]
- (39).Costa RD; Aragó J; Ortí E; Pappenfus TM; Mann KR; Matczyszyn K; Samoc M; Zafra JL; López Navarrete JT; Casado J Impact of the Synergistic Collaboration of Oligothiophene Bridges and Ruthenium Complexes on the Optical Properties of Dumbbell-Shaped Compounds. Chem. - Eur. J 2013, 19, 1476–1488. [DOI] [PubMed] [Google Scholar]
- (40).Liu Y; De Nicola A; Reiff O; Ziessel R; Schanze KS Photophysics and Photoinduced Electron-Transfer Reactivity of Ruthenium(II) Complexes with Oligo(Thiophene-Bipyridine) Ligands †, 1. J. Phys. Chem. A 2003, 107, 3476–3485. [Google Scholar]
- (41).Becker RS; Seixas de Melo J; Maçanita AL; Elisei F Comprehensive Evaluation of the Absorption, Photophysical, Energy Transfer, Structural, and Theoretical Properties of α-Oligothiophenes with One to Seven Rings. J. Phys. Chem 1996, 100, 18683–18695. [Google Scholar]
- (42).de Melo JS; Silva LM; Arnaut LG; Becker RS Singlet and Triplet Energies of α-Oligothiophenes: A Spectroscopic, Theoretical, and Photoacoustic Study: Extrapolation to Polythiophene. J. Chem. Phys 1999, 111, 5427–5433. [Google Scholar]
- (43).Ford WE; Rodgers MAJ Reversible Triplet-Triplet Energy Transfer within a Covalently Linked Bichromophoric Molecule. J. Phys. Chem 1992, 96, 2917–2920. [Google Scholar]
- (44).Ogilby PR; Foote CS Chemistry of Singlet Oxygen. 42. Effect of Solvent, Solvent Isotopic Substitution, and Temperature on the Lifetime of Singlet Molecular Oxygen (1.DELTA.g). J. Am. Chem. Soc 1983, 105, 3423–3430. [Google Scholar]
- (45).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; et al. Ru(II) Dyads Derived from Alpha-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- (46).O’Brien J; Wilson I; Orton T; Pognan F Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem 2000, 267, 5421–5426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.