

Abstract
The oxidation of carbon-carbon triple bonds by cytochrome P450 produces ketene metabolites that are hydrolyzed to acetic acid derivatives or are trapped by nucleophiles. In the special case of 17α-ethynyl sterols, D-ring expansion and de-ethynylation have been observed as competing pathways. The oxidation of acetylenic groups is also associated with mechanism-based inactivation of cytochrome P450 enzymes. One mechanism for this inactivation is reaction of the ketene metabolite with cytochrome P450 residues essential for substrate binding or catalysis. However, in the case of monosubstituted acetylenes, inactivation can also occur by addition of the oxidized acetylenic function to a nitrogen of the heme prosthetic group. This addition reaction is not mediated by the ketene metabolite, but rather occurs during oxygen transfer to the triple bond. In some instances, a detectable intermediate is formed that is most consistent with a ketocarbene-iron heme complex. This complex can progress to the N-alkylated heme or revert back to the unmodified enzyme. The ketocarbene complex may intervene in the formation of all the N-alkyl heme adducts, but is normally too unstable to be detected.
Keywords: Acetylene oxidation, ketene formation, cytochrome P450 inactivation, iron-carbene complexes, oxirene, ethynylsterols
Introduction
The reactions catalyzed by cytochrome P450, with some exceptions, fall into three classes: (i) hydroxylations in which an oxygen is inserted between a hydrogen and a heavier element, usually carbon or nitrogen; (ii) heteroatom oxidations, in which the oxygen atom is added to the free electron pair of a heteroatom, usually nitrogen or sulfur; and (iii) oxidation of double, triple, or aromatic π-bonds. This review focuses exclusively on the oxidation of carbon-carbon triple bonds (i.e., acetylenic groups). The oxidation of acetylenes resembles in some, but not all respects the oxidation of olefinic bonds. The oxidation of triple bonds has appeared in general reviews on the mechanism of cytochrome P450 enzymes (e.g., Ortiz de Montellano 2015) and the mechanism-based inactivation of cytochrome P450 enzymes (e.g., Correia and Hollenberg 2015), but the last review dealing extensively with the oxidation of acetylenes was published more than 30 years ago (Ortiz de Montellano 1985). This review updates our understanding of the oxidation of acetylenes in the light of the information obtained in the quarter of a century since that last review. The discussion of the formation of metabolites and the inactivation of cytochrome P450 enzymes includes a critical reevaluation of the putative role of oxirenes as reaction intermediates, the oxidation of disubstituted triple bonds, and the nature of “reversible” inhibitory complexes.
Oxidation of monosubstituted acetylenes to acetic acid derivatives
In 1958, El Masri et al. reported that phenyl acetylene was slowly converted in vivo by rabbits to phenaceturic acid, the N-glycine conjugate of phenylacetic acid (Figure 1), although most of the dose was excreted unchanged (El Masri et al. 1958). They postulated that this transformation involved addition of water to the triple bond to give the enol form of phenylacetaldehyde, followed by oxidation of the aldehyde to phenylacetic acid. Conjugation with glycine then yielded the isolated metabolite. This transformation is now known to involve an oxidative rather than hydrolytic process (Chan et al. 1993)
Figure 1.

In vivo conversion of phenylacetylene to phenylacetic acid and its phenylaceturic acid derivative.
Subsequent studies of the in vivo oxidation of 4-ethynylbiphenyl by rats and rabbits led to the isolation of two metabolites: 4-biphenylacetic acid (2-10% of the dose) and 4’-hydroxy-4-biphenylacetic acid (90-95% of the dose) (Figure 2, R = H) (Wade et al. 1979). In vitro oxidation of 4-ethynylbiphenyl by rat liver microsomes also yielded the acetic acid metabolite (Wade et al. 1980). A fluorinated biphenyl derivative was similarly oxidized in vivo by rats to the corresponding acetic acid metabolites (Sullivan et al. 1979). Many examples are now known of the oxidation of terminal acetylenes to carboxylic acids.
Figure 2:

Cytochrome P450 catalyzed oxidation of 4-ethynylbiphenyl (R = H) or its 2’-fluoro substituted analogue (R = F) to the acetic acid metabolites.
Wade et al. suggested that the triple bond was converted to the acetic acid group via initial “epoxidation” to give an oxirene (Figure 3, path c), which spontaneously rearranged to give a ketene that reacted with water to give the acetic acid product (Wade et al. 1979; Wade et al. 1980). The validity of this proposal is discussed below. An alternative mechanism envisions insertion of oxygen into the acetylenic C-H bond, followed by rearrangement to the ketene (Figure 3, path b) (McMahon et al. 1981). However, this latter mechanism is highly disfavored by the C-H bond energy, which is much higher than that of a vinylic C-H bond and very much higher than that of a secondary alkyl C-H bond: compare the dissociation energies (DH298 in kcal/mol) for acetylene (131.3 ± 0.7), ethylene (109.8 ± 0.8), and the secondary C-H of propane (98.6 ± 0.4) (Blanksby and Ellison 2003; Ervin et al. 1990).
Figure 3.

Theoretical mechanistic alternatives for oxidation of the triple bond.
Direct evidence that oxidation of the triple bond involves reaction of the oxygen with one of the acetylenic π-bonds rather than insertion into the acetylenic C-H bond is provided by the finding that when the acetylenic proton of 4-ethynylbiphenyl is replaced by a deuterium, the deuterium migrates quantitatively to the adjacent carbon in the acetic acid product. Loss of deuterium would be expected for C-H hydroxylation. Furthermore, when the biphenyl-substituted carbon is 13C-labeled, the 13C-labeled carbon remains attached to the biphenyl in the product. These results establish that the hydrogen rather than the biphenyl is the moiety that migrates during the oxidative process. In addition, a substantial deuterium isotope effect is observed on the rate of ketene formation upon replacement of the acetylenic hydrogen by a deuterium (Ortiz de Montellano and Komives 1985; Komives and Ortiz de Montellano 1987). Finally, the ketene oxygen has been shown in the case of biphenylacetylene to derive from molecular oxygen, in accord with oxygen transfer from a cytochrome P450 enzyme (Ortiz de Montellano and Komives 1985). The same hydrogen shift has been shown to occur when the triple bond is chemically oxidized by meta-chloroperbenzoic acid. In the chemical experiments, direct evidence for formation of the ketene was provided by isolation of the methyl ester rather than free acid when the oxidation was carried out in the presence of methanol (Ortiz de Montellano and Kunze 1980a, 1981a).
Metabolic formation of a ketene intermediate is confirmed by experiments on the metabolism of the anticancer drug erlotinib (Zhao et al. 2018). Oxidation of the drug by human liver microsomes resulted in covalent binding to proteins of a ketene intermediate that could also be trapped by reaction with 4-bromobenzylamine (Figure 4).
Figure 4.

Oxidation of erlotinib to a ketene that can react with a protein (Protein-XH) or be trapped by reaction with 4-bromobenzylamine.
Similarly, the oxidation of selegiline by CYP2B6 was shown to result in a glutathione-trappable ketene metabolite that also acylated the P450 protein (Sridar et al. 2012). Oxidation of the triple bond to a ketene was preceded by hydroxylation of the aromatic ring, as shown in Figure 5.
Figure 5.

Oxidation of selegiline to a hydroxylated derivative (R = H -> R = OH) is followed by oxidation of the acetylenic group to a ketene that can be trapped by glutathione (GSH) or can acylate the P450 protein (Protein-XH).
As a final example, the ketenes formed in the CYP3A4-catalyzed oxidation of 7-coumarin propargyl ether and 7-(4-trifluoromethyl)coumarin propargyl ether have similarly been trapped with glutathione (Sridar et al. 2008).
Are oxirenes intermediates in the oxidation of carbon-carbon triple bonds?
Epoxides are well-known, stable molecules, although they are more sensitive to acids and more reactive with nucleophiles than conventional ethers. Their relative instability is due to the ring tension caused by constraining a nominally sp3 tetrahedral carbon, in which the bonding orbitals subtend an angle of 109°, into a geometry that forces the bonds to an angle of 60°. This results in bent or “banana” bonds that are much weaker than those in a normal ether molecule. Nevertheless, epoxides are stable enough to be routinely formed as isolable metabolites in the oxidation of olefins (Ortiz de Montellano 1985, 2015).
The carbon in an oxirene, however, is nominally in the sp2 state, which means that the bonding orbitals ideally subtend an angle of approximately 120° degrees, whereas geometry still constrains the bonds to an angle to 60° degrees. Furthermore, the π-bond electrons and those of one of the oxygen orbitals constitute an antiaromatic 4π-electron system, which further destabilizes the system. Computational methods are inconclusive as to whether an oxirene is truly a molecule, in the sense that it resides at the bottom of a localized energy well, or is simply a transition state between more stable species (Vacek et al. 1994; Mawhinney and Goddard 2003). More importantly, despite the use of sophisticated methods and low temperatures, there is little evidence for the existence of an oxirene as a discrete molecular entity (Lewars 1983; Zeller et al. 2004). In view of this, the formation of an oxirene as an intermediate with a finite lifetime in the cytochrome P450-catalyzed oxidation of acetylenes is highly unlikely. Thus, the reaction is best formulated as one in which delivery by the enzyme of the reactive oxygen to the acetylenic π-bond occurs concurrently with hydrogen migration to give the ketene (or whatever other reaction is required to give the observed products), as shown in Figure 6 for a monosubstituted acetylene. This scheme rationalizes all the information on triple bond oxidation, including the observation of an isotope effect when the terminal hydrogen is replaced by a deuterium.
Figure 6.

Oxidation of a terminal triple bond with concomitant migration of the hydrogen via a transition state that does not generate an oxirene intermediate. The Fe in a box of nitrogens represents the activated species of cytochrome P450.
Ring expansion reactions
In 1969, Abdel-Aziz and Williams reported that in vivo metabolism of 17α-ethynylestradiol by rabbits yielded, inter alia, a D-homoannulated product with one carbon of the acetylenic group incorporated into an expanded D-ring, the other acetylenic carbon being lost (Figure 7) (Abdel-Aziz and Williams 1969). This ring-expanded product was postulated to involve oxidation of the triple bond with concomitant migration of the ring carbon to give the ring expansion, resulting in an enol alcohol that undergoes further oxidation to the acid and then decarboxylative loss of carbon dioxide. D-Homoannulation was also found among the metabolites of 17α-ethynylestradiol by women (Abdel Aziz and Williams 1970), and in the metabolism by women of both norgestrel (Sisenwine et al. 1973, 1979) and moxestrol (Salmon et al. 1983). Formation of the D-homoannulated metabolite of 17α-ethynylestradiol by hepatic microsomes from rhesus monkeys was accompanied by the formation of carbon dioxide, as proposed in the scheme (Schmid et al. 1983). In these substrates carbon migration facilitated by the 17-hydroxy group is apparently faster than terminal hydrogen migration to give the ketene.
Figure 7.

D-Homoannulation of 17α-ethynylestradiol (Abdel-Aziz and Williams 1969; Schmid et al. 1983) and a revised mechanism for this transformation that does not invoke an oxirene intermediate. The heme and activated oxygen of P450 are represented by the square of nitrogens with an iron atom.
A ring expansion also occurs in the oxidative metabolism of the AIDS drug efavirenz (Figure 8). In this reaction, the cyclopropyl ring expands to a cyclobutyl ring by migration of one of its carbon atoms to the outside carbon of the triple bond. The ring expansion is driven by release of ring strain and transfer of the incipient cation from a vinylic to a secondary carbon atom. The resulting conjugated ketone can then react with glutathione or thiol groups on proteins. The authors proposed the formation of an oxirene as the initial product formed by cytochrome P450-catalyzed oxidation of the acetylene function (Mutlib 2000). However, as already discussed, oxirenes are not expected as actual metabolites due to their high energy. A shift of the cyclopropyl carbon atom to give the cyclobutyl ring concomitant with oxygen transfer to the triple bond, without formation of a stable intermediate, is a more attractive explanation of the observed reaction.
Figure 8.

Cytochrome P450-catalyzed oxidation of efavirenz to a ring-expanded metabolite by a concerted mechanism not involving formation of an oxirene metabolite. The cytochrome P450 activated species is represented as in Figure 6.
De-ethynylation
Palmer et al. reported that incubation of norethindrone with a 10,000 × g supernatant of rabbit liver produced, among others, the 17-keto derivative from which the acetylenic group had been eliminated (Figure 9) (Palmer et al. 1969). Analogous loss of the ethynyl group in the oxidative metabolism of 17α-ethynylestradiol has also been reported (Williams et al. 1975; Helton et al. 1977; Raitano et al. 1981; Kent et al. 2002). This de-ethynylation process has so far only been described in the oxidation of 17-hydroxy-17-ethynyl sterols, suggesting that the specific substructure in these molecules is important for the reaction to occur.
Figure 9.

De-ethynylation of (top) norethindrone and (bottom) 17α-ethynylestradiol.
The mechanism for de-ethynylation is unclear, but a mechanism based on a reverse aldol reaction subsequent to trapping of the ketene metabolite by a thiol agent, shown in Figure 10 as RSH, provides a possible explanation. The intervention of cysteine and glutathione in the oxidative metabolism of acetylenes, resulting inter alia in cleavage of the triple bond to leave behind an aldehyde group, has been reported (Subramanian et al. 2011), but these reactions do not seen to relate to the process of de-ethynylation described here.
Figure 10:

A possible mechanism for the de-ethynylation of 17-hydroxy-17-ethynyl sterols.
Oxidation of disubstituted carbon-carbon triple bonds
The cytochrome P450-catalyzed oxidation of disubstituted triple bonds is relatively rare, although hydroxylation of the alkyl substituents themselves is a common reaction. The clearest evidence for disubstituted triple bond oxidation is provided by the metabolism of 1-(4-biphenyl)-1-propyne to 2-(4-biphenyl)propionic acid, a reaction that requires the methyl to migrate during the oxidation to give a ketene product (Figure 11) (Foroozesh et al. 1997). The reaction is analogous to the hydrogen migration illustrated in Figure 6, except that the migrating group is a methyl. This is one of the few instances in which the structure of the product implicating an alkyl group shift during the oxidation has been well characterized.
Figure 11.

Oxidation of 1-biphenyl-1-propyne to 2-biphenylylpropionic acid by CYP1A1 and CYP1A2.
Based on the structure of the glutathione adduct, a similar methyl shift to give a ketene occurs in the oxidation of mifepristone by CYP3A5 (Figure 12) (Lin et al. 2009). The authors proposed initial formation of an oxirene but, as already discussed, the shift is likely to be concerted with oxygen transfer to the triple bond.
Figure 12.

Oxidation of mifepristone to a glutathione conjugate (GSH = glutathione).
In vivo studies have shown that [14C]-dichloroethyne yields a glutathione conjugate as the major metabolite and dichloroacetic acid as a minor metabolite (Figure 13). The formation of dichloroacetic acid clearly suggests that one of the two chlorine atoms migrates during oxidation of the triple bond, yielding dichloroketene, which is then hydrolyzed to the observed dichloroacetic acid (Kanhai et al. 1991).
Figure 13.

In vivo oxidation of dichloroacetylene to dichloroacetic acid and the mechanism proposed for its formation.
An unusual desilylation reaction has been reported in the cytochrome P450-catalyzed oxidation of a family of proinsecticides (Figure 14) (Palmer et al. 1991). However, the mechanism of this transformation is obscure.
Figure 14.

Oxidative desilylation of a proinsecticide to an insecticidal terminal acetylene, where Pr stands for propyl.
Prosthetic heme alkylation by acetylene and monosubstituted acetylenes
The oxidation of a carbon-carbon triple bond often results in time-dependent inactivation of the cytochrome P450 catalyzing the reaction. The ability of acetylenes to inhibit cytochrome P450 enzymes was implied in the 1960s by the identification of a series propargyloxy compounds that acted as synergists for carbamate insecticides, although this activity was attributed at the time to complexation by the synergists of an essential metal cofactor (Sacher et al. 1968; Felig et al. 1970). The inhibition of cytochrome P450 enzymes by acetylenic compounds was more definitively established by the confirmed inhibitory activity of 17-ethynyl sterols (e.g., O’Malley et al. 1972; Freudenthal et al. 1974; Herz et al. 1978). Some of the cytochrome P450 inhibition of acetylenes is due simply to competitive binding (as, for example, in Shimada et al. 2007), but the potency of compounds with terminal ethynyl groups is now known to stem from irreversible mechanism-based inactivation of the P450 enzymes. Three distinct but related mechanisms can contribute to this cytochrome P450 inactivation: prosthetic heme alkylation, formation of a carbene-iron complex, and protein acylation. All three mechanisms cause time-dependent enzyme inactivation, require catalytic turnover, as indicated by a requirement for cytochrome P450 reductase and NADPH, and result in covalent modification of the enzyme.
The first mechanism, which only applies to acetylene itself and monosubstituted acetylenes, results in proportionate loss of both heme and spectroscopically detectable cytochrome P450. Most importantly, these losses are associated with accumulation in the liver (or in incubations with tissue microsomes) of a green, red-fluorescing pigment (Tables 1 and 2). Isolation and characterization by UV-visible, mass spectrometric and NMR methods of several of these pigments has shown that in all of them the acetylene is covalently attached to one of the nitrogen atoms of protoporphyrin IX, the framework of the prosthetic heme group of the enzyme (e.g., Ortiz de Montellano et al. 1979, 1982a, 1982b; Ortiz de Montellano and Kunze 1980b, 1981b; Kunze et al. 1983). In these adducts, the terminal carbon of the acetylene is attached to a porphyrin ring nitrogen atom and the internal carbon of the acetylene is present as a carbonyl group, as shown for the adduct obtained with propyne in Figure 15. Studies with 18O2 demonstrated that the oxygen in the ketone group derives from molecular oxygen and is therefore inserted by cytochrome P450 in the inactivation process (Kunze et al. 1983). The heme iron atom is lost during purification of the adduct (e.g., White 1981; 1982), a process during which the two carboxylic acid groups are converted to their methyl esters.
Table 1:
Examples of the time-dependent inhibition of cytochrome P450 by acetylenes involving addition to the prosthetic heme group
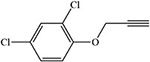
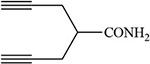
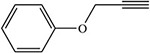
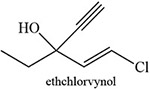
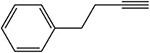
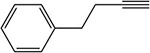
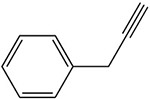
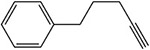
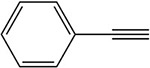
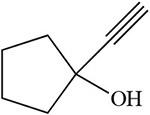
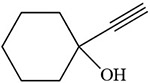
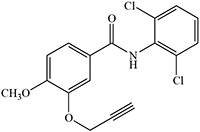
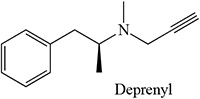
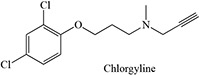
Table 2:
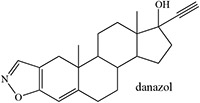
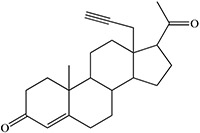
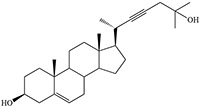
Time-dependent inhibition of cytochrome P450 by steroidal acetylenes with formation of heme adducts
| Compound | Reference | Compound | Reference |
|---|---|---|---|
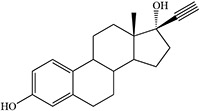 |
Blakey and White 1986 | 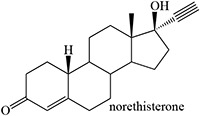 |
White 1978, 1981; Ortiz de Montellano et al. 1979; Blakey and White 1986 |
 |
White 1978 | 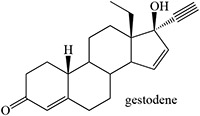 |
Guengerich 1990 |
 |
Delorme et al. 1997 | 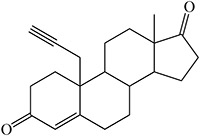 |
Metcalf et al. 1981; Johnston et al. 1991 |
Figure 15.

The structure of the adduct formed with propyne (Ortiz de Montellano et al. 1981b) after demetallation and esterification of the carboxyl groups. The four pyrrole rings of the porphyrin are labeled A-D. In this instance, the covalent bond was formed exclusively with pyrrole ring A, but other pyrrole rings can be alkylated in other adducts (e.g., Ortiz de Montellano et al. 1982a).
CYP102 (P450BM3) oxidizes 17-octadecynoic acid (17-ODYA) to the ω-2 hydroxylated derivative, 16-hydroxy-17-octadecynoic acid, rather than oxidizing the triple bond (Shirane et al. 1993). However, the 16-hydroxylated metabolite, whether generated in situ or added exogenously, is able to inactivate the enzyme in a mechanism-dependent manner to give a porphyrin adduct comparable to that in Figure 15. The inactivation and heme loss are not prevented by glutathione, as expected if the inactivating species is not a diffusible intermediate. The fact that covalent binding to the heme group is not prevented by agents such as N-acetylcysteine or glutathione was first reported in a related case for the inactivation mediated by 1-octyne (White et al. 1984).
The mechanism which covalently attaches acetylenic inhibitors to the cytochrome P450 prosthetic heme group does not proceed through a ketene metabolite, although a ketene may also be formed. If a ketene is formed, it may contribute to enzyme inactivation, as discussed below, but that inactivation reflects covalent binding to the protein rather than the heme group. The structure of the heme adduct itself establishes that it was not formed by addition of a heme nitrogen to the ketene, as that would produce an N-acyl (N-COCH2R) rather than N-alkyl (N-CH2COR) adduct. This inference is confirmed by the finding that replacement of the acetylenic hydrogen by a deuterium gives rise to a significant isotope effect on ketene formation, but no isotope effect on inactivation of the P450 enzyme (Ortiz de Montellano and Komives 1985; Komives and Ortiz de Montellano 1987). Furthermore, the oxidation of a series of substituted phenylacetylenes gives rise to a linear free energy correlation with ρ = −2.2 for formation of the ketene, whereas the rate of inactivation is the same for all the analogues (Komives and Ortiz de Montellano 1987). In consequence, the partition ratio (i.e., turnovers/inactivation event) decreases from 38 to 4 in going from para-methyl- to para-nitro-phenylacetylene. Heme N-alkylation is thus most consistent with addition of the cytochrome P450 activated oxygen to the internal, not terminal, carbon of the acetylenic group in a process in which the porphyrin nitrogen atom is simultaneously bound to the terminal carbon, as shown schematically in Figure 16.
Figure 16.

Schematic mechanism for autocatalytic alkylation of the heme prosthetic group of cytochrome P450 in the oxidation of a terminal acetylene. Delivery of the reactive oxygen to the substituted acetylene terminus leads to heme alkylation rather than ketene formation.
The relationship between the carbon atom to which the activated oxygen is delivered and the outcome of the reaction is highlighted in the inactivation of CYP1A1 by 1-ethynylpyrene and phenylacetylene (Chan et al. 1993). Although both agents are converted to the acetic acid derivatives by oxidation of the triple bond, only phenylacetylene detectably inactivates the enzyme through formation of a heme adduct. However, quantitative analysis shows that 1-ethynylpyrene inactivates the enzyme by binding covalently to the P450 protein in essentially a 1:1 ratio. The factors that favor heme alkylation with phenylacetylene, but protein alkylation with 1-ethynylpyrene, are not clear, but the results indicate that oxygen delivery to the substituted carbon of the triple bond leads to heme alkylation. In contrast, oxygen delivery to the unsubstituted triple bond carbon leads to the ketene (and therefore carboxylic acid) with both substrates, but only in the case of 1-ethynylpyrene is the ketene primarily responsible for enzyme inactivation.
Reversible carbene complexes
The inactivation of CYP2E1 and its T303A mutant by tert-butyl-acetylene (tBA) and tert-butyl 1-methyl-2-propynyl ether (tBMP) uncovered another nuance in the inactivation process (Blobaum et al. 2002). Inactivation of CYP2E1 by these agents resulted in (i) loss of the ability to deethylate 7-ethoxy-4-(trifluoromethyl)coumarin, (ii) a proportionate loss of the P450 heme chromophore and heme content, (iii) the formation of two isomeric heme adducts and, to some extent, (iv) covalent binding of the acetylenic substrates to the protein. Comparable results were obtained with the CYP2E1 T303A mutant, particularly with tBA, except that no protein binding was detected. The surprise, however, was that dialysis resulted in reversal of these inhibition-related parameters, albeit only with the T303A mutant and not the parent CYP2E1 protein. Furthermore, a mutation is not essential for reversible inhibition to occur, as tBMP inactivated CYP2B4 with the formation of N-alkyl heme adducts, although in this instance the loss of activity was greater than the loss of P450 chromophore or heme content (von Weyman et al. 2004a). Detailed analysis revealed the formation of an inhibitory complex that could be partially reversed by dialysis, but which converted irreversibly to an N-alkylporphyrin adduct when exposed to acid (Blobaum et al. 2005). A T302A mutation in CYP2B4 increased the formation of this inhibitory complex. To explain these results the authors proposed the formation of a reversible peroxy complex (Figure 17) that could revert to the intact enzyme via a mechanism proceeding through an oxirene intermediate (Blobaum et al. 2004a; Blobaum 2006).
Figure 17.

Structures of tBA and tBMP, and a structure proposed for the reversible intermediate formed in the reaction of the CYP2E1 T303A mutant with tBA and tBMP (Blobaum 2006).
Key clues to the nature of the observed reversible activity and heme loss are provided by (a) the finding that the inactivation reaction was associated with the appearance of a broad heme chromophore with a maximum at ~485 nm (Blobaum et al. 2004b), and (b) that the N-alkylporphyrin heme adducts were not obtained if the protein sample was not acidified (Blobaum et al. 2004a; Blobaum et al. 2005). These results, and particularly the highly red shifted Soret band, are incompatible with the formation of the proposed peroxyiron complex and are much better explained by the formation of a carbene complex.
The formation of heme iron-carbene complexes is well established in, for example, the reactions of polyhalo alkanes and diazo esters with hemoproteins (Simonneaux and Le Maux 2006; Guilard and Kadish 1988). In the case of P450 enzymes, these complexes have long wavelength absorption maxima in the 450 to 480 nm range (Simonneaux and Le Maux 2006; Wolf et al. 1977; Battioni et al. 1983) that are consistent with the broad absorbance at 485 nm for the reversible complex reported by Blobaum et al. (2004b). In contrast, no cytochrome P450 oxy anion-iron heme complex has such a red-shifted absorption maximum. The crystal structure of a ketocarbene complex formed by a diazoacetate with a hemoprotein (albeit not a P450 enzyme) has been reported (Lewis et al. 2018), and the cytochrome P450-catalyzed insertion of 2-diazoester derived carbenes into olefins has developed into a broad synthetic strategy (Coelho et al. 2013; Su and Liu 2018). Furthermore, the shift of carbene complexes to give N-alkyl heme adducts has been extensively investigated (e.g., Simonneaux and Le Maux 2006; Guilard and Kadish 1988). Finally, ketocarbenes are part of the manifold of theoretical species potentially formed by triple bond oxidation (Figure 18) (Lewars 1983). These considerations suggest that acetylene oxidation yields a carbene complex that can either undergo a proton-assisted shift to give the observed heme adducts or, in the absence of proton donation by Thr303 (or acidification), undergo time-dependent hydrolytic decomposition to regenerate the parent heme (Figure 19).
Figure 18.

The theoretically accessible manifold of intermediates in the oxidation of a triple bond.
Figure 19.

Proposed formation of a carbene-iron complex in the oxidation of tBA (R = tert-butyl) by the CYP2E1 T303A mutant. With proton donation, the complex migrates to give a nitrogen alkylated heme adduct, but in its absence it can decompose to regenerate the parent heme.
The possibility exists that all the heme N-alkylations associated with oxidation of a triple bond proceed via carbene iron complexes that are too unstable towards migration to a nitrogen of the heme to be detectable except in a few situations, such as those reported by Blobaum (2006).
Protein adducts formed with monosubstituted acetylenes.
The final general mechanism for catalysis-dependent cytochrome P450 inactivation follows naturally from the formation of highly reactive ketene metabolites in the oxidation of acetylenes. Covalent binding of the ketene to a protein was mentioned above in the context of the CYP2B6-catalyzed oxidation of selegiline (Figure 5), a compound that irreversibly inactivates CYP2A6 (Siu and Tyndale 2008). If the ketene binds covalently to the P450 protein within the active site, or in some other manner that interferes with substrate binding or catalysis, irreversible inhibition is the result.
11-Dodecynoic acid (11-DDYA) and 10-undecynoic acid (10-UDYA) (Figure 20) inactivate cytochrome P450 enzymes in liver microsomes that oxidize lauric acid to its ω- and ω-1 hydroxylated derivatives (Ortiz de Montellano and Reich 1984; Cajacob et al. 1988). This inactivation is accomplished without causing a measurable decrease in the spectroscopically observable cytochrome P450 concentration or in benzphetamine or N-methyl p-chloroaniline N-demethylase activities, oxidations presumably catalyzed by other P450 enzymes. Quantitation of the process with liver microsomes from clofibrate-induced rats incubated with 10-UDYA reveals a highly efficient process in which 1 mol of 1,11-undecandioic acid is produced per mole of inactivated enzyme, indicating that on average each enzyme is inactivated by the second substrate molecule that it oxidizes (Cajacob et al. 1988). SDS-PAGE analysis of the incubation of radiolabeled 10-UDYA with the microsomes shows that covalent binding of the labeled fatty acid to the P450 protein accounts for its inactivation.
Figure 20.

Structures of 11-dodecynoic acid (11-DDYA) and 10-undecynoic acid (10-UDYA) and the diacid metabolites formed from them by cytochrome P450.
The lauric acid ω-hydroxylase from Vicia sativa is similarly inactivated by 11-DDYA in a time- and concentration-dependent process that simultaneously produces 1,12-dodecanedioic acid (Salaün et al. 1988; Helvig et al. 1997). However, no loss of heme chromophore was observed, in agreement with a protein binding mechanism. Incubation in the presence of β-mercaptoethanol decreased overall covalent binding of the radiolabeled 11-DDYA to microsomal protein, but did not prevent the loss of ω-hydroxylase activity, as expected for a true mechanism-based inhibitor. Nevertheless, the results suggest that some of the ketene intermediate diffused out of the active site and bound covalently to other proteins, but the covalent binding responsible for enzyme inactivation occurred before diffusion from the active site.
In contrast, as mentioned earlier, 17-octadecynoic acid (17-ODYA), a compound closely related to 11-DDYA except for the additional length of the hydrocarbon chain, inactivates CYP102 via formation of a heme, not protein, covalent adduct (Shirane et al. 1993). Whether heme or protein alkylation occurs, or both, is therefore a function of both the specific inhibitor and the specific cytochrome P450 enzyme.
2-Ethynylnaphthalene is a mechanism-based inactivator of rat and rabbit, but not human, CYP1A2 (Hammons et al. 1989; Yun et al. 1992). Using radiolabeled 2-ethynylnaphthalene, single radiolabeled peptides were isolated from the rat and rabbit enzymes, with the sequences (L(S)QQYGDVLQIR) and (FQELMAAVGR), respectively (Yun et al. 1992). 2-Ethynylnaphthalene also inactivates CYP2B1 in a process that approaches 1:1 covalent binding of the radiolabeled agent to the protein without loss of the spectroscopically observable cytochrome P450 chromophore (Roberts et al. 1993). In this case the radiolabeled peptide had the sequence ISLLSLFFAGTETSSTTLRYGFLLM. Mass spectrometric analysis indicated that 2-ethynylnaphthalene was specifically attached to the peptides (Roberts et al. 1994, 1995a, 1995b). Other aryl acetylenes, including 9-ethynylphenanthrene and 7-ethynylcoumarin, have been shown to bind covalently to the protein of various P450 enzymes (Hopkins et al. 1992; Roberts et al. 1995a, 1997; Regal et al. 2000).
The well-characterized inactivation of CYP2B4 by 4-tert-butylphenylacetylene offers a particularly clear insight into cytochrome P450 inactivation via protein modification. This particular inactivation reaction is highly efficient in that every oxidation of the inhibitor results in covalent modification of the protein, i.e., every ketene molecule that is formed binds to the protein, inhibiting the enzyme (Zhang et al. 2009, 2011). Quantitative analysis indicates that in each protein one molecule of the inhibitor is bound to Thr302, causing a 5 nm shift of the Soret band. The addition of benzphetamine, a normal substrate, to the inactivated protein fails to cause either the normal spin shift or the greatly enhanced reduction of the protein normally observed on binding of benzphetamine. The conclusion that covalent attachment of the inhibitor to Thr302 sterically interferes with productive substrate binding is supported by a resonance Raman studies (Mak et al. 2010). An even more precise view of this steric interference is provided by a crystal structure of the inactivated protein, which shows the oxidatively activated 4-tert-butylphenylacetylene molecule attached via an ester link to the oxygen of Thr302 (Gay et al. 2011). This is the adduct expected from reaction of the threonine hydroxyl with the ketene formed from the acetylene group. A second crystal structure shows that 9-ethynylphenanthrene also inactivates CYP2B4 by binding covalently through an ester bond to Thr302 (Zhang et al. 2013).
Covalent binding of the ketene metabolite from 4-tert-butylphenylacetylene to Thr302 in CYP2B1 and CYP2B6 establishes the importance of this residue in the inactivation of these homologous proteins (Lin et al. 2010, 2011). However, even these proteins can be inactivated by covalent attachment of the ketene to other residues, as illustrated by the inactivation of CYP2B1 and CYP2B6 by 17α-ethynylestradiol (Kent et al. 2006, 2008). Thus, mass spectrometric analysis of digests of the inactivated CYP2B1 protein demonstrated that the ketene was bound to Ser360. However, mutation of Ser360 to an alanine did not prevent inactivation, indicating that alternative reaction sites were available. Mutating CYP1B1 Thr205 to a valine protected the enzyme from inactivation by 17α-ethynylestradiol and made it more resistant to inactivation by 2-ethynylnapthalene (Lin et al. 2003). CYP1B1 formed three identifiable metabolites from this substrate, one of which correlated with inactivation of the enzyme, and the T205V mutation suppressed its formation (Lin et al. 2004). This finding suggests that Thr205 may help to position 17α-ethynylestradiol so that oxidation of the triple bond occurs. Other mutations also protect the enzyme from inactivation, possibly in a similar manner (Von Weyman et al. 2004b).
CYP3A4 and CYP3A5 are also inactivated by 17α-ethynylestradiol (Guengerich 1988; Lin et al. 2002; Lin and Hollenberg 2007), but with these enzymes the inhibitor is covalently bound to both the heme and the protein. Analysis of the metabolites trapped by glutathione led to the suggestion that a sufficiently stable oxirene was formed that could be trapped by glutathione addition to one or the other of the oxirene carbon atoms (Lin and Hollenberg 2007). As already discussed, oxirenes are unlikely to form as actual intermediates in the reaction manifold leading to ketenes or carbenes. Heme and protein adducts are also observed in the inactivation of CYP2J2 by 17-octadecynoic acid (Lin et al. 2017, 2018). In instances where both protein and heme adducts are formed, the heme adducts are formed by covalent attachment to a porphyrin nitrogen, whereas formation of a ketene metabolite accounts for the protein binding.
Protein modification by disubstituted acetylenes
Disubstituted acetylenes can also inactivate cytochrome P450 enzymes (Table 3), but their ability to do so is less predictable, as illustrated by the examples in Table 4 of agents that cause no inactivation. One factor that contributes to the lesser ability of disubstituted acetylenes to inactivate P450 enzymes is that oxidation of the substituents may compete more effectively with triple bond oxidation. A second important factor is that there are no validated examples of covalent binding to the heme group due to the oxidation of a disubstituted acetylene, presumably because steric factors disfavor the reaction.
Table 3.
Examples of the inactivation of cytochrome P450 enzymes by disubstituted acetylenes
| Compound | Reference | Compound | Reference |
|---|---|---|---|
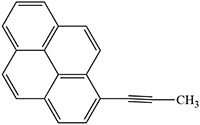 |
Foroozesh et al. 1997 | 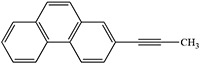 |
Foroozesh et al.. 1997 |
 |
Burger et al. 1987 | 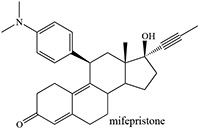 |
Reilly et al. 1999; He et al. 1999; Lin et al. 2009; Khan et al. 2002 |
Table 4.
Examples of disubstituted acetylenes not causing detectable cytochrome P450 inactivation
| Compound | Reference | Compound | Reference |
|---|---|---|---|
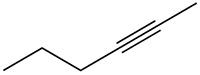 |
Ortiz de Montellano and Kunze 1980b | 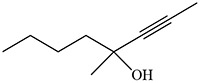 |
Ortiz de Montellano and Kunze 1980b |
| White 1980 | 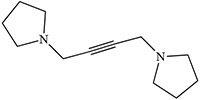 |
White 1980 |
Nevertheless, disubstituted acetylenes can inactivate P450 enzymes through oxidation of the triple bond with concurrent migration of one of the alkyl group substituents to give a ketene, as illustrated in Figure 11 for the oxidation of 1-biphenyl-1-propyne. The ketene can then inactivate the enzyme by covalent attachment to the protein, as occurs with monosubstituted acetylenes. This may be the mechanism for the inactivation of cytochrome P450 by compounds with disubstituted triple bonds in Table 3 and in other studies (e.g., Nagahisa et al. 1983; Foroozesh et al. 1997). However, in the absence of direct evidence implicating the ketene metabolite in protein binding, an alternative mechanism must be considered, as illustrated by the oxidation of 1-octyne described below.
1-Octyne, a monosubstituted acetylene, was reported to inactivate hepatic cytochrome P450 with the formation of both protein and heme covalent adducts (White et al. 1984). The substrate itself was oxidized in the incubation system to 1-octyne-3-one, which was readily trapped by thiol agents via a Michael addition reaction. Synthetic 1-octyne-3-one did not react with the P450 heme group, as expected, but did react with thiols. Furthermore, thiol agents had no effect on heme adduct formation, but did decrease protein binding. The formation of 1-octyne-3-one could therefore account for protein binding without requiring oxidation of the triple bond, although ketene formation may also have occurred and may have contributed to protein covalent binding. The chemical reactivity of conjugated acetylenic ketones with thiols is illustrated by the formation of glutathione conjugates with acalabrutinib, an anticancer agent with a conjugated ketone function that covalently binds to a cysteine thiol in Bruton tyrosine kinase, the target protein of the drug (Podoll et al., 2019). These results suggest a mechanism for protein alkylation and P450 inactivation that does not involve triple bond oxidation. In this vein, 11-DDYA (Figure 20) and its in-chain analogue 10-dodecynoic acid have been shown to inactivate and bind covalently to CYP2B4 (Helvig et al. 1997). The mechanism of action of 11-DDYA, a terminal acetylene, is straightforward and has already been discussed, but the inhibitory activity of 10-dodecynoic acid, which has a disubstituted triple bond, is less clear. Metabolism experiments identified 12-hydroxy-10-dodecynoic acid as the only metabolite of 10-dodecynoic acid. If the terminal hydroxyl was further oxidized to the aldehyde, the resulting Michael acceptor may have been responsible for protein binding and P450 inactivation, although oxidation of the triple bond to a ketene cannot be excluded. In this context, it is important to note that the formation of diffusible reactive metabolites of acetylenes, whether ketenes or triple bonds conjugated to carbonyl groups, can lead to protein alkylation reactions and toxicities unrelated to the inactivation of cytochrome P450 enzymes.
A final anomaly is provided by the inactivation of cytochrome P450scc (CYP11A1) by a disubstituted acetylenic sterol. CYP11A1, which specifically cleaves the side-chain of cholesterol between carbons 20 and 22 to give pregnenolone, is inactivated by substrates with a triple bond between carbons 22 and 23 (Figure 21) (Nagahisa et al. 1983). Pregnenolone is formed in these reactions, but the exact nature of the oxidation and P450 inactivation is unclear, although the de-ethynylation reaction in Figure 10 may be instructive in this regard. Nevertheless, the results suggest that the triple bond is oxidized to an intermediate that can inactivate the enzyme and may or may not be involved in the formation of pregnenolone.
Figure 21.

Oxidation of disubstituted acetylenes to pregnenolone by CYP11A1.
Specificity
The oxidation of carbon-carbon triple bonds and the associated cytochrome P450 inactivation must satisfy two initial conditions: (i) the molecule must bind to the cytochrome P450 enzyme, and (ii) the triple bond must be positioned so that its oxidation is competitive with alternative oxidative reactions. Addition of the activated oxygen to the internal carbon of a monosubstituted triple bond will then result in heme alkylation, with the caveat that a carbene-iron complex may precede formation of the N-alkyl heme adduct. In contrast, addition of the activated oxygen to the unsubstituted acetylenic carbon, with few exceptions, produces a ketene metabolite. In the case of disubstituted acetylenes, heme alkylation does not occur, but ketene formation may occur. The factors that determine to which carbon of a triple bond the reactive oxygen is added remain unclear.
If a ketene metabolite participates in mechanism-based P450 inactivation, it must reside within the active site or entrance channel long enough for protein acylation to occur. If the residence time is low, or there is no suitably reactive amino acid, the ketene may diffuse out of the active site to react more randomly with nucleophilic protein residues, with agents such as glutathione, or with water to give an acetic acid derivative. The binding affinity of the parent acetylene and the ketene metabolite to the P450 protein are therefore relevant to the efficiency of inactivation by ketene metabolites.
The interplay of the factors that govern triple bond oxidation can be illustrated with several examples. As noted earlier, 2-ethynylnaphthalene inactivates rat and rabbit, but not human, CYP1A2 despite ~80% sequence identity among the three proteins (Yun et al. 1992). Similarly, although there is 83% sequence identity between the protein sequences of CYP3A4 and CYP3A5, only CYP3A4 is inactivated by mifepristone, a disubstituted acetylene (Table 3). Furthermore, mutation of individual CYP3A4 active site residues to those found in CYP3A5 did not confer protection against mifepristone inactivation, the CYP3A4 single-mutant proteins being inactivated as effectively as the wild-type. The authors argued that orientation of the substrate to favor oxidation of the triple bond was not achieved in CYP3A5, and therefore mifepristone did not inactivate that protein (Khan et al. 2002). In the same vein, 7-coumarin propargyl ether inactivates CYP3A4, but not CYP3A5 (Sridar et al. 2008). In the CYP3A4 reaction there was a comparable loss of activity and heme, suggesting inactivation through N-alkylation of the heme group, but glutathione-trapped ketene metabolites were also obtained.
Norethisterone and MDL 18,962 are sterols that differ critically in that an ethynyl group is attached to C17 in the former and a propynyl to C10 in the latter (structures in Table 2) (Johnston et al. 1991). Pretreatment of rats with norethisterone increased pentobarbital sleeping time and caused the accumulation of hepatic N-alkylated porphyrins, a marker of prosthetic heme alkylation, whereas these effects were not observed with MDL 18,962. The acetylene function in MDL 18,962 is apparently not appropriately placed for catalytic activation. In contrast, MDL 18,962 does inactivate human CYP19, showing that there is no inherent difficulty to oxidation of its triple bond (Metcalf et al. 1981).
Finally, CYP2B4 and CYP2B5, which differ by 12 amino acid residues, are differentially inactivated by 2-ethynylnaphthalene (Strobel et al. 1999). 2-Ethynylnaphthalene inhibits CYP2B4, whereas CYP2B5 oxidizes this compound to 2-naphthylacetic acid without being inactivated. Based on molecular dynamics simulations, it was postulated that the ketene in CYP2B5 was more loosely held and more rapidly hydrolyzed to the acid. Mutation of Ile363 in CYP2B4 to a valine, the residue in CYP2B5, conferred protection against inactivation by 2-ethynylnaphthalene, whereas mutation of the corresponding Val363 in CYP3A5 to an isoleucine made that protein susceptible to inactivation by the same agent.
Acknowledgments
Funding
This work was financially supported by grant AI074824 from the National Institutes of Health.
Footnotes
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Abdel-Aziz MT, Williams KIH. 1969. Metabolism of 17α-ethynylestradiol and its 3-methyl ether by the rabbit; an in vivo d-homoannulation. Steroids. 13:809–820. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz MT, Williams KIH. 1970. Metabolism of radioactive 17α-ethynylestradiol by women. Steroids 15:695–710. [DOI] [PubMed] [Google Scholar]
- Battioni P, Mahy JP, Delaforge M, Mansuy P. 1983. Reaction of monosubstituted hydrazines and diazenes with rat-liver cytochrome P450. Eur. J. Biochem 134:241–248. [DOI] [PubMed] [Google Scholar]
- Blakey DC, White INH. 1986. Destruction of cytochrome P-450 and formation of green pigments by contraceptive steroids in rat hepatocyte suspensions. Biochem Pharmacol. 35:1561–1567. [DOI] [PubMed] [Google Scholar]
- Blanksby SJ, Ellison GB. 2003. Bond dissociation energies of organic molecules. Accts Chem Res. 36:255–263. [DOI] [PubMed] [Google Scholar]
- Blobaum AL. 2006. Mechanism-based inactivation and reversibility: is there a new trend in the inactivation of cytochrome P450 enzymes? Drug Metab Disp. 34:1–7. [DOI] [PubMed] [Google Scholar]
- Blobaum AL, Kent UM, Alworth WL, Hollenberg PF. 2002. Mechanism-based inactivation of cytochromes P450 2E1 and 2E1 T303A by tert-butyl acetylenes: characterization of reactive intermediate adducts to the heme and apoprotein. Chem Res Toxicol. 15:1561–1571. [DOI] [PubMed] [Google Scholar]
- Blobaum AL, Kent UM, Alworth WL, Hollenberg PF. 2004a. Novel reversible inactivation of cytochrome P450 2E1 T303A by tert-butyl acetylene: the role of threonine 303 in proton delivery to the active site of cytochrome P450 2E1. J Pharmacol Exp Therap. 310:281–290. [DOI] [PubMed] [Google Scholar]
- Blobaum AL, Lu Y, Kent UM, Wang S, Hollenberg PF. 2004b. Formation of a novel reversible cytochrome P450 spectral intermediate: role of threonine 303 in P450 2E1 inactivation. Biochemistry 43:11942–11952. [DOI] [PubMed] [Google Scholar]
- Blobaum AL, Harris DL, Hollenberg PF. 2005. P450 active site architecture and reversibility: inactivation of cytochromes P450 2B4 and 2B4 T302A by tert-butyl acetylenes. Biochemistry. 44:3831–3844. [DOI] [PubMed] [Google Scholar]
- Burger A, Roussel JP, Colobert F, Kappler C, Hetru C, Luu B, Hoffmann JA. 1987. In vitro studies on potential selective and irreversible inhibitors of enzymes involved in the biosynthesis of ecdysone. Pest Biochem Physiol. 29:197–208. [Google Scholar]
- CaJacob CA, Chan WK, Shephard E, Ortiz de Montellano PR. 1988. The catalytic site of rat hepatic lauric acid ω-hydroxylase. Protein versus prosthetic heme alkylation in the ω-hydroxylation of acetylenic fatty acids. J Biol Chem. 263:18640–18649. [PubMed] [Google Scholar]
- Chan WK, Sui Z, Ortiz de Montellano PR. 1993. Determinants of protein modification versus heme alkylation: Inactivation of cytochrome P450 1A1 by 1-ethynylpyrene and phenylacetylene. Chem Res Toxicol. 6:38–45. [DOI] [PubMed] [Google Scholar]
- Coelho PS, Brustad EM, Kannan A, Arnold FH. 2013. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science. 339:307–310. [DOI] [PubMed] [Google Scholar]
- Correia MA, Hollenberg PF. 2015. Inhibition of cytochrome P450 enzymes, In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed. New York: Springer; p. 177–260. [Google Scholar]
- Cutler AJ, Rose PA, Squires TM, Loewen MK, Shaw AC, Quail JW, Krochko JE, Abrams SR. 2000. Inhibitors ofabscisic acid 8’-hydroxylase. Biochemistry. 39:13614–13624. [DOI] [PubMed] [Google Scholar]
- Delorme C, Piffeteau A, Sobrio F, Marquet A. 1997. Mechanism-based inactivation of bovine cytochrome P-45011β by 18-unsaturated progesterone derivatives. Eur J Biochem. 248:252–260. [DOI] [PubMed] [Google Scholar]
- El Masri AM, Smith JN, Williams RT. 1958. The metabolism of alkylbenzenes: phenylacetylene and phenylethylene (styrene). Biochem J. 68:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervin KM, Gronert S, Barlow SE, Gilles MK, Harrison AG, Bierbaum VM, DePuy CH, Lineberger WC, Ellison GB. 1990. Bond strengths of ethylene and acetylene. J Am Chem Soc. 112:5750–5759. [Google Scholar]
- Fan PW, Gu C, Marsh SA, Stevens JC. 2003. Mechanism-based inactivation of cytochrome P450 2B6 by a novel terminal acetylene inhibitor. Drug Metab Dispos. 31:28–36. [DOI] [PubMed] [Google Scholar]
- Felig J, Barnes JR, Rachlin AI, O’Brien JP, Focella A. 1970. Substituted phenyl 2-propynyl ethers as carbamate synergists. J Agric Food Chem. 18:78–80. [Google Scholar]
- Foroozesh M, Primrose G, Guo Z, Bell LC, Alworth WL, Guengerich FP. 1997. Aryl acetylenes as mechanism-based inhibitors of cytochrome P450-dependent monooxygenase enzymes. Chem Res Toxicol. 10:91–102. [DOI] [PubMed] [Google Scholar]
- Freudenthal RI, Amerson E, Martin J, Wall ME. 1974. The effect of norethynodrel, norethindrone and ethynodiol diacetate on hepatic microsomal drug metabolism. Pharmacol Res Commun. 6:457–468. [DOI] [PubMed] [Google Scholar]
- Gay SC, Zhang H, Wilderman PR, Roberts AG, Liu T, Li S, Lin H, Zhang Q, Woods VL, Stout CD, Hollenberg PF, Halpert JR. 2011. Structural analysis of mammalian cytochrome P450 2B4 covalently bound to the mechanism-based inactivator tert-butylphenylacetylene: Insight into partial enzymatic activity. Biochemistry. 50:4903–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP. 1988. Oxidation of 17α-ethynylestradiol by human liver cytochrome P450. Molec Pharmacol. 33:500–508. [PubMed] [Google Scholar]
- Guengerich FP. 1990. Mechanism-based inactivation of human liver microsomal cytochrome P-450 IIIA4 by gestodene. Chem Res Toxicol. 3:363–371. [DOI] [PubMed] [Google Scholar]
- Guilard R, Kadish M. 1988. Some aspects of organometallic chemistry in metalloporphyrin chemistry: synthesis, chemical reactivity, and electrochemical behavior of porphyrins with metal-carbon bonds. Chem Rev. 88:1121–1146. [Google Scholar]
- Hammons GJ, Alworth WL, Hopkins NE, Guengerich FP, Kadlubar FF. 1989. 2-Ethynylnaphthalene as a mechanism-based inactivator of the cytochrome P450 catalyzed N-oxidation of 2-naphthylamine. Chem Res Toxicol. 2:367–374. [DOI] [PubMed] [Google Scholar]
- He K, Woolf TF, Hollenberg PF. 1999. Mechanism-based inactivation of cytochrome P-450-3A4 by mifepristone (RU486). J Pharmacol Exp Therap. 288:791–797. [PubMed] [Google Scholar]
- Helton ED, Williams MC, Goldzieher JW. 1977. Oxidative metabolism and de-ethynylation of 17α-ethynylestradiol by baboon liver microsomes. Steroids. 30:71–83. [DOI] [PubMed] [Google Scholar]
- Helvig C, Alayrac C, Mioskowski C, Koop D, Poullain D, Durst F, Salaün JP. 1997. Suicide inactivation of cytochrome P450 by midchain and terminal acetylenes. A mechanistic study of inactivation of a plant lauric acid ω-hydroxylase. J Biol Chem. 272:414–421. [DOI] [PubMed] [Google Scholar]
- Herz R, Koelz HR, Haemmerli UP, Benes I, Blum AL. 1978. Inhibition of hepatic demethylation of aminopyrine by oral contraceptive steroids in humans. Eur J Clin Invest. 8:27–30. [DOI] [PubMed] [Google Scholar]
- Hopkins NE, Foroozesh MK, Alworth WL. 1992. Suicide inhibitors of cytochrome P450 1A1 and P450 2B1. Biochem Pharmacol. 44:787–796. [DOI] [PubMed] [Google Scholar]
- Johnston JO, Wright CL, Leeson GA. 1991. Regioselectivity of metabolic activation of acetylenic steroids by hepatic cytochrome P450 isozymes. Steroids. 56:180–184. [DOI] [PubMed] [Google Scholar]
- Kanhai W, Koob M, Dekant W, Henschler D. 1991. Metabolism of 14C-dichloroethyne in rats. Xenobiotica. 21:905–916. [DOI] [PubMed] [Google Scholar]
- Kent UM, Mills DE, Rajnarayaanan RV, Alworth WL, Hollenberg PF. 2002. Effect of 17-α-ethynylestradiol on activities of cytochrome P450 2B (P450 2B0 enzymes: Characterization of inactivation of P450s 2B1 and 2B6 and identification of metabolites. J Pharmacol Exp Therap. 300:549–558. [DOI] [PubMed] [Google Scholar]
- Kent UM, Lin H, Mills DE, Regal KA, Hollenberg PF. 2006. Identification of 17α-ethynylestradiol-modified active site peptides and glutathione conjugates formed during metabolism and inactivation of P450s 2B1 and 2B6. Chem Res Toxicol. 19:279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent UM, Sridar C, Spahlinger G, Hollenberg PF. 2008. Modification of serine 360 by a reactive intermediate of 17α-ethynylestradiol results in mechanism-based inactivation of cytochrome P450s 2B1 and 2B6. Chem Res Toxicol. 21:1956–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan KK, He YQ, Correia MA, Halpert JR. 2002. Differential oxidation of mifepristone by cytochromes P450 3A4 and 3A5: selective inactivation of P450 3A4. Drug Metab Dispos. 30:985–990. [DOI] [PubMed] [Google Scholar]
- Komives EA, Ortiz de Montellano PR. 1987. Mechanism of oxidation of π bonds by cytochrome P450. Electronic requirements of the transition state in the turnover of phenylacetylenes. J Biol Chem. 262:9793–9802. [PubMed] [Google Scholar]
- Kunze KL, Mangold BLK, Wheeler C, Beilan HS, Ortiz de Montellano PR. 1983. The cytochrome P-450 active site. Regiospecificity of the prosthetic heme alkylation by olefins and acetylenes. J Biol Chem. 258:4202–4207. [PubMed] [Google Scholar]
- Lewars EG. 1983. Oxirenes. Chem Rev. 83:519–534. [Google Scholar]
- Lewis RD, Garcia-Borrás M, Chalkley MJ, Buller AR, Houk KN, Kan SBJ, Arnold FH. 2018. Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase. Proc Natl Acad Sci USA. 115:7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HL, Hollenberg PF. 2007. The inactivation of cytochrome P450 3A5 by 17α-ethynylestradiol is cytochrome b5-dependent: metabolic activation of the ethynyl moiety leads to the formation of glutathione conjugates, a heme adduct, and covalent binding to the apoprotein. J Pharmacol Exp Therap. 321:276–287. [DOI] [PubMed] [Google Scholar]
- Lin HL, Kent UM, Hollenberg PF. 2002. Mechanism-based inactivation of cytochrome P450 3A4 by 17α-ethynylestradiol: Evidence for heme destruction and covalent binding to protein. J Pharmacol Exp Therap. 301:160–167. [DOI] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Waskell L, Hollenberg PF. 2003. Threonine-205 in the F helix of P450 2B1 contributes to androgen 16β-hydroxylation activity and mechanism-based inactivation. J Pharmacol Exp Therap. 306:744–751. [DOI] [PubMed] [Google Scholar]
- Lin HL, Kent UM, Zhang H, Waskell L, Hollenberg PF. 2004. The functional role of threonine-205 in the mechanism-based inactivation of P450 2B1 by two ethynyl substrates: the importance of the F helix in catalysis. J Pharmacol Exp Therap. 311:855–863. [DOI] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Hollenberg PF. 2009. Metabolic activation of mifepristone [RU486; 17β-hydroxy-11β-(4-dimethylaminophenyl)-17α-(1-propynyl)-estra-4,9-dien-3-one] by mammalian cytochromes P450 and the mechanism-based inactivation of human CYP2B6. J Pharmacol Exp Therap. 329:26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Jushchyshyn M, Hollenberg PF. 2010. Covalent modification of Thr302 in cytochrome P450 2B1 by the mechanism-based inactivator 4-tert-butylphenylacetylene. J Pharmacol Exp Therap. 333:663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Pratt-Hyatt MJ, Hollenberg PF. 2011. Thr302 is the site for the covalent modification of human cytochrome P450 2B6 leading to mechanism-based inactivation by tert-butylphenylacetylene. Drug Metab Dispos 39:2431–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Walker V, D’Agostino J, Hollenberg PF. 2017. Heme modification contributes to the mechanism-based inactivation of human cytochrome P450 2J2 by two terminal acetylenic compounds. Drug Metab Dispos. 45:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HL, Zhang H, Hollenberg PF. 2018. Formation of both heme and apoprotein adducts contributes to the mechanism-based inactivation of human CYP2J2 by 17α-ethynylestradiol. Drug Metab Dispos. 46:813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak PJ, Zhang H, Hollenberg PF, Kincaid JR. 2010. Defining the structural consequences of mechanism-based inactivation of mammalian cytochrome P450 2B4 using resonance Raman spectroscopy. J Am Chem Soc. 132:1494–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawhinney RC, Goddard JD. 2003. Assessment of density functional theory for the prediction of the nature of the oxirene stationary point. J Molec Struct (Theochem). 629:263–270. [Google Scholar]
- McMahon RE, Turner JC, Whitaker GW, Sullivan HR. 1981. Deuterium-isotope effect in the biotransformation of 4-ethynylbiphenyls to 4-biphenylacetic acids by rat hepatic microsomes. Biochem Biophys Res Commun. 99:662–667. [DOI] [PubMed] [Google Scholar]
- Metcalf BW, Wright CL, Burkhart JP, Johnston JO. 1981. Substrate-induced inactivation of aromatase by allenic and acetylenic steroids. J Am Chem Soc. 103:3221–3222. [Google Scholar]
- Mutlib A, Chen H, Shockcor J, Espina R, Chen S, Cao K, Du A, Nemeth G, Prakash S, Gan L-S. 2000. Characterization of novel glutathione adducts of a non-nucleoside reverse trascriptase inhibitor, (S)-6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-3,4-dihydro-2(1H)-quinazolinone (DPC 961), in rats. Possible formation of an oxirene metabolic intermediate from a disubstituted alkyne. Chem Res Toxicol. 13:775–784. [DOI] [PubMed] [Google Scholar]
- Nagahisa A, Spencer RW, Orme-Johnson WH. 1983. Acetylenic mechanism-based inhibitors of cholesterol side chain cleavage by cytochrome P-450scc. J Biol Chem. 258:6721–6723. [PubMed] [Google Scholar]
- O’Malley K, Stevenson IH, Crooks J. 1972. Impairment of human drug metabolism by oral contraceptive steroids. Clin Pharmacol Therap. 13:552–557. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR. 1985. Alkenes and Alkynes In: Anders MW editor. Bioactivation of Foreign Compounds. New York: Academic Press, p. 121–155. [Google Scholar]
- Ortiz de Montellano PR. 2015. Substrate oxidation by cytochrome P450 enzymes In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed. New York: Springer, p. 111–176. [Google Scholar]
- Ortiz de Montellano PR, Komives EA. 1985. Branchpoint for heme alkylation and metabolite formation in the oxidation of arylacetylenes by cytochrome P-450. J Biol Chem. 260:3330–3336. [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL. 1980a. Occurrence of a 1,2-shift during enzymatic and chemical oxidation of a terminal acetylene. J Am Chem Soc. 102:7373–7375. [Google Scholar]
- Ortiz de Montellano PR, Kunze KL. 1980b. Self-catalyzed inactivation of hepatic cytochrome P-450 by ethynyl substrates. J Biol Chem. 255:5678–5685. [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL. 1981a. Shift of the acetylenic hydrogen during chemical and enzymatic oxidation of the biphenylacetylene triple bond. Arch Biochem Biophys. 209:710–712. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL. 1981b. Cytochrome P-450 inactivation: structure of the prosthetic heme adduct with propyne. Biochemistry. 20:7266–7271. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Reich NO. 1984. Specific inactivation of hepatic fatty acid hydroxylases by acetylenic fatty acids. J Biol Chem. 259:4136–4141. [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL, Yost GS, Mico BA. 1979. Self-catalyzed destruction of cytochrome P-450: Covalent binding of ethynyl sterols to prosthetic heme. Proc Natl Acad Sci USA. 76:746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Beilan HS, Mathews JM. 1982a. Alkylation of the prosthetic heme in cytochrome P-450 during oxidative metabolism of the sedative-hypnotic ethchlorvynol. J Med Chem. 25:1174–1179. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL, Beilan HS, Wheeler C. 1982b. Destruction of cytochrome P-450 by vinyl fluoride, fluroxene, and acetylene. Evidence for a radical intermediate in olefin oxidation. Biochemistry. 21:1331–1339. [DOI] [PubMed] [Google Scholar]
- Palmer KH, Feierabend JF, Baggett B, Wall ME. 1969. Metabolic removal of a 17α-ethynyl group from the antifertility steroid, norethindrone. J Pharmacol Exp Therap. 167:217–222. [PubMed] [Google Scholar]
- Palmer CJ, Cole LM, Smith IH, Moss MDV, Casida JE. 1991. Silylated 1-(4-ethynyl]phenyl)-2,6,7-trioxabicyclo[2,2,2]octanes: Structural features and mechanisms of proinsecticidal action and selective toxicity. J Agric Food Chem. 39:1335–1341. [Google Scholar]
- Podoll T, Pearson PG, Evarts J, Ingallinera T., Bibikova E., Sun H, Gohdes M, Cardinal K, Sanghvi M, Slatter JG. 2019. Bioavailability, biotransformation, and excretion of the covalent Bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab Dispos 47:145–154. [DOI] [PubMed] [Google Scholar]
- Raitano LA, Slikker W Jr, Hill DE, Hadd HE, Cairns T, Helton ED. 1981. Ethynyl cleavage of 17α-ethynylestradiol in the Rhesus monkey. Drug Metab Dispos. 9:129–134. [PubMed] [Google Scholar]
- Regal KA, Schrag ML, Kent UM, Wienkers LC, Hollenberg PF. 2000. Mechanism-based inactivation of cytochrome P450 2B1 by 7-ethynylcoumarin: verification of apo-P450 adduction by electrospray ion trap mass spectrometry. Chem Res Toxicol. 13:262–270. [DOI] [PubMed] [Google Scholar]
- Reilly PEB, Gomi RJ, Mason SR. 1999. Mechanism-based inhibition of rat liver microsomal diazepam C3-hydroxylase by mifepristone associated with loss of spectrally detectable cytochrome P450. Chemico-Biol Interact. 118:39–49. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Hopkins NE, Alworth WL, Hollenberg PF. 1993. Mechanism-based inactivation of cytochrome P450 2B1 by 2-ethynylnaphthalene: identification of an active-site peptide. Chem Res Toxicol. 6:470–479. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Hopkins NE, Zaluzec EJ, Gage DA, Alworth WL, Hollenberg PF. 1994. Identification of active-site peptides from 3H-labeled 2-ethynylnaphthalene-inactivated P450 2B1 and 2B4 using amino acid sequencing and mass spectrometry. Biochemistry. 33:3766–3771. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Hopkins NE, Zaluzec EJ, Gage DA, Alworth WL, Hollenberg PF. 1995a. Mechanism-based inactivation of cytochrome P4502B1 by 9-ethynylphenanthrene. Arch Biochem Biophys. 323:295–302. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Ballou DP, Hopkins NE, Alworth WL, Hollenberg PF. 1995b. Mechanistic studies of 9-ethynylphenanthrene-inactivated cytochrome P450 2B1. Arch Biochem Biophys. 323:303–312. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Hopkins NE, Foroozesh M, Alworth WL, Halpert JR, Hollenberg PF. 1997. Inactivation of cytochrome P450s 2B1, 2B4, 2B6, and 2B11 by arylalkynes. Drug Metab Dispos. 25:1242–1248. [PubMed] [Google Scholar]
- Roberts ES, Alworth WL, Hollenberg PF. 1998. Mechanism-based inactivation of cytochromes P450 2E1 and 2B1 by 5-phenyl-1-pentyne. Arch Biochem Biophys. 354:295–302. [DOI] [PubMed] [Google Scholar]
- Rose PA, Cutler AJ, Irvine NM, Shaw AC, Squires TM, Loewen MK, Abrams SR. 1997. 8’-Acetylene ABA: an irreversible inhibitor of ABA 8’-hydroxylase. Bioorg Med Chem Lett. 7:2543–2546. [Google Scholar]
- Sacher RM, Metcalf RL, Fukuto TR. 1968. Propynyl naphthyl ethers as selective carbamate synergists. J Agric Food Chem. 16:779–786. [Google Scholar]
- Salaün JP, Simon A, Durst F, Reich NO, Ortiz de Montellano PR. 1988. Differential inactivation of plant lauric acid ω -and in-chain hydroxylases by terminally unsaturated fatty acids. Arch Biochem Biophys. 260:540–545. [DOI] [PubMed] [Google Scholar]
- Salmon J, Coussediere D, Cousty C, Raynaud JP. 1983. Pharmacokinetics and metabolism of moxestrol in humans. J Steroid Biochem. 19:565–573. [DOI] [PubMed] [Google Scholar]
- Schmid SE, Au WYW, Hill DE, Kadlubar FF, Slikker W. 1983. Cytochrome P-450-dependent oxidation of the 17α-ethynyl group of synthetic steroids. D-Homoannulation or enzyme inactivation. Drug Metab Disp. 11:531–536. [PubMed] [Google Scholar]
- Sharma U, Roberts ES, Hollenberg PF. 1996. Inactivation of cytochrome P4502B1 by the monoamine oxidase inhibitors R-(−)-deprenyl and clorgyline. Drug Metab Dispos. 24:669–675. [PubMed] [Google Scholar]
- Shimada T, Murayama N, Okada K, Funae Y, Yamazaki H, Guengerich FP. 2007. Different mechanisms for inhibition of human cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic inhibitors. Chem Res Toxicol. 20:489–496. [DOI] [PubMed] [Google Scholar]
- Shirane N, Sui Z, Peterson JA, Ortiz de Montellano PR. 1993. Cytochrome P450BM-3 (CYP102): Regiospecificity of oxidation of ω-unsaturated fatty acids and mechanism-based inactivation. Biochemistry. 32:13732–13741. [DOI] [PubMed] [Google Scholar]
- Simonneaux G, Le Maux P. 2006. Carbene complexes of heme protein and iron porphyrin models. Top Organomet Chem. 17:83–122. [Google Scholar]
- Sisenwine SF, Kimmel HB, Liu AL, Ruelius HW. 1973. Urinary metabolites of DL-norgestrel in women. Acta Endocrinol. 75:91–104. [DOI] [PubMed] [Google Scholar]
- Sisenwine SF, Kimmel HB, Liu AL, Ruelius HW. 1979. The metabolic disposition of norgestrel in female rhesus monkeys. Drug Metab Dispos 7:1–6. [PubMed] [Google Scholar]
- Siu ECK, Tyndale RF, 2008. Selegiline is a mechanism-based inactivator of CYP2A6 inhibiting nicotine metabolism in humans and mice. J Pharmacol Exp Therap, 324:992–999. [DOI] [PubMed] [Google Scholar]
- Sridar C, Kent UM, Noon K, McCall A, Alworth B, Foroozesh M, Hollenberg PF. 2008. Differential inhibition of cytochromes P450 3A4 and 3A5 by the newly synthesized coumarin derivatives 7-coumarin propargyl ether and 7-(4-trifluoromethyl)coumarin propargyl ether. Drug Metab Dispos 36:2234–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridar C, Kenaan C, Hollenberg PF. 2012. Inhibition of bupropion metabolism by selegiline: mechanism-based inactivation of human CYP2B6 and characterization of glutathione and peptide adducts. Drug Metab Disp. 40:2256–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel SM, Szklarz GD, He YQ, Foroozesh M, Alworth WL, Roberts ES, Hollenberg PF, Halpert JR. 1999. Identification of selective mechanism-based inactivators of cytochromes P-450 2B4 and 2B5, and determination of the molecular basis for differential susceptibility. J Pharmacol Exp Therap. 290:445–451. [PubMed] [Google Scholar]
- Su H, Ma G, Liu Y. 2018. Theoretical insights into the mechanism and stereoselectivity of olefin cyclopropanation catalyzed by two engineered cytochrome P450 enzymes. Inorg Chem. 57:11738–11745. [DOI] [PubMed] [Google Scholar]
- Subramanian R, Tam J, Aidasani D, Reid DL, Skiles GL. 2011. Novel cytochrome P450 bioactivation of a terminal phenyl acetylene group: formation of a one-carbon loss benzaldehyde and other oxidative products in the presence of N-acetyl cysteine or glutathione. Chem Res Toxicol. 16:677–686. [DOI] [PubMed] [Google Scholar]
- Sullivan HR, Roffey P, McMahon RE. 1979. Biotransformation of 4’-ethynyl-2-fluorobiphenyl in the rat. In vitro and in vivo studies. Drug Metab Dispos. 7:76–80. [PubMed] [Google Scholar]
- Vacek G, Galbraith JM, Yamaguchi Y, Schaefer HF III, Nobes RH, Scott AP, Radom L. 1994. Oxirene: to be or not to be? J Phys Chem. 98:8660–8665. [Google Scholar]
- von Weyman LB, Blobaum AL, Hollenberg PF. 2004a. The mechanism-based inactivation of P450 2B4 by tert-butyl 1-methyl-2-propynyl ether: structural determination of the adducts to the P450 heme. Arch Biochem Biophys. 425:95–105. [DOI] [PubMed] [Google Scholar]
- von Weyman LB, Sridar C, Hollenberg PF. 2004b. Identification of amino acid residues involved in the inactivation of cytochrome P450 2B1 by two acetylenic compounds: the role of three residues in nonsubstrate recognition sites. J Pharmacol Exp Therap. 311:71–79. [DOI] [PubMed] [Google Scholar]
- Wade A, Symons AM, Martin L, Parke DV. 1979. Metabolic oxidation of the ethynyl group in 4-ethynylbiphenyl. Biochem J. 184:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade A, Symons AM. Martin L, Parke DV. 1980. The metabolic oxidation of the ethynyl group in 4-ethynylbiphenyl in vitro. Biochem J. 188:867–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White INH. 1978. Metabolic activation of acetylenic substituents to derivatives in the rat causing the loss of hepatic cytochrome P-450 and haem. Biochem J. 174:853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White INH. 1980. Structure-activity relationships in the destruction of cytochrome P-450 mediated by certain ethynyl-substituted compounds in rats. Biochem Pharmacol. 29:3253–3255. [DOI] [PubMed] [Google Scholar]
- White INH. 1981. Biochem J. 196:575–583. Destruction of liver haem by norethindrone. Conversion into green pigments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White INH. 1982. Biochem Pharmacol. 31:1337–1342. Biliary excretion of green pigments produced by norethindrone in the rat. [DOI] [PubMed] [Google Scholar]
- White INH, Campbell JB, Farmer PB, Bailey E, Nam NH, Thang DC. 1984. Metabolic activation of acetylenes. Covalent binding of [1,2-14C]octyne to protein, DNA, and haem in vitro and the protective effects of certain thiol compounds. Biochem J. 220:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MC, Helton ED, Goldzieher JW. 1975. The urinary metabolites of 17α-ethynylestradiol-9α,11ε-3H in women. Chromatographic profiling and identification of ethynyl and non-ethynyl compounds. Steroids. 25:229–246. [DOI] [PubMed] [Google Scholar]
- Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, Ullrich V. 1977. The reduction of polyhalogenated methanes by liver microsomal cytochrome P450. Molec Pharmacol. 13:698–705. [PubMed] [Google Scholar]
- Yun CH, Martin MV, Hopkins NE, Alworth WL, Hammons GJ, Guengerich FP. 1992. Modification of cytochrome P4501A2 enzymes by the mechanism-based inactivator 2-ethynylnaphthalene. Biochemistry. 31:10556–10563. [DOI] [PubMed] [Google Scholar]
- Zeller KP, Blocher A, Haiss P. 2004. Oxirene participation in the photochemical Wolff rearrangement. Mini-Reviews Org Chem, 1:291–308. [Google Scholar]
- Zhang H, Lin H, Walker VJ, Hamdane D, Hollenberg PF. 2009. tert-Butylphenylacetylene is a potent mechanism-based inactivator of cytochrome P450 2B4: inhibition of cytochrome P450 catalysis by steric hindrance. J Pharmacol Exp Therap. 331:1011–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lin H, Kenaan C, Hollenberg PF. 2011. Targeting of the highly conserved threonine 302 residue of cytochromes P450 2B family during mechanism-based inactivation by aryl acetylenes. Arch Biochem Biophys. 507:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gay SC, Shah MB, Foroozesh M, Liu J, Osawa Y, Zhang Q, Stout CD, Halpert JR, Hollenberg PF. 2013. Potent mechanism-based inactivation of cytochrome P450 2B4 by 9-ethynylphenanthrene: implications for allosteric modulation of cytochrome P450 catalysis. Biochemistry. 52:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Li S, Yang Z, Peng Y, Chen X, Zheng J 2018. Identification of ketene-reactive intermediate of erlotinib possibly responsible for inactivation of P450 enzymes. Drug Metab Dispos. 46:442–450. [DOI] [PubMed] [Google Scholar]