

Abstract
The base excision repair (BER) pathway historically has been associated with maintaining genome integrity by eliminating nucleobases with small chemical modifications. In the past several years, however, BER was found to play additional roles in genome maintenance and metabolism, including sequence-specific restriction modification and repair of bulky adducts and interstrand crosslinks. Central to this expanded biological utility are specialized DNA glycosylases, enzymes that selectively excise damaged, modified, or mismatched nucleobases. In this review, we discuss the newly identified roles of the BER pathway and examine the structural and mechanistic features of the DNA glycosylases that enable these functions.
Keywords: DNA Damage, DNA Repair, Base Excision Repair, DNA Glycosylase, Interstrand Crosslink, Secondary Metabolite
Recognition and Repair of DNA Damage
DNA is a reactive molecule that is continually challenged by both endogenous and exogenous insults [1, 2]. Cellular metabolites and their by-products, environmental toxins, and radiation alter the chemical structure of DNA, producing a wide spectrum of DNA damage. Single and double-strand breaks (DSBs) are generated by hydrolysis of the phosphodeoxyribose backbone, nucleotide mismatches are introduced by replication errors, and nucleobases are modified by alkylation, oxidation, and deamination (Figure 1A). Chemical adducts range in size from a single non-hydrogen atom (e.g., 8-oxoguanine, 3-methyladenine) to bulky lesions with helix-distorting properties, such as those produced by polyaromatic hydrocarbons and crosslinking agents. These chemically diverse lesions interfere with normal cellular processes through inhibition of replication, transcription, and chromosome maturation, leading to chromosome rearrangements and instability, cell death, ageing, and diseases including cancer [3].
Figure 1. Base Excision Repair of Damaged Nucleobases.

(A) Common DNA lesions resulting from alkylation, oxidation, and deamination of nucleobases. 3mA, 3-methyl-2’-deoxyadenosine; 7mG, 7-methyl-2’-deoxyguanosine; 8oxoG, 8-oxo-2’-deoxyguanosine; εA, 1,N6-etheno-2’-deoxyadenosine; mFapyG, methyl derivative of N6-(2’-deoxyribosyl)-2,6-diamino-4-oxo-5-formamidopyrimidine (FapyG); dU, 2’-deoxyuridine. (B) General steps in the base excision repair (BER) pathway. BER is initiated by lesion-specific DNA glycosylases, which remove damaged nucleobases to create an apurinic/apyrimidinic (AP) site. An AP endonuclease (or a bifunctional DNA glycosylase) then incises the modified strand, producing a single-strand break. As necessary, the break is processed by one of several enzymes to create a gap with a 3’-hydroxyl group and a 5’-phosphoryl group. A DNA polymerase fills the gap with new DNA, and a DNA ligase seals the strand to complete repair. In eukaryotes, if strand incision is performed by an AP endonuclease, repair synthesis occurs before end processing, displacing the AP site.
Several DNA repair pathways exist to eliminate specific types of damage from the genome [3]. Pathway choice is dictated in part by the enzymes that recognize or initiate repair of a particular type of damage. By and large, the base excision repair (BER) pathway (Figure 1B) eliminates nucleobases with small modifications, abasic sites, and single-strand breaks, while nucleotide excision repair (NER) (see Glossary) removes bulky, helix-destabilizing lesions. BER is initiated by lesion-specific DNA glycosylases that excise the modified nucleobase from the DNA by catalyzing hydrolysis of the N-glycosidic bond (Figure 1B and Figure 2A). The resulting apurinic/apyrimidinic (AP) site is incised by an AP endonuclease (or a bifunctional DNA glycosylase), which generates a 3’-hydroxyl group needed for polymerase-dependent synthesis of new DNA [for a detailed overview of BER, see 4, 5, 6]. Almost every DNA glycosylase, regardless of its specificity or structural architecture, uses a similar overall strategy in which the aberrant nucleotide is flipped out of the duplex and trapped in a nucleobase binding pocket on the protein surface, while the resulting void left in the DNA is filled by one or more intercalating residues that stabilize the extrahelical conformation (Figure 2B,C) [7–9]. Remodeling of the DNA substrate through bending of the helical axis and widening of the minor groove promotes base flipping by decreasing the energetic barrier to base pair opening, while also inducing strain that allows the glycosylase to detect altered base stacking, base pairing, or solvation resulting from chemical modification of the nucleobase [7, 10–13]. In addition to allowing for more stringent substrate recognition, base flipping facilitates excision by enabling catalytic activation of the lesion through chemical complementarity between the active site and the target nucleobase and improving the reaction geometry between the glycosidic bond and an attacking water molecule (Box 1) [9]. Nucleobase binding pockets are generally too small to accommodate more than a small modification, and thus the discovery of DNA glycosylases capable of removing bulky or crosslinked lesions associated with other types of repair, as well as non-toxic or unmodified bases, has been unexpected. This review describes these new glycosylases with a focus on the structural mechanisms that enable their activities.
Figure 2. Initiation of Base Excision Repair by DNA Glycosylases.

(A) Base excision and strand incision reactions performed by monofunctional and bifunctional DNA glycosylases. Monofunctional enzymes catalyze only base excision, wherein the glycosidic bond between the nucleobase and the phosphodeoxyribose backbone is hydrolyzed, removing the nucleobase and creating an AP site. Bifunctional enzymes catalyze both base excision and strand incision (lyase activity). During removal of the nucleobase, most bifunctional glycosylases form an iminium intermediate, which covalently links the protein and the DNA. Some bifunctional enzymes, however, initially hydrolyze the glycosidic bond to create an AP site before then converting the AP site to an iminium intermediate. Following base excision, all bifunctional DNA glycosylases incise the strand on the 3’-side of the AP site (β-elimination), generating a single-strand break with a 3’-phospho-α,β-unsaturated aldehyde (PUA) group and a 5’-phosphoryl group. Some bifunctional enzymes also subsequently incise the strand on the 5’-side of the PUA (δ-elimination), leaving a 3’-phosphate, which must also be removed prior to repair synthesis, requiring the phosphatase activity of a separate enzyme. Alternatively, if β-elimination occurs following strand incision by an AP endonuclease, a gap is generated with the 3’-hydroxyl and 5’-phosphoryl groups necessary for synthesis and ligation. (B) X-ray crystal structures of the bifunctional DNA glycosylase Fpg (green) bound to DNA (orange and yellow) containing an 8oxoG lesion (red; PDB ID: 3GO8, 3GPY) [82]. After base flipping, Met77, Arg112, and Phe114 (not shown) fill the void in the duplex. (C) An extrahelical 8oxoG lesion in the nucleobase binding pocket of Fpg. Hydrogen-bonding interactions are indicated with dashed lines.
Box 1. The Chemistry of Base Excision.
Two chemical mechanisms have been proposed for base excision, which differ primarily in the number of steps and the presence or absence of a chemical intermediate [98, 99]. By a concerted mechanism (one step), the nucleobase is displaced by an attacking nucleophile, and both groups simultaneously have some degree of bonding character. By a stepwise mechanism (two steps), the nucleobase departs before the nucleophile attacks, generating a short-lived but discrete oxocarbenium intermediate (Figure IA). To date, all studies of glycosidic bond hydrolysis in DNA have been consistent with a stepwise mechanism, both for enzymatic and non-enzymatic reactions [99, 100]. As the glycosidic bond is broken, electron density is pulled away from the sugar and onto the nucleobase [29, 98, 99]. For neutral lesions, this creates a negatively charged and usually unstable nucleobase leaving group, as well as a positively charged and highly unstable oxocarbenium intermediate (Figure IA). Stabilization of these developing charges is a key factor in how DNA glycosylases catalyze base excision. Depending on the enzyme and the lesion, transfer of electron density to the nucleobase is stabilized by one of two mechanisms [98, 100]. By acid catalysis, the nucleobase is protonated prior to excision, creating a positively charged substrate with a nucleobase that departs as a stable, neutral leaving group. Conversely, by electrostatic catalysis, the nucleobase is not protonated before excision, and departs as an anionic leaving group. Interactions within the nucleobase binding pocket stabilize the increase in electron density on the nucleobase. In both mechanisms, the corresponding loss of electron density on the sugar is stabilized through electrostatic interactions. Carboxylate (aspartate/glutamate) or carboxamide (asparagine/glutamate) side chains located in the active site and near the anomeric carbon (C1’) provide electron donation that offsets the loss of electron density to the nucleobase (Figure IB) [9, 100]. In monofunctional DNA glycosylases, these side chains also position the water nucleophile that attacks the oxocarbenium intermediate to produce an AP site. In bifunctional DNA glycosylases, an amine functional group (lysine ε-NH2 or an N-terminal valine or proline α-NH2) positions the water nucleophile to generate an AP site, or more commonly, attacks the oxocarbenium intermediate to form an iminium crosslink. Recent studies have proposed an alternative mechanism for bifunctional glycosylases, whereby the amine group attacks the sugar before departure of the nucleobase [81, 101, 102]. In this scenario, instead of breaking the glycosidic bond, nucleophilic addition opens the deoxyribose ring between C1’ and O4’, requiring protonation of O4’ by a catalytic carboxylic acid side chain. By this “ribose-protonated mechanism”, the glycosidic bond remains intact until the iminium crosslink is fully formed.
Self-Resistance to Genotoxic Secondary Metabolites
Bacterial secondary metabolites are often used as defense mechanisms in microbial warfare. To withstand the toxicity of their own natural products, antibiotic-producing bacteria require self-resistance mechanisms, such as sequestration, efflux, and degradation of the toxin, and protection and repair of the target (e.g., DNA) [14]. Two target-repair mechanisms for the genotoxins yatakemycin (YTM) and azinomycin B (AZB) were recently found to involve the evolutionarily unrelated DNA glycosylases YtkR2 and AlkZ [15, 16], which are encoded by genes embedded within the ytk and azi biosynthesis clusters [15, 17]. Homologs of these enzymes are present in diverse bacterial species, but only some of which are known to produce antibiotics [16, 18–20]. Homologs of YtkR2 are also present in archaea and lower eukaryotes. It is unclear if these organisms encounter compounds similar to YTM and AZB in their environments, or if these homologs have evolved to perform different functions.
Excision of Bulky Adducts by YtkR2
Streptomycetes produce a staggering number and variety of secondary metabolites, including many highly genotoxic antibiotics [21]. Among the most potent are those in the spirocyclopropylcyclohexadienone family, which consists of CC-1065, duocarmycin A and SA, and YTM [22, 23]. These bulky molecules undergo binding-induced conformational changes in the minor groove of AT-rich regions of DNA that increase their reactivity, resulting in sequence-specific alkylation of adenine at the N3 position (Figure 3A) [24]. While only a single covalent bond is formed, a large number of non-covalent contacts, principally C-H/π interactions, are created between the antibiotic and deoxyribose groups in both DNA strands. Individually each of these interactions is weak, but together they create a strong energetic barrier to duplex unwinding that inhibits both transcription and replication, while also making these lesions poor substrates for the NER pathway [25, 26].
Figure 3. Removal of Bulky Adducts by AlkD.

(A) Bulky adducts excised by AlkD. 7POBG, 7-pyridyloxobutyl-2’-deoxyguanosine; O2-POBC, O2-pyridyloxobutyl-2’-deoxycytidine; YTMA, 3-yatakemycinyl-2’-deoxyadenosine. AlkD eliminates bulky lesions with modifications located in either the major (7POBG) or the minor (O2-POBC and YTMA) groove. (B,D) X-ray crystal structure of AlkD (cyan) in complex with free 3-methyladenine (3mAde) nucleobase (purple) and DNA (orange and yellow) containing a tetrahydrofuran (THF) spacer (purple) to mimic an AP site (PDB ID: 5CLE) [29]. Water molecules located in the large cavity between the protein and the DNA are depicted as red spheres. (C,E) X-ray crystal structure of AlkD in complex with an excised yatakeymycinyladenine (YTMAde) nucleobase (purple) and DNA containing an AP site (purple; PDB ID: 5UUF) [26]. Hydrogen-bonding interactions are indicated with dashed lines. Unlike DNA glycosylases that use a traditional base-flipping mechanism, the catalytic residues (Trp109, Asp113, and Trp187) present in AlkD are located on the protein surface, not recessed in a nucleobase binding pocket. Without a catalytic requirement for base flipping, the lack of protein-DNA contacts in the major groove and the large solvent-filled cavity between the protein and the minor groove allow AlkD to recognize and excise nucleobases with bulky modifications at any position.
The YTM-producer Streptomyces sp. TP-A0356 has evolved multiple resistance mechanisms to minimize YTM toxicity [15, 27]. Within the gene cluster for YTM synthesis are seven resistance genes that play roles in efflux and degradation of YTM and repair of YTM-DNA adducts. YtkR2 is a DNA glycosylase that hydrolyzes the glycosidic bond of 3-yatakemycinyl-2’-deoxyadenosine (YTMA) lesions (Figure 3A), enabling the BER pathway to provide self-resistance to YTM toxicity [15]. Similarly, the CC-1065 biosynthesis cluster of Streptomyces zelensis contains a homolog of YtkR2 (C10R5) that likely provides resistance to CC-1065 [19].
YtkR2 was identified as a YTM resistance protein by its similarity to AlkD, a previously discovered DNA glycosylase with the ability to excise bulky pyridyloxobutyl (POB) adducts of guanine and cytosine (Figure 3A) [18, 28]. Despite being present in Bacillus cereus, which is not a producer of YTM, AlkD efficiently removes YTMA lesions in vitro [26, 29]. As such, its structural and mechanistic characterization is a basis for understanding how YtkR2 provides self-resistance to YTM. AlkD has a structural architecture distinct from those of other DNA glycosylases. The HEAT-like repeat (HLR) fold lacks both a nucleobase binding pocket and apparent intercalating residues [9, 18, 30, 31]. Accordingly, studies using either methylated or yatakemycinylated nucleobases showed that AlkD recognizes and excises lesions without first sequestering the modified base from the DNA duplex [26, 29]. AlkD instead interacts with the phosphodeoxyribose backbone of the lesion, while leaving the nucleobase, which is not contacted by the enzyme, stacked in the duplex and paired with the opposing nucleotide (Figure 3B–E). In this conformation, the deoxyribose moiety of the lesion is in contact with three residues: T rp109, Asp113, and T rp187. Surprisingly, all three, including the two tryptophan residues, are catalytic [26, 32, 33]. As with catalytic residues in other DNA glycosylases (Box 1), Asp113 stabilizes the sugar as positive charge develops during cleavage of the glycosidic bond, while also pre-organizing the water nucleophile. Uniquely, Trp109 and Trp187 form C-H/π interactions with the deoxyribose group. Electrostatic in nature, these interactions become stronger as the C-H bond becomes increasingly polarized [29], such as when positive charge develops on the sugar. As such, the CH/π interactions formed with Trp109 and Trp187 preferentially stabilize the transition state, reducing the activation barrier and accelerating excision.
AlkD was crystallized in two product complexes, containing either 3-methyladenine (3mAde) or 3-yatakemycinyladenine (YTMAde) nucleobase (Figure 3B–E) [26, 29]. In both complexes, the only contacts between the protein and the modified nucleobases are with the bulky YTM moiety, which is located in an extended cavity between the enzyme surface and the minor groove (Figure 3CE). In the complex with the much smaller 3mAde nucleobase, this cavity is filled by solvent, as is the major groove in both complexes (Figure 3B–E). This lack of steric restraint is likely what allows AlkD to remove nucleobases with bulky YTM or POB modifications located in either groove (Figure 3A). However, in contrast to DNA glycosylases that flip the lesion into a more restrictive nucleobase binding pocket, which contains catalytic residues that activate the base for excision (Box 1), AlkD is limited to removing positively charged lesions with nucleobases that are inherently good leaving groups. Moreover, this lack of steric restriction seems to make DNA glycosylases in this family well-suited to remove diverse lesions, limited primarily by the mechanistic requirement for a positively charged substrate.
Unhooking of Interstrand Crosslinks by AlkZ
Along with the spirocyclopropylcyclohexadienone compounds, bifunctional alkylating agents are among the most potent antibiotics produced by Streptomycetes [34]. AZB is a genotoxic nonribosomal peptide/polyketide secondary metabolite with a densely functionalized backbone that scaffolds electrophilic aziridine and epoxide moieties [35, 36]. This arrangement creates a molecule that is ideally suited to bind in the major groove of GNC and GNT sequences, and to undergo nucleophilic addition at the N7 positions of guanine and adenine (Figure 4A) [37–39]. By covalently tethering opposing strands, bifunctional alkylating agents generate interstrand crosslinks (ICLs), which block transcription and replication, and generally necessitate repair by a combination of repair pathways [40–42]. In bacteria, the primary mechanism of ICL repair requires the combined actions of NER and either translesion synthesis (TLS) or homologous recombination (HR) [1, 43, 44]. Unlike the primary mechanisms of ICL repair in vertebrates, which involve DNA structures generated only during replication [45–48], ICL repair in bacteria occurs in the context of duplex DNA [1, 43, 44]. The lesion is unhooked from one strand by the NER machinery, the gap filled by either HR-dependent synthesis or TLS, and the monoadduct repaired by a second round of NER. Recently, however, a DNA glycosylase was found to unhook ICLs produced by AZB, implicating BER in bacterial ICL repair [16].
Figure 4. Unhooking of Interstrand Crosslinks by AlkZ.

(A) Interstrand crosslink (ICL) formed between two 2’-deoxyguanosine nucleotides by azinomycin B (AZB). AZB preferentially reacts in GNC sequences to form {1–3} crosslinks. (B) Hypothetical models of AlkZ (green and yellow) bound to DNA (gray and blue) containing an AZB ICL (red). The models were constructed by rigid-body docking of an X-ray crystal structure of AlkZ (PDB ID: 5UUJ) [50] and a computationally derived model of DNA containing an AZB ICL [38]. The DNA could be docked equally well in either of two binding orientations by placing the β11/β12 hairpin into the minor groove across from the ICL. Each orientation positions one of the two modified nucleotides (G1 or G2) near Gln39 in the putative active site of AlkZ. (C,D) Hypothetical model of an AlkZ dimer bound to DNA containing an AZB ICL. Docking two molecules of AlkZ with a single AZB ICL creates a network of salt bridges (Glu152 and Arg153), hydrogen bonds (Ser304 and Arg308), and hydrophobic contacts (Ala309 and Pro340) at the protein interface. (E) Alternate mechanisms of AZB ICL processing by the BER pathway. Concerted unhooking of both strands, consistent with the hypothetical dimeric complex, produces two closely spaced AP sites, potentially leading to a double-strand break (DSB). Sequential unhooking and repair of each strand avoids the concurrence of multiple AP sites and minimizes the possibility of a DSB.
Much like Streptomyces sp. TP-A0356, the AZB-producer Streptomyces sahachiroi has evolved multiple mechanisms for self-resistance, including sequestration and efflux of AZB, and repair of AZB ICLs [16, 49]. Originally annotated as a putative winged helix-turn-helix (wHTH) transcription factor, AlkZ is a novel DNA glycosylase that excises ICLs formed by AZB, cleaving the glycosidic bonds of both modified nucleotides [16]. The resultant AP sites, despite their proximity on opposing strands, were shown to be substrates for the bacterial AP endonuclease Endo IV, suggesting AZB ICLs can be repaired by the BER pathway alone. Consistent with this role, genetic knockouts of alkZ were highly susceptible to AZB toxicity, and resistance could be restored with a plasmid expressing AlkZ [16]. In addition to its repair role, AlkZ has been proposed to provide self-resistance to AZB by blocking target sites, thereby preventing crosslinks from forming [16]. However, the experiments that served as the basis for this proposal were not designed to distinguish between protection of target sites and unhooking of ICLs. Both activities would allow for unwinding of double-stranded DNA after incubation with AZB, which was the experimental observation. Given the proven excision activity of AlkZ, unhooking seems to be the more probable explanation.
Even more so than excision of bulky adducts, unhooking of ICLs, which are tethered to both DNA strands, is incompatible with the base-flipping mechanisms used by most DNA glycosylases. Correspondingly, the structural architecture of AlkZ most closely resembles that of AlkD, although the two folds are distinct [26, 50]. Like AlkD, AlkZ is C-shaped with a positively charged concave surface ideal for binding DNA, and lacks a nucleobase binding pocket and apparent DNA intercalating residues. AlkZ is the defining member of the HTH_42 superfamily of proteins, which are predicted to contain tandem wHTH motifs [16]. Interestingly, the structure of AlkZ shows that the enzyme uses three such motifs to create the DNA binding scaffold, although none seem to engage DNA in the same manner as wHTH motifs found in transcription factors [50]. While no structure of an AlkZ-DNA complex is available, mutational analysis indicated three essential catalytic elements within the putative substrate binding cleft, including a β-hairpin (β11/β12) and two glutamine residues (Gln37 and Gln39). Docking of a computationally derived model of an AZB ICL revealed two nearly equivalent DNA orientations, related by the pseudosymmetric nature of the crosslink relative to the dyad axis, and suggested possible roles for the three catalytic elements (Figure 4B) [38, 50]. In each of the binding orientations, Gln37 interacts with a phosphate group in the DNA backbone, while the β11/β12 hairpin inserts into the minor groove and interacts with the backbones of both strands and one of the modified nucleobases. Gln39 is located just outside the minor groove and is ideally positioned to catalyze hydrolysis of the glycosidic bond (Box 1).
As modeled, no contacts are present between AlkZ and AZB in the hypothetical protein-DNA complexes, which could otherwise favor one binding orientation over the other. This is consistent with biochemical data indicating excision of both modified nucleotides [16]. Strikingly, the two binding orientations are not mutually exclusive, allowing for the possibility of dimer formation at the crosslink (Figure 4C,D) [50]. Moreover, docking of two AlkZ molecules on a single AZB ICL revealed remarkable self-complementarity at the protein interface, including a potential network of salt bridges (Glu152 and Arg153), hydrogen bonds (Ser304 and Arg308), and hydrophobic contacts (Ala309 and Pro340). However, there is currently no available experimental data to support or refute dimerization. Further work will be necessary to determine whether dimer formation plays a role in recognition or excision of AZB ICLs, as well as the mechanism by which downstream enzymes in the BER pathway process the AP products generated by AlkZ (Figure 4E).
Restart of Stalled Replication Forks
By preventing the replisome from unwinding duplex DNA, ICLs stall progression of the replication fork, which in turn results in genomic instability [41, 42]. As in prokaryotes, the conventional mechanism of ICL repair in eukaryotes involves the combined activities of NER, TLS, and HR. In higher eukaryotes, these activities are coordinated by the Fanconi anemia (FA) pathway and are coupled with DNA replication [51, 52]. Following convergence of two replication forks at a crosslink, eukaryotic ICL repair is initiated by unloading of replisome components from the DNA template [46, 47, 53]. Fanconi proteins then recruit endonucleases associated with the NER pathway to create incisions in one strand flanking the lesion [45, 48, 54]. These incisions generate a DSB in one sister chromatid and leave a monoadduct on the other. The former is repaired by HR, and the latter bypassed by TLS. Recently, however, the vertebrate DNA glycosylases NEIL1 and NEIL3 were found to unhook ICLs, as well as to excise bulky adducts, providing an alternative mechanism to restart stalled replication forks [54–57].
Identified by sequence similarity to bacterial DNA glycosylases in the Fpg/Nei superfamily, the Nei-like enzymes NEIL1, NEIL2, and NEIL3 are the most recent DNA glycosylases to be discovered in vertebrates [58–61]. Like the related bacterial enzymes, NEIL1–3 were originally linked to repair of oxidized nucleobases [62]. However, several subsequent findings have called into question whether this is the primary function of the vertebrate enzymes. First, the small oxidative lesions excised by NEIL1–3 are also removed by other DNA glycosylases present in higher eukaryotes, namely oxidized pyrimidines by NTHL1 and oxidized purines by OGG1 [60]. Second, NEIL1 and NEIL3 form specific interactions with key proteins required for replication, and NEIL2 interacts with a number of proteins necessary for transcription, including RNA polymerase II [63–65]. Third, expression of NEIL1 and NEIL3 is cell-cycle dependent, and induced during S phase [66–68]. Fourth, NEIL2 and NEIL3 preferentially excise lesions from bubble, fork, and single-stranded DNA structures [64, 69–72]. Together, these findings suggest roles for NEIL1 and NEIL3 in replication-associated repair, and for NEIL2 in transcription-coupled repair [73].
The different functions of NEIL1 and NEIL3 during replication were first suggested when NEIL1 was shown to remove psoralen crosslinks from triplex DNA (Figure 5A,B) [74, 75]. During normal ICL repair, crosslinked triplex structures are generated following unhooking of the ICL by NER and bypass of the resulting monoadduct by TLS (Supplemental Figure S1). While NER is able to repair these structures [76, 77], the activity of NEIL1 suggests BER may be an alternative [57, 75, 78]. Importantly, NEIL1 is not able to perform the initial unhooking of the ICL in duplex DNA, or in the X-shaped structures formed when replication forks converge at a crosslink (Figure 5C) [55, 74, 75]. Conversely, NEIL3 was recently shown to unhook both psoralen and AP crosslinks at convergent replication forks (Figure 5A,C), providing an incision-independent alternative for repair of ICLs [54]. Both incision-dependent and -independent repair take place at replication forks that have converged at a crosslink (Figure 5C, Supplementary Figure S1) [47, 54]. Pathway choice is determined by ubiquitinylation of replisome components (Supplemental Figure S1) [46]. Short polyubiquitin chains recruit NEIL3 to the ICL, whereas longer chains, which form in the absence of unhooking by NEIL3, lead to unloading of the replisome and unhooking of the ICL by structure-specific endonucleases. NEIL3 recruitment and unhooking prior to initiation of the incision-dependent pathway suggests the glycosylase-mediated pathway is the preferred mechanism of ICL repair during S-phase in vertebrates [46, 54]. One key distinction between the two pathways is that the incision-independent mechanism avoids the DSBs that are integral to the incision-dependent mechanism (Supplemental Figure S1). However, as a bifunctional DNA glycosylase, NEIL3 possesses AP lyase activity (Figure 2A), which could potentially generate a DSB at convergent replication forks, negating the apparent benefit of an otherwise incision-independent repair pathway. The AP lyase activity of NEIL3, however, is weak relative to those of NEIL1 and NEIL2 [70], and appears to be further reduced in this context, as DSBs were not observed when ICL repair was initiated by NEIL3 in nuclear extracts [54].
Figure 5. Excision of Bulky Adducts and Interstrand Crosslinks by NEIL1 and NEIL3.

(A) Bulky adducts and interstrand crosslinks removed by NEIL1 or NEIL3. NM-FapyG, nitrogen mustard derivative of FapyG; AFBrFapyG, aflatoxin B1 derivative of FapyG; PSO, psoralen. Both psoralen and AP sites form {1–2} crosslinks by modifying nucleobases on opposing strands. (B) Triplex substrate for NEIL1. The crosslink is colored red and nascent DNA produced by translesion synthesis is gray. Triplex structures are generated during replication-associated ICL repair (Supplemental Figure S1). (C) Convergent fork substrate for NEIL3. Nascent DNA generated during replication is colored gray. NEIL3 is recruited to ICLs after fork convergence and replisome ubiquitinylation (Supplemental Figure S1). (D) X-ray crystal structure of NEIL1 (cyan) bound to DNA (orange and yellow) containing a thymine glycol (Tg) lesion (red; PDB ID: 5ITY) [81]. Three intercalating residues (Met81, Arg118, and Phe120) stabilize the extrahelical conformation of the DNA substrate, while a flexible capping loop allows for accommodation and excision of bulky lesions. (E) Crystal structure of the glycosylase domain of NEIL3 (PDB ID: 3W0F) [73]. NEIL3 lacks the two intercalating residues that interact with the undamaged strand in the NEIL1 complex, as well as the flexible capping loop that contacts the Tg lesion. Consistent with a preference for non-duplex substrates, NEIL3 also lacks the basic residue (Arg274) that forms a salt bridge with the undamaged strand in the NEIL1 complex.
The mechanisms by which NEIL1 and NEIL3 excise bulky adducts and unhook ICLs are unclear. Structures of NEIL1 are available only with DNA containing small lesions [79–81], and the single available structure of NEIL3 lacks DNA (Figure 5D,E) [73]. Nonetheless, comparison of the two structures, as well as structures of the bacterial enzymes, provides a basis for speculation. Both NEIL1 and NEIL3 share the same structural architecture and arrangement of N-terminal catalytic residues (Pro2/Val2 and Glu3) as Fpg and Nei. Like the bacterial enzymes [9], NEIL1 also utilizes three intercalating residues to stabilize the extrahelical conformation of the DNA substrate (Figure 2B and Figure 5D). The capping loop in NEIL1, which contacts the flipped lesion in the bacterial enzymes, is similar to that of Nei [9, 79–81]. In all structures of Nei and most structures of NEIL1, this loop is largely disordered (Figure 5D), suggesting an inherent flexibility that may allow for recognition and excision of diverse lesions. In contrast to the well-ordered loop in Fpg [82], which contacts extrahelical nucleobases through amide groups in the protein backbone (Figure 2B,C), the partially disordered loop in NEIL1 interacts with the lesion through the sidechain of a conserved arginine residue [81]. The flexibility of this loop may enable expansion of the nucleobase binding pocket to accommodate bulky adducts, such as nitrogen mustard and aflatoxin B1 derivatives of N6-(2′-deoxyribosyl)-2,6-diamino-4-oxo-5-formamidopyrimidine (FapyG) and psoralen monoadducts (Figure 5A) [56, 74]. This flexibility may also allow NEIL1 to bind triplex substrates, by pulling the short, third strand away from the duplex and binding the duplex and the crosslink in the same manner as a bulky adduct in double-stranded DNA.
In contrast, NEIL3 shares fewer structural features with the bacterial enzymes [9, 73]. Unlike Fpg and Nei, NEIL3 appears to lack intercalating residues, with the possible exception of Met99 (Figure 5E) [73]. In NEIL1 and the bacterial enzymes, the two other intercalating residues primarily interact with the complementary DNA strand in duplex substrates [9, 81]. The absence of these residues is consistent with the established preference of NEIL3 for lesions in single-stranded DNA [71, 73], and suggests NEIL3, like AlkZ, may unhook ICLs using a non-base-flipping mechanism. However, the non-base-flipping mechanisms employed by AlkZ and AlkD/YtkR2 limit these proteins to excising positively charged lesions, which do not require activation of the nucleobase through contacts in the nucleobase binding pocket (Box 1). NEIL3 acts on neutral lesions and crosslinks [54, 56, 62], which would seemingly require activation by the enzyme. Yet, the capping loop that performs this function in NEIL1, Nei, and Fpg is strikingly short in NEIL3, and seems unlikely to contact even a fully flipped nucleotide (Figure 5E). While such an open nucleobase binding pocket is consistent with removal of bulky adducts, including nitrogen mustard derivatives of FapyG [56], how NEIL3 activates neutral lesions for excision is unclear, and will require additional structural experiments with appropriate DNA substrates. Similarly, mechanistic understanding of NEIL2 has been severely limited by a lack of available structural information. However, the primary structure of NEIL2 shares all features of NEIL3 that seem to enable unhooking of ICLs, and NEIL2, like NEIL3, also shares a preference for non-duplex structures [62]. Based on these common features and the interaction of NEIL2 with RNA polymerase II [65], we speculate NEIL2 may be involved in transcription-coupled repair of ICLs.
Protection from Foreign DNA
Restriction-modification systems protect prokaryotes from the potentially deleterious effects of foreign DNA, regulating genetic exchange among bacteria and guarding against infection by phage [83–85]. At a minimum, these systems are comprised of two components: a restriction endonuclease that recognizes a specific DNA sequence and introduces one or more strand breaks, and a DNA methyltransferase that modifies the same sequence to render it resistant to the endonuclease. Type II restriction endonucleases use a Mg2+-dependent mechanism to cleave both strands of DNA, usually within a short palindromic sequence, producing a DSB [86]. However, genomic analyses recently identified a novel type II enzyme in hyperthermophilic archaea—with homologs in both thermophilic and mesophilic bacteria—that generates strand breaks without requiring Mg2+ [87–89]. Unlike other type II enzymes, which directly hydrolyze the phosphodiester backbone, R.PabI hydrolyzes the glycosidic bond of 2’-deoxyadenosine, producing nearby AP sites on opposing strands. A DSB is then generated by heat-promoted β-elimination or enzymatic AP endonuclease or AP lyase activities (Figure 6A) [90–92]. Similar to restriction endonucleases, R.PabI cuts exclusively within palindromic GTAC sequences, making it the only known sequence-specific DNA glycosylase. To avoid formation of DSBs in the genome, R.PabI is coexpressed with the DNA methyltransferase M.PabI, which forms N6-methyl-2’-deoxyadenosine (N6mA) within the same GTAC sequences, preventing excision by R.PabI [91, 93].
Figure 6. Sequence-Specific Excision of Adenine by R.Pabl.

(A) Double-strand break resulting from dual base excision and subsequent incision of AP sites on opposing strands. (B) X-ray crystal structure of tetrameric R.PabI (blue, green, and white) bound to non-specific DNA (orange and yellow; PDB ID: 5IFF) [94]. Four salt bridges formed by Arg70 and Asp71 are the only interactions between the two R.PabI dimers in the tetrameric search complex. (C) Crystal structure of dimeric R.PabI bound to specific DNA after dual excisions of adenine (Ade) nucleobase (red) to create AP sites (red) on opposing strands (PDB ID: 3WAZ) [90]. Binding of the recognition sequence induces a transition to a dimeric excision complex in which the GTAC base pairs are pulled apart and the void created in the duplex is stabilized by insertion of Gln155 and Arg156. (D) Hydrogen-bonding interactions in the dimeric product complex. DNA binding residues are colored according to protein subunit. R.PabI forms 13 sequence-specific and 21 non-sequence-specific hydrogen-bonding interactions with each strand in the palindromic product. (E) Recognition of Ade in the active site of R.PabI (stereodiagram). Hydrogen-bonding interactions are indicated with dashed lines. N6-methyl-2’-deoxyadenosine, N6-mA. Hydrogen bonds between Ade and backbone atoms in Ile66 and Val164 select for an N7-protonated substrate to catalyze excision. Sequence-specific methylation of Ade by M.PabI introduces steric clashes with these same backbone atoms to prevent catalytically productive binding and excision of N6-mA.
The unique ability of R.PabI to specifically excise adenine from a palindromic sequence is based on a novel structural architecture, distinct from not only other DNA glycosylases but other proteins [88]. In the absence of DNA, R.PabI forms a dimer with a central, highly twisted β-sheet, comprised of six β-strands from each subunit. This structural element is the defining feature of the HALFPIPE superfamily and plays a critical role in substrate recognition, providing much of the positively charged DNA binding interface [88, 90]. In the presence of non-specific DNA, R.PabI dimers associate to form a homotetramer that fully encircles the DNA duplex (Figure 6B} [94]. The interface between dimers is small, consisting only of four salt bridges between Arg70 and Asp71. Nonetheless, tetramerization appears to facilitate the search for GTAC sequences, as mutation of Arg70 decreased the relative rate of adenine excision in the presence of increasing amounts of non-specific DNA and reduced the proportion of higher oligomeric species bound to DNA lacking the recognition sequence [94]. As in other complexes between DNA glycosylases and DNA [9], nearly all DNA interactions in the tetrameric search complex are non-specific and mediated by hydrogen-bonds between R.PabI and phosphoryl groups in the DNA backbone [94]. Perturbation of the duplex in this complex is modest, consisting of a 20° bend along the helical axis and a slight widening of the minor groove. However, upon locating the recognition sequence, R.PabI transitions to a far more disruptive binding mode, in which one of the two dimers is displaced as the duplex is bent by nearly 90° and the minor groove is stretched until the GTAC base pairs are pulled apart (Figure 6C) [90]. This highly distorted DNA conformation is stabilized by insertion of Gln155 and Arg156 into the melted region of the duplex (Figure 6C). The unpaired nucleotides themselves form a large number of contacts with the remaining R.PabI dimer, including 13 sequence-specific hydrogen bonds with each strand in the palindromic sequence (Figure 6D). Such a large number of contacts is possible because both the 2’-deoxyguanosine nucleotide and the excised adenine nucleobase in the product complex are pulled into adjacent but separate nucleobase binding pockets (Figure 6C,E). Only the adenine binding pocket possesses the catalytic residues necessary for base excision. Asp214, which forms a hydrogen bond with the AP site in the product complex (Figure 6E), is positioned to stabilize the sugar as positive charge develops during cleavage of the glycosidic bond, while also pre-organizing the water nucleophile. The manner in which the nucleobase is activated for excision is less apparent (Box 1). No obvious general acid is present in the active site. Yet, N7 of adenine appears to form a hydrogen bond with the backbone carbonyl of Ile66, which would require N7 to be protonated (Figure 6E). Like the plant toxin ricin, which excises adenine from 28S rRNA [95, 96], R.PabI may select for substrates that are protonated at N7 prior to entering the nucleobase binding pocket. However, given the high acidity of N7-protonated adenosine (pKa = 2.2) [97], the fraction of adenine likely to be activated for excision at neutral pH is exceedingly small. Conceivably, R.PabI—as well as ricin—could facilitate protonation prior to or during flipping of the substrate into the active site.
R.PabI and its homologs are the only DNA glycosylases known to function in a restriction-modification system, a role that requires sequence-specific excision of unmodified nucleobases [90]. The specificity that R.PabI exhibits for GTAC sequences comes from a large number of sequence-specific contacts present in a homodimeric structure that are ideally suited for recognition of palindromic sequences. The residues involved in these contacts are highly conserved among homologs, suggesting all enzymes in the HALFPIPE superfamily recognize GTAC sequences. This is in stark contrast to other DNA glycosylases, which display little to no sequence selectivity, and only form sequence-specific contacts with the lesion and, in some instances, the nucleotide complementary to the lesion [9]. As with restriction endonucleases, the sequence-specific activity of R.PabI has potential biotechnology applications, which could be expanded by modulating the specificity of the enzyme through mutation of residues involved in DNA sequence recognition. However, it is currently unclear what features of the recognition sequence induce the tetrameric search complex to further remodel the DNA substrate and transition to the dimeric excision complex.
Concluding Remarks
Our understanding of how the BER pathway is involved in alternative biological roles has moved forward on two fronts, the discovery of new DNA glycosylases and the identification of new functions associated with previously known enzymes. In the past 12 years, three superfamilies of DNA glycosylase have been discovered, each with its own unique structural architecture and mechanism of lesion recognition and excision. While structurally unrelated, both YtkR2 and AlkZ employ non-base-flipping mechanisms to excise bulky adducts and ICLs, respectively, providing bacteria with self-resistance to genotoxic natural products. NEIL3, which is structurally distinct from both enzymes, likely also utilizes some form of a non-base-flipping mechanism to unhook ICLs, providing an incision-independent pathway for restart of stalled replication forks in vertebrates. Conversely, R.PabI flips not one but four nucleotides into nucleobase binding pockets, enabling recognition of a short palindromic sequence and sequence-specific excision of adenine, providing prokaryotes with protection from foreign DNA as part of a unique restriction-modification system. Together, these enzymes illustrate how deviation from a traditional base-flipping mechanism broadens the range of substrates amenable to excision by DNA glycosylases, and ultimately how these enzymes expand the biological utility of the BER pathway.
Despite our advances in understanding the structural and mechanistic features of these unique DNA glycosylases, many questions remain unanswered (see Outstanding Questions). We are only beginning to speculate how the products of these specialized enzymes are processed in the downstream steps of the BER pathway, and how multiple repair pathways may be coordinated to perform these alternative roles. As we work to answer these questions, additional DNA glycosylases, potentially with new functions, are likely to be discovered. Without question, we still have much to learn about a pathway once thought only to eliminate small nucleobase modifications from the genome.
OUTSTANDING QUESTIONS.
Three new families of DNA glycosylase, each with a unique structural architecture and a novel function, have been discovered in the past ten years. Are there more DNA glycosylases, particularly with new functions, that have yet to be identified?
Two of the three recently discovered families of DNA glycosylase play roles in self-resistance to genotoxic secondary metabolites. Are YtkR2 and AlkZ representative of a more widespread mechanism of bacterial self-resistance to natural products? That is, do other bacteria that produce genotoxins also employ specialized DNA repair enzymes that have not been discovered? If so, are these enzymes, like YtkR2 and AlkZ, encoded within biosynthetic gene clusters?
Base excision of non-traditional substrates (e.g., ICLs) generates non-traditional products. It is unclear if these products can be processed by downstream enzymes in the BER pathway. If not, have specialized enzymes evolved, or are other canonical DNA repair pathways involved?
Many DNA glycosylases remove a range of nucleobase substrates. Moreover, every DNA glycosylase that has been shown to excise bulky adducts or interstrand crosslinks also acts upon nucleobases with small modifications. Do any of the currently known DNA glycosylases that have been shown to excise traditional substrates also process unidentified substrates or perform unknown functions?
Supplementary Material
Figure I. Base Excision by DNA Glycosylases.
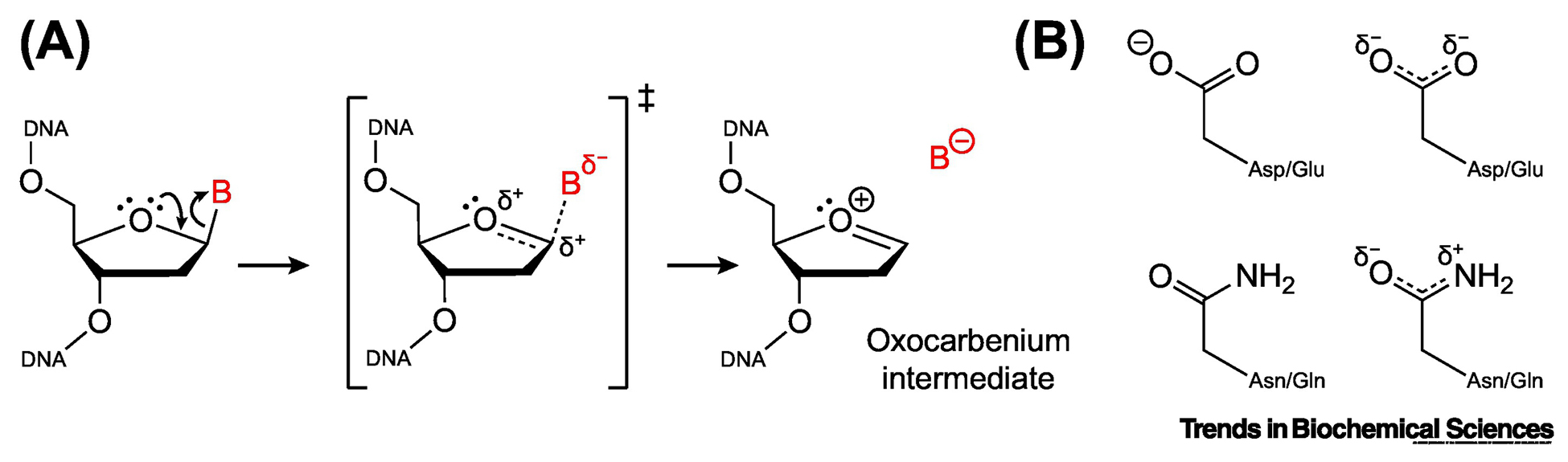
(A) Putative mechanism of glycosidic bond cleavage. Addition of a water molecule or an amine group to the oxocarbenium intermediate would produce an AP site or an iminium crosslink, respectively. (B) Key catalytic residues required for base excision. Carboxylate (aspartate/glutamate) and carboxamide (asparagine/glutamine) functional groups are depicted both as lowest-energy resonance forms (left) and as resonance hybrids (right).
HIGHLIGHTS.
Recent studies revealed new and unexpected functions for diverse DNA glycosylases, expanding the known roles of the base excision repair pathway.
Two specialized bacterial DNA glycosylases excise bulky adducts and interstrand crosslinks to provide self-resistance to genotoxic secondary metabolites.
A vertebrate DNA glycosylase previously associated with excision of oxidative lesions performs replication-coupled unhooking of interstrand crosslinks to restart stalled replication forks.
An archaeal DNA glycosylase originally annotated as a restriction endonuclease uses sequence-specific excision of adenine to induce double-strand breaks in foreign DNA.
Acknowledgments
Research on DNA glycosylases in our laboratory is supported by the National Science Foundation (MCB-1517695). A.A.R. and N.P.B. are supported by the National Science Foundation Graduate Research Fellowship Program (DGE-1445197).
GLOSSARY
- C-H/π interaction
a weak electrostatic interaction between the dipole of a C-H bond and the quadrupole of a π system
- Fanconi anemia (FA) pathway
a complex network for coordination of DNA repair pathways, often associated with repair of interstrand crosslinks and found only in vertebrates
- Genotoxin
a chemical agent that damages DNA
- Homologous recombination (HR)
a type of genetic recombination in whichnucleotide sequences are exchanged between two similar or identical molecules of DNA, commonly used by cells to repair double-strand breaks
- Intercalating residue
a protein residue that is inserted between stacked bases in the DNA duplex, often associated with base flipping
- Interstrand crosslink (ICL)
a highly toxic lesion that covalently links opposing strands of DNA, preventing separation into the single-stranded templates that are necessary for transcription and replication
- Monoadduct
a lesion in which the modifying agent is covalently attached to a single nucleotide, often used in association with compounds capable of simultaneously and covalently attaching to two nucleotides, thereby generating an interstrand crosslink
- Natural product
a chemical compound produced by a living organism
- Nucleotide excision repair (NER)
a versatile mechanism for removal of diverse bulky and helix-distorting/destabilizing lesions in which a short segment of the damaged strand is excised and replaced with newly synthesized DNA
- Replication fork
a Y-shaped DNA structure formed by separation of double-stranded DNA into two single-stranded templates during replication
- Replisome
a large multiprotein complex assembled at replication forks and comprised of the enzymes necessary to replicate the genome, including a DNA helicase to unwind the parental duplex, multiple DNA polymerases to synthesize the daughter strands, and numerous accessory proteins with various functions
- Secondary metabolite
an organic compound produced by a microorganism but not required for normal growth or reproduction, frequently associated with interspecies defense mechanisms
- Sister chromatid
one of two identical copies of a chromosome formed during replication
- Translesion synthesis (TLS)
error-prone DNA synthesis by a low-fidelity polymerase to bypass DNA damage in the template strand, often introducing a mutation in the daughter strand
- Ubiquitinylation
conjugation of one or more molecules of the small regulatory protein ubiquitin with a target protein to modulate its function or to mark it for processing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Friedberg EC et al. (2006) DNA Repair and Mutagenesis, 2nd edn., ASM Press. [Google Scholar]
- 2.Gates KS (2009) An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem. Res. Toxicol 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson SP and Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krokan HE and Bjoras M (2013) Base excision repair. Cold Spring Harb. Perspect. Biol 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zharkov DO (2008) Base excision DNA repair. Cell. Mol. Life Sci 65, 1544–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robertson AB et al. (2009) Base excision repair: The long and short of it. Cell. Mol. Life Sci 66, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stivers JT (2004) Site-specific DNA damage recognition by enzyme-induced base flipping. Prog. Nucleic Acid Res. Mol. Biol 77, 37–65. [DOI] [PubMed] [Google Scholar]
- 8.Hitomi K et al. (2007) The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair (Amst.) 6, 410–428. [DOI] [PubMed] [Google Scholar]
- 9.Brooks SC et al. (2013) Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta 1834, 247–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramstein J and Lavery R (1988) Energetic coupling between DNA bending and base pair opening. Proc. Natl. Acad. Sci. U.S.A 85, 7231–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts RJ and Cheng X (1998) Base flipping. Annu. Rev. Biochem 67, 181–198. [DOI] [PubMed] [Google Scholar]
- 12.Jiang YL et al. (2001) Turning on uracil-DNA glycosylase using a pyrene nucleotide switch. J. Biol. Chem 276, 42347–42354. [DOI] [PubMed] [Google Scholar]
- 13.Yang W (2006) Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair (Amst.) 5, 654–666. [DOI] [PubMed] [Google Scholar]
- 14.Tenconi E and Rigali S (2018) Self-resistance mechanisms to DNA-damaging antitumor antibiotics in actinobacteria. Curr. Opin. Microbiol 45, 100–108. [DOI] [PubMed] [Google Scholar]
- 15.Xu H et al. (2012) Self-resistance to an antitumor antibiotic: A DNA glycosylase triggers the base-excision repair system in yatakemycin biosynthesis. Angew. Chem. Int. Ed 51, 10532–10536. [DOI] [PubMed] [Google Scholar]
- 16.Wang S et al. (2016) Characterization of a novel DNA glycosylase from S. sahachiroi involved in the reduction and repair of azinomycin B induced DNA damage. Nucleic Acids Res 44, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Q et al. (2008) Characterization of the azinomycin B biosynthetic gene cluster revealing a different iterative type I polyketide synthase for naphthoate biosynthesis. Chem. Biol 15, 693–705. [DOI] [PubMed] [Google Scholar]
- 18.Rubinson EH et al. (2010) An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature 468, 406–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S et al. (2017) Unified biosynthetic origin of the benzodipyrrole subunits in CC-1065. ACS Chem. Biol 12, 1603–1610. [DOI] [PubMed] [Google Scholar]
- 20.Shi R et al. (2018) Structural biology of the HEAT-like repeat family of DNA glycosylases. Bioessays 40, e1800133. [DOI] [PubMed] [Google Scholar]
- 21.Jacob C and Weissman KJ (2017) Unpackaging the roles of Streptomyces natural products. Cell Chem. Biol 24, 1194–1195. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi Y et al. (2003) Yatakemycin, a novel antifungal antibiotic produced by Streptomyces sp. TP-A0356. J. Antibiot. (Tokyo) 56, 107–113. [DOI] [PubMed] [Google Scholar]
- 23.Tichenor MS et al. (2007) Systematic exploration of the structural features of yatakemycin impacting DNA alkylation and biological activity. J. Am. Chem. Soc 129, 10858–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacMillan KS and Boger DL (2009) Fundamental relationships between structure, reactivity, and biological activity for the duocarmycins and CC-1065. J. Med. Chem 52, 5771–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunz D et al. (1996) Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J. Biol. Chem 271, 25089–25098. [DOI] [PubMed] [Google Scholar]
- 26.Mullins EA et al. (2017) Toxicity and repair of DNA adducts produced by the natural product yatakemycin. Nat. Chem. Biol 13, 1002–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan H et al. (2017) GyrI-like proteins catalyze cyclopropanoid hydrolysis to confer cellular protection. Nat. Commun 8, 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alseth I et al. (2006) A new protein superfamily includes two novel 3-methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol. Microbiol 59, 1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mullins EA et al. (2015) The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions. Nature 527, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalhus B et al. (2007) Structural insight into repair of alkylated DNA by a new superfamily of DNA glycosylases comprising HEAT-like repeats. Nucleic Acids Res 35, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubinson EH et al. (2008) A new protein architecture for processing alkylation damaged DNA: The crystal structure of DNA glycosylase AlkD. J. Mol. Biol 381, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullins EA et al. (2014) The substrate binding interface of alkylpurine DNA glycosylase AlkD. DNA Repair (Amst.) 13, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsons ZD et al. (2016) A catalytic role for C-H/π interactions in base excision repair by Bacillus cereus DNA glycosylase AlkD. J. Am. Chem. Soc 138, 11485–11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawley PD, and D.H. Phillips (1995) DNA Adducts from Chemotherapeutic Agents. Mutation Research 355, 13–40. [DOI] [PubMed] [Google Scholar]
- 35.Hata T et al. (1954) Carzinophilin, a new tumor inhibitory substance produced by streptomyces. I. J. Antibiot. (Tokyo) 7, 107–112. [PubMed] [Google Scholar]
- 36.Armstrong RW et al. (1992) Novel interstrand cross-links induced by the antitumor antibiotic carzinophilin/azinomycin B. J. Am. Chem. Soc 114, 3144–3145. [Google Scholar]
- 37.Fujiwara T et al. (1999) Highly efficient DNA interstrand crosslinking induced by an antitumor antibiotic, carzinophilin. Tetrahedron Lett 40, 315–318. [Google Scholar]
- 38.Alcaro S and Coleman RS (2000) A molecular model for DNA cross-linking by the antitumor agent azinomycin B. J. Med. Chem 43, 2783–2788. [DOI] [PubMed] [Google Scholar]
- 39.Coleman RS et al. (2002) Studies on the mechanism of action of azinomycin B: definition of regioselectivity and sequence selectivity of DNA cross-link formation and clarification of the role of the naphthoate. J. Am. Chem. Soc 124, 13008–13017. [DOI] [PubMed] [Google Scholar]
- 40.McVey M (2010) Strategies for DNA interstrand crosslink repair: insights from worms, flies, frogs, and slime molds. Environ. Mol. Mutagen 51, 646–658. [DOI] [PubMed] [Google Scholar]
- 41.Deans AJ and West SC (2011) DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clauson C et al. (2013) Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol 5, a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noll DM et al. (2006) Formation and repair of interstrand cross-links in DNA. Chem. Rev 106, 277–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y and Li L (2013) DNA crosslinking damage and cancer - a tale of friend and foe. Transl. Cancer Res 2, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J and Walter JC (2014) Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst.) 19, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu RA et al. (2019) TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J et al. (2015) DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol 22, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amunugama R et al. (2018) Replication fork reversal during DNA interstrand crosslink repair requires CmG unloading. Cell Rep 23, 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foulke-Abel J et al. (2011) Characterization of AziR, a resistance protein of the DNA cross-linking agent azinomycin B. Mol. Biosyst 7, 2563–2570. [DOI] [PubMed] [Google Scholar]
- 50.Mullins EA et al. (2017) Structure of a DNA glycosylase that unhooks interstrand cross-links. Proc. Natl. Acad. Sci. U.S.A 114, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moldovan GL and D’Andrea AD (2009) How the fanconi anemia pathway guards the genome. Annu. Rev. Genet 43, 223–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kee Y and D’Andrea AD (2010) Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev 24, 1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Long DT et al. (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 56, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Semlow DR et al. (2016) Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 167, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin PR et al. (2017) The human DNA glycosylases NEIL1 and NEIL3 excise psoralen-induced DNA-DNA cross-links in a four-stranded DNA structure. Sci. Rep 7, 17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minko IG et al. (2019) Processing of N(5)-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair (Amst.) 73, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z et al. (2017) A role for the base excision repair enzyme NEIL3 in replication- dependent repair of interstrand DNA cross-links derived from psoralen and abasic sites. DNA Repair (Amst.) 52, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bandaru V et al. (2002) A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst.) 1, 517–529. [DOI] [PubMed] [Google Scholar]
- 59.Hazra TK et al. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morland I et al. (2002) Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res 30, 4926–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takao M et al. (2002) Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J 21, 3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu M et al. (2013) Neil3, the final frontier for the DNA glycosylases that recognize oxidative damage. Mutat. Res 743–744, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hazra TK et al. (2002) Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem 277, 30417–30420. [DOI] [PubMed] [Google Scholar]
- 64.Dou H et al. (2003) Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem 278, 49679–49684. [DOI] [PubMed] [Google Scholar]
- 65.Banerjee D et al. (2011) Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem 286, 6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Torisu K et al. (2005) Hematopoietic tissue-specific expression of mouse Neil3 for endonuclease VIII-like protein. J. Biochem 138, 763–772. [DOI] [PubMed] [Google Scholar]
- 67.Hildrestrand GA et al. (2009) Expression patterns of Neil3 during embryonic brain development and neoplasia. BMC Neurosci 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neurauter CG et al. (2012) Release from quiescence stimulates the expression of human NEIL3 under the control of the Ras dependent ERK-MAP kinase pathway. DNA Repair (Amst.) 11, 401–409. [DOI] [PubMed] [Google Scholar]
- 69.Bandaru V et al. (2007) Human endonuclease VIII-like (NEIL) proteins in the giant DNA Mimivirus. DNA Repair (Amst.) 6, 1629–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takao M et al. (2009) Human Nei-like protein NEIL3 has AP lyase activity specific for single-stranded DNA and confers oxidative stress resistance in Escherichia coli mutant. Genes Cells 14, 261–270. [DOI] [PubMed] [Google Scholar]
- 71.Liu M et al. (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A 107, 4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu M et al. (2012) Expression and purification of active mouse and human NEIL3 proteins. Protein Expr. Purif 84, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu M et al. (2013) Structural characterization of a mouse ortholog of human NEIL3 with a marked preference for single-stranded DNA. Structure 21, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Couvé-Privat S et al. (2007) Psoralen-induced DNA adducts are substrates for the base excision repair pathway in human cells. Nucleic Acids Res 35, 5672–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Couvé S et al. (2009) The human oxidative DNA glycosylase NEIL1 excises psoralen- induced interstrand DNA cross-links in a three-stranded DNA structure. J. Biol. Chem 284, 11963–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheng S et al. (1988) Use of psoralen-modified oligonucleotides to trap three-stranded RecA-DNA complexes and repair of these cross-linked complexes by ABC excinuclease. J. Biol. Chem 263, 15110–15117. [PubMed] [Google Scholar]
- 77.Kaushik Tiwari M et al. (2016) Triplex structures induce DNA double strand breaks via replication fork collapse in NER deficient cells. Nucleic Acids Res 44, 7742–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilson DM III and Seidman MM (2010) A novel link to base excision repair? Trends Biochem. Sci 35, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doublié S et al. (2004) The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc. Natl. Acad. Sci. U.S.A 101, 10284–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prakash A et al. (2014) Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: Activity, structure, and the effect of editing. DNA Repair (Amst.) 14, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu C et al. (2016) Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc. Natl. Acad. Sci. U.S.A 113, 7792–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qi Y et al. (2009) Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature 462, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bickle TA and Kruger DH (1993) Biology of DNA restriction. Microbiol. Rev 57, 434–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murray NE (2002) Immigration control of DNA in bacteria: Self versus non-self. Microbiology 148, 3–20. [DOI] [PubMed] [Google Scholar]
- 85.Samson JE et al. (2013) Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol 11, 675–687. [DOI] [PubMed] [Google Scholar]
- 86.Pingoud A et al. (2005) Type II restriction endonucleases: Structure and mechanism. Cell. Mol. Life Sci 62, 685–707. [DOI] [PubMed] [Google Scholar]
- 87.Ishikawa K et al. (2005) Discovery of a novel restriction endonuclease by genome comparison and application of a wheat-germ-based cell-free translation assay: PabI (5’GTA/C) from the hyperthermophilic archaeon Pyrococcus abyssi. Nucleic Acids Res 33, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miyazono K et al. (2007) Novel protein fold discovered in the PabI family of restriction enzymes. Nucleic Acids Res 35, 1908–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Humbert O and Salama NR (2008) The Helicobacter pylori HpyAXII restriction-modification system limits exogenous DNA uptake by targeting GTAC sites but shows asymmetric conservation of the DNA methyltransferase and restriction endonuclease components. Nucleic Acids Res 36, 6893–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miyazono K et al. (2014) A sequence-specific DNA glycosylase mediates restriction- modification in Pyrococcus abyssi. Nat. Commun 5, 3178. [DOI] [PubMed] [Google Scholar]
- 91.Fukuyo M et al. (2015) Restriction-modification system with methyl-inhibited base excision and abasic-site cleavage activities. Nucleic Acids Res 43, 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y et al. (2017) Restriction glycosylases: Involvement of endonuclease activities in the restriction process. Nucleic Acids Res 45, 1392–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Watanabe M et al. (2006) Hyperthermophilic DNA methyltransferase M.PabI from the archaeon Pyrococcus abyssi. Appl. Environ. Microbiol 72, 5367–5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang D et al. (2016) Tetrameric structure of the restriction DNA glycosylase R.PabI in complex with nonspecific double-stranded DNA. Sci. Rep 6, 35197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen X et al. (2000) Transition-state analysis for depurination of DNA by ricin A-chain. J. Am. Chem. Soc 122, 6527–6534. [Google Scholar]
- 96.Ho MC et al. (2009) Transition state analogues in structures of ricin and saporin ribosome inactivating proteins. Proc. Natl. Acad. Sci. U.S.A 106, 20276–20281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kapinos LE et al. (2011) Understanding the acid-base properties of adenosine: the intrinsic basicities of N1, N3 and N7. Chem. Eur. J 17, 8156–8164. [DOI] [PubMed] [Google Scholar]
- 98.Stivers JT and Jiang YL (2003) A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev 103, 2729–2759. [DOI] [PubMed] [Google Scholar]
- 99.Drohat AC and Maiti A (2014) Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem 12, 8367–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Drohat AC and Coey CT (2016) Role of base excision “repair” enzymes in erasing epigenetic marks from DNA. Chem. Rev 116, 12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sadeghian K et al. (2014) Ribose-protonated DNA base excision repair: A combined theoretical and experimental study. Angew. Chem. Int. Ed 53, 10044–10048. [DOI] [PubMed] [Google Scholar]
- 102.Sadeghian K and Ochsenfeld C (2015) Unraveling the base excision repair mechanism of human DNA glycosylase. J. Am. Chem. Soc 137, 9824–98231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.