

Abstract
Rett syndrome (RTT) is a severe neurological disorder usually caused by mutations in the MECP2 gene. Since the MECP2 gene is located on the X chromosome, X chromosome inactivation (XCI) could play a role in the wide range of phenotypic variation of RTT patients; however, classical methylation-based protocols to evaluate XCI could not determine whether the preferentially inactivated X chromosome carried the mutant or the wild-type allele. Therefore, we developed an allele-specific methylation-based assay to evaluate methylation at the loci of several recurrent MECP2 mutations. We analyzed the XCI patterns in the blood of 174 RTT patients, but we did not find a clear correlation between XCI and the clinical presentation. We also compared XCI in blood and brain cortex samples of two patients and found differences between XCI patterns in these tissues. However, RTT mainly being a neurological disease complicates the establishment of a correlation between the XCI in blood and the clinical presentation of the patients. Furthermore, we analyzed MECP2 transcript levels and found differences from the expected levels according to XCI. Many factors other than XCI could affect the RTT phenotype, which in combination could influence the clinical presentation of RTT patients to a greater extent than slight variations in the XCI pattern.
Subject terms: Genetics, Molecular medicine
Introduction
Rett syndrome (RTT, OMIM #312750) is a severe neurodevelopmental disorder characterized by a period of normal development until 6-18 months of age followed by a regression of neurological traits. RTT features include compromised brain functions, severe mental retardation, epilepsy, regression of purposeful hand use and language, breathing disturbances, gait apraxia and repetitive stereotyped hand movements1–3. RTT has an incidence of 1:10,000–20,000 live births and affects mainly young females4, being the second most common cause of severe mental retardation in females after Down syndrome.
The association of RTT with mutations in methyl-CpG binding protein 2 (MECP2; Xq28; OMIM *300005) gene was recognized in 19992. Since then, more than 800 different mutations in MECP2 have been identified in more than 95% of patients with classic RTT5,6. There are also some atypical RTT variants, such as the early onset seizure variant and the congenital variant, which have been associated with mutations in cyclin-dependent kinase-like 5 (CDKL5; Xp22; OMIM *300203) and forkhead box protein G1 (FOXG1; 14q12; OMIM *164874), respectively7,8. However, the vast majority of RTT patients have a de novo mutation in MECP2, and there are 8 mutation hotpots with recurrent mutations (p.Thr158Met, p.Arg255*, p.Arg168*, p.Arg306Cys, p.Arg294*, p.Arg270*, p.Arg133Cys and p.Arg106Trp), which are responsible for over 60% of all RTT cases9,10.
Increasing experience has shown that RTT patients present a large degree of phenotypic variation2. Patients with truncating mutations in MECP2 tend to show a more severe phenotype than those with missense mutations4, and there are also phenotypical presentation differences between patients with the same mutation11–13.
These clinical differences have been attributed, at least in part, to X chromosome inactivation (XCI). Through the XCI process, mammalian female cells inactivate one of the two X chromosomes to compensate for gene dosage. XCI is a stochastic process that takes place in the initial stages of the embryogenesis, causing a mosaic expression of X-linked genes in the adult organism3,14,15. Since MECP2 is located on the X chromosome, the severity of RTT could be theoretically regulated by XCI, showing a more severe phenotype as more cells express the mutated MECP214.
Some cases of healthy carriers of RTT-causing mutations with highly skewed XCI patterns have been documented14,16,17, as have cases of RTT patients with milder symptoms who also presented a skewed XCI pattern13,17,18. However, in most XCI studies in RTT, the phase of the two X chromosomes was not determined, so the XCI pattern could only be classified as either skewed or random. Therefore, no evidence of whether the preferentially inactivated chromosome was the mutant or the wild-type (WT) could be obtained.
We have developed an allele-specific methylation-based assay to evaluate methylation on the loci of several recurrent MECP2 mutations, allowing for evaluation of the XCI pattern while taking into account which is the mutant and which is the wild-type allele. We compared the results from the classical androgen receptor assay for evaluating X chromosome inactivation (XCI-AR) with the allele-specific X chromosome inactivation (XCI-AS) assay we developed. We also compared all XCI results with a score of clinical severity of the clinical presentation of RTT to determine if we could correlate the XCI pattern with milder or more severe forms of RTT. Our cohort included 221 RTT patients with several recurrent mutations and two deletions in MECP2, for whom we could evaluate XCI patterns in blood. Moreover, we also assessed XCI in brain samples of two patients and compared the XCI status to blood to determine if it could be used as an accurate predictor. Finally, we measured MECP2 RNA levels in brain samples to determine whether they correlated with the XCI pattern detected.
Results
Allele-specific X chromosome inactivation and XCI skewing in blood samples
For each patient, we performed an XCI-AR and the corresponding XCI-AS when blood samples were available (174/221 patients), and we also calculated the global score of the clinical presentation when clinical data were available (181/221 patients). The reference values for considering an XCI pattern as skewed in the literature are usually established at an 80:20 ratio14,19, so we also used that threshold to allow the comparison of our results with previous studies. The entire list of XCI results and clinical scores for all patients can be found in Supplementary Table S1.
The overall tendency of our cohort was to have random XCI. However, 9.8% of our patients showed a skewed XCI pattern (80:20 or higher; Table 1), which is similar to what was found in other studies13,20. No patients with p. R152R, p.T158M or p. P225R mutations showed skewed XCI patterns in either XCI-AR or XCI-AS.
Table 1.
Proportion of patients per mutation with a skewed XCI pattern according to at least one of the two techniques used for assessing XCI (XCI-AR and XCI-AS).
| Mutation | Type of mutation | MeCP2 region | Number of patients with skewed XCI | % of patients with skewed XCI |
|---|---|---|---|---|
| c.455C > G (p.P152R) | Missense | MBD | 0/6 | 0% |
| c.473C > T (p.T158M) | Missense | MBD | 0/33 | 0% |
| c.502C > T (p.R168X) | Nonsense | IDR | 5/29 | 17.2% |
| c.674C > G (p.P255R) | Missense | TRD | 0/2 | 0% |
| c.763C > T (p.R255X) | Nonsense | TRD | 4/36 | 11.1% |
| c.806delG (p.G269fs) | Frameshift | TRD-NLS | 1/11 | 9.1% |
| c.808C > T (p.R270X) | Nonsense | TRD-NLS | 4/20 | 20% |
| c.880C > T (p.R294X) | Nonsense | TRD | 1/20 | 5% |
| c.916C > T (p.R306C) | Missense | TRD | 1/15 | 6.7% |
| Large deletions | Deletion | Exons 3-4 | 1/2 | 50% |
| All | — | — | 17/174 | 9.8% |
When we applied the 80:20 skewing threshold, 17 out of 174 patients presented a skewed XCI pattern according to at least one of the two XCI assays performed (Table 2). We compared these patients’ clinical severity scores with the average clinical score of RTT patients with the same mutation. We found that, when the clinical score was available, in the majority of cases this value was included in the interval of µ ± σ (central 68% of individuals in a normal distribution) of the patients with the same mutation.
Table 2.
Data of patients with skewed XCI according to at least one of the two assays.
| Patient Number | XCI-AR | XCI-AS | Global Score | |
|---|---|---|---|---|
| WT | Mut | |||
| Patients with c.502C > T (p.Arg168*) mutation | = 13.12 (SD = 3.361) | |||
| P47 | n.i. | 81.5 | 18.5 | 13 |
| P60 | 84:16 | 28 | 72 | 16 |
| P68 | 75:25 | 15.5 | 84.5 | NA |
| P70 | 85:15 | 35 | 65 | NA |
| P74 | 81:19 | 55.5 | 44.5 | NA |
| Patients with c.763C > T (p.Arg255*) mutation | = 15.21 (SD = 3.213) | |||
| P83 | 85:15 | 57 | 43 | NA |
| P84 | 87:13 | 55.5 | 44.5 | 13 |
| P85 | 80:20 | 28 | 72 | 14 |
| P107 | 87:13 | 68 | 32 | 11 |
| Patients with c.806delG (p.Gly269fs) mutation | = 14.29 (SD = 4.112) | |||
| P139 | 82:18 | 58 | 42 | NA |
| Patients with c.808C > T (p.Arg270*) mutation | = 14.69 (SD = 3.846) | |||
| P143 | 97:3 | 16 | 84 | 18 |
| P144 | 84:16 | 21 | 79 | NA |
| P145 | 81:19 | 30 | 70 | 9 |
| P146 | 80:20 | 73 | 27 | 13 |
| Patients with c.880C > T (p.Arg255*) mutation | = 10.46 (SD = 2.993) | |||
| P191 | 89:11 | 49 | 51 | NA |
| Patients with c.916C > T (p.Arg306Cys) mutation | = 11.18 (SD = 3.065) | |||
| P195 | 89:11 | 59.5 | 40.5 | 9 |
| Patients with deletions in MECP2 | ||||
| P220 | 88:12 | 6.73 | 93.27 | NA |
The XCI-AR column shows the results of the AR XCI assay (percentage of inactivation of each allele). The XCI-AS WT and Mut columns show the results of the allele-specific XCI assay (percentage of inactivation of each allele, mean of two replicates n = 2 or three replicates n = 3 in the cases of the deletions). The Global Score column shows the average () score and its standard deviation (SD) in brackets for the patients of our cohort with each mutation. Bold formatting indicates patients with a clinical score lower than the interval µ-σ for the average clinical score of their mutation. n.i. = polymorphism noninformative for the assay. NA = clinical data not available.
There were only two patients who had a clinical score lower than the interval µ-σ for their mutation (P107 and P145, Table 2, in bold). In the case of patient P107, the preferentially inactivated allele was the WT allele, while in the case of patient P145 the mutant allele was inactivated. The results from patient P145 seem to be consistent with the theory that when the chromosome that harbors the MECP2 mutation is preferentially inactivated, the clinical presentation of RTT may be milder.
Allele-specific X chromosome inactivation and XCI skewing in brain samples
We also performed XCI-AR and XCI-AS assays in samples of several brain regions of two patients with the c.763C > T mutation (Table 3). The XCI-AS assay was useful for assessing the XCI pattern in both patients, but especially in patient P119, since in this case, the polymorphism in the AR locus was noninformative for the XCI-AR assay.
Table 3.
Data of patients P109 and P119 with the c.763C > T mutation.
| Sample | XCI-AR | XCI-AS | |
|---|---|---|---|
| WT | Mut | ||
| Patient 109 (Clinical score = 20) | |||
| Frontal Cortex | 65:35 | 26 | 74 |
| Occipital Cortex | 58:42 | 59 | 41 |
| Parietal Cortex | 64:36 | 40 | 60 |
| Temporal Cortex | 60:40 | 32 | 68 |
| White matter | 59:41 | 23 | 77 |
| Brain stem | 59:41 | 31 | 69 |
| Striatum | 61:39 | 51 | 49 |
| Cerebellum | 55:45 | 43 | 57 |
| Blood | 73:27 | 64 | 36 |
| Patient 119 (Clinical score = 19) | |||
| Frontal Cortex | n.i. | 48 | 52 |
| Occipital Cortex | n.i. | NA | NA |
| Parietal Cortex | n.i. | 56 | 44 |
| Temporal Cortex | n.i. | 73 | 27 |
| White matter | n.i. | 46 | 54 |
| Brain stem | n.i. | 38 | 62 |
| Striatum | n.i. | 50 | 50 |
| Cerebellum | n.i. | 50 | 50 |
| Blood | n.i. | 34 | 66 |
The XCI-AR column shows the results of the AR XCI assay (percentage of inactivation of each allele). The XCI-AS WT and Mut columns show the results of the allele-specific XCI assay (percentage of inactivation of each allele). n.i. = polymorphism noninformative for the assay. NA = data not available.
Although no samples showed skewed XCI by either assay, there was no clear homogeneity among blood and brain samples. Some samples, such as the frontal cortex or the white matter sample of patient P109, showed an XCI pattern closer to the skewing threshold than other regions, such as the cerebellum, of the same patient. In patient P119, the vast majority of samples were close to the random XCI pattern, but the temporal cortex sample showed an XCI pattern closer to the skewing threshold.
Brain RNA analysis
Finally, we analyzed frontal and occipital cortex RNA samples. We performed RT-PCR to obtain cDNA samples so that we could perform Sanger sequencing to check if we could detect the presence of one allele over the other (Fig. 1).
Figure 1.

Brain RNA Sanger Sequencing. cDNA analysis of brain samples. Electropherograms obtained from Sanger sequencing of frontal and occipital cortex cDNA samples. Blue peaks correspond to the C allele (WT), while red peaks correspond to the T allele (mutated), and the red box highlights the locus of the c.763C > T mutation in heterozygosis. Inactivation ratios are shown as inactivation WT:inactivation Mut.
In cDNA samples from patient P109, the T allele (mutated allele) was overrepresented, while in samples from patient P119, the C allele (WT allele) was overrepresented. However, both patients presented a severe form of RTT, with clinical scores of 20 and 19, respectively.
The cDNA analysis was not conclusive since Sanger sequencing is not the best technique for quantifying the RNA of each allele. However, the sequencing analysis seemed to indicate that one allele was more frequently present than the other, although the XCI assay results showed inactivation patterns that did not reach the threshold for classifying the XCI pattern as skewed in any of the two patients and regions.
We later confirmed our findings in the frontal cortex samples by qRT-PCR, a more suitable technique for quantifying RNA levels (Fig. 2a,b). We found that in samples from patient P109, the mutated allele was overexpressed, while in samples from patients P119, the WT allele was overexpressed.
Figure 2.
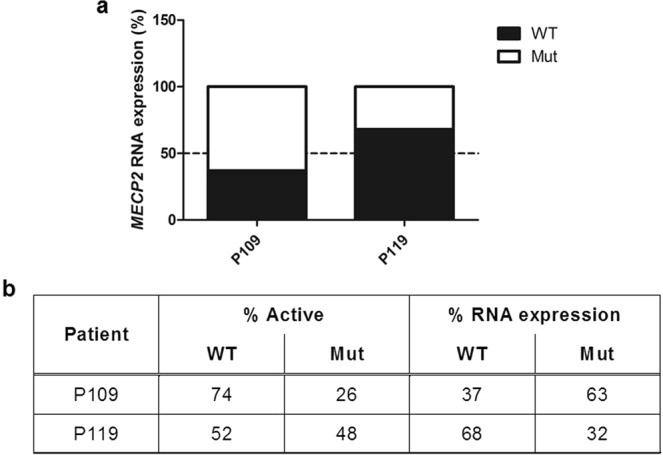
Brain RNA qRT-PCR analysis and comparison with XCI-AS assay results. (a) cDNA analysis of brain samples. The results obtained by qRT-PCR of frontal cortex RNA samples (% of expression of each allele). The discontinuous line indicates 50% of the expression of each allele (each allele is equally present in the sample). (b) Comparison of XCI and qRT-PCR data from patients P109 and P119 with the c.763C > T mutation. Data are shown as % of activation of each X chromosome (% Active) and % RNA expression measured by qRT-PCR.
Discussion
The XCI-AS assay allowed us to describe the XCI patterns of patients previously classified as noninformative by the classical XCI-AR assay and to identify which MECP2 allele (mutated or WT) was preferentially inactivated in cases of skewed XCI pattern.
Differences between the XCI patterns obtained by both techniques can be explained because in each technique, the methylation status is only analyzed at a single locus, and the methylation of a single cytosine residue may not be representative of the inactivation status of the entire X chromosome21,22. Different studies have shown that when methylation in several loci of the X chromosome is assessed, different ratios of XCI can be obtained, with up to 27% of variation21,22. Therefore, the use of several loci for characterizing XCI would indicate the true XCI pattern more consistently21.
Gathering data from both XCI assays performed with samples of 174 patients, we found that 9.8% of patients had skewed XCI patterns (80:20 XCI ratio or higher). Other studies have found either similar results13,20 or a considerably higher incidence of skewing, up to 43%, among RTT patients23. Some authors claim that most of the patients who meet the diagnostic criteria for RTT have a random XCI pattern, while those with skewed XCI patterns may not meet all the criteria and therefore are not included in some RTT studies18.
However, the percentage of patients in our cohort with skewed XCI patterns varied among different types of mutations. Mutations that produce a truncated protein result in a more severe phenotype than missense mutations23, and skewed XCI patterns were more common in RTT patients with deletions and nonsense mutations than in those with missense mutations. This could be due to a protective effect related to the severity of the mutation. It is possible that mutations producing a less functional, truncated protein (deletions and nonsense mutations) cause cells to preferentially inactivate the X chromosome harboring the mutation. It has been shown that skewed XCI can be caused by a selective advantage of cells with a particular active X chromosome proliferating faster than cells where the other X chromosome is active15,24,25. This type of skewing has been described in up to 50% of familial cases of X-linked mental retardation disorders26.
This skewed proliferation could be the case for patient P220 (Table 2), who had a large deletion in MECP2 and showed a skewed XCI pattern (88:12) by the XCI-AR assay. In this patient, the XCI-AS assay confirmed an extremely skewed XCI pattern and that the preferentially inactivated allele was the mutated allele at a ratio of 93:7. We also found this tendency in several patients with p.Arg168* (P60, P68, P70; Table 2) p.Arg255* (P85; Table 2) and p.Arg270* (P143, P144, P145; Table 2) mutations. However, there were other patients with these same mutations with skewed XCI according to the XCI-AR assay who showed a preferential inactivation of the WT allele when the XCI-AS assay was performed, such as P146 (Table 2). Patient P47 (Table 2), who was noninformative for the XCI-AR assay, also showed a preferential inactivation of the WT allele at a ratio of 81:19 when the XCI-AS assay was performed. These last patients do not support the abovementioned hypothesis.
We found no substantial correlation between the XCI patterns in blood and the clinical presentation of RTT following the scale of evaluation of the RTT phenotype by Monrós, et al.27 (data not shown). We did not observe consistent increases or decreases in the clinical score of RTT patients with a preferential inactivation of the WT or mutated alleles in blood samples.
It has been published that XCI patterns can vary among different tissues22,28. Indeed, we compared the XCI patterns of blood and brain samples of the same patient, and they did not show homogeneous XCI patterns. Although they were small, there was also a slight difference in the XCI patterns between different brain regions of the same patient.
Moreover, it has been shown that blood is especially prone to XCI skewing29 because of the proliferation of different clones of lymphocytes under different conditions22,29. In fact, blood XCI patterns have shown variations at different time points in different studies14. For two of the patients included in the study (P9 and P199; Table S5), we compared two different blood samples from two different extractions. Both patients showed some differences in the results of the XCI assays in the two extraction samples.
The lack of a direct correlation between the XCI patterns in blood and the clinical presentation of RTT could be explained by different reasons. First, we observed that the XCI patterns in blood and different regions of the brain are not necessarily homogeneous. Therefore, if RTT symptoms are caused mainly by the lack of MECP2 function in the brain, it is expected that the severity of the phenotype will be more related to the XCI pattern in the brain than to the XCI pattern in the blood.
Moreover, there are many other factors that can influence the presentation of the RTT phenotype, such as other polymorphisms and genetic variants, the expression levels of other genes and environmental conditions4. It is likely that the combination and addition of these additional factors can influence the phenotype to a greater extent than only the XCI pattern in the brain.
RTT symptoms arise from either a partial or a complete loss of function of MECP2 in neurons13,30. RTT affects mainly females, partly because a complete loss of function of MECP2 in males is so damaging that it can cause death in the first months of life or even before birth. The severity of the male phenotype points towards a dose-dependent mechanism of action of MECP2, where the expression of the mutant MECP2 in a high proportion of cells causes the RTT phenotype13,31. It is possible that in females, slight deviations from random 50:50 XCI ratios do not cause sufficient changes in the levels of the mutant MECP2 in the brain to be translated into a different phenotype.
However, it is possible that in more extreme cases, the effect is more remarkable. This could be similar to the case of female carriers of the MECP2 duplication who show an extremely skewed XCI pattern with the mutant chromosome inactivated in most of their cells. In these cases, where a greater number of cells have inactivated the mutant chromosome, the effects of the XCI pattern are more important and cause the carrier of the MECP2 duplication not to present the MECP2 duplication syndrome. The same phenomenon could occur with pathogenic mutations in MECP2. If there is an extremely skewed XCI pattern in the brain, where a greater number of cells express the WT copy of MECP2, a threshold of MECP2 function could be reached, and the RTT phenotype would therefore not be expressed. In some familial cases of RTT, it has been observed that a healthy mother with extremely skewed XCI can be a carrier of a pathogenic mutation responsible for causing RTT in her offspring16–18, although she remains asymptomatic. Some authors have claimed that these familial cases of RTT are only possible due to the presence of two coincident traits: RTT and the trait for skewed XCI, which would be genetically determined14,16.
The differences between the XCI patterns measured and the levels of each allele observed in Sanger sequencing and qRT-PCR could be due to RNA degradation, both in the postmortem interval and during life due to the nonsense-mediated mRNA decay (NMD) pathway, which could degrade mutant mRNA because of its potential to be translated into a truncated protein.
However, brain RNA levels of each allele seemed to show discrepancies with the XCI patterns identified in our XCI assays. Some authors have noticed discordances between the XCI pattern according to the XCI-AR assay and the quantification of the AR gene expression32. These discrepancies suggest, first, that the methylation assay may not always be representative of XCI and, second, that gene transcript levels may be regulated by more factors than XCI.
The difference between the XCI pattern and the final RNA levels of each allele suggests that the levels of MECP2 are not directly determined by the XCI pattern and that there could be mechanisms other than XCI involved in regulating MECP2 transcript levels. Consistent with what we have discussed, there might be other genes involved in regulating MECP2 transcription and/or RNA degradation, causing changes in the final levels of functional MECP210. Therefore, XCI may not necessarily be determining the severity of the clinical presentation of RTT, which would be more related to the levels of functional MECP2 in the brain30,31.
Nevertheless, it is important to keep in mind that we are measuring MECP2 transcript levels from brain bulk RNA. Since different neuronal types have showed diverse transcriptional profiles in several studies33, the levels of the MECP2 transcripts we measured do not necessarily reflect these transcript’s levels in neurons relevant for RTT pathophysiology.
Although one patient showed higher levels of the MECP2 mutant transcript than the other, the clinical severity scores of both patients were not dissimilar (20 vs 19). This score similarity supports the hypothesis that slight deviations from a 1:1 ratio of each allele produce little to no change in the RTT phenotype. It is possible that more consistent differences would be noticeable if one allele was more prevalent than the other, such as in asymptomatic carriers with an XCI pattern close to the 100:0 ratio.
In conclusion, our results show that the relationship between XCI and the severity of the RTT phenotype is not straightforward. Factors other than XCI can influence MECP2 transcript levels, and presumably many additional factors, such as genetic polymorphisms and the expression of other genes, may influence the final clinical presentation of RTT. Therefore, probably only extremely skewed XCI patterns affecting neurons can be correlated with milder forms of RTT or asymptomatic carriers.
Materials and Methods
Sample material
The study cohort consisted of 221 RTT patients with one of the 9 following recurrent mutations in the MECP2 gene: c.455C > G-p.Pro152Arg (6 patients), c.473C > T-p.Thr158Met (36 patients), c.502C > T-p.Arg168* (38 patients), c.674C > G-p.Pro225Arg (2 patients), c.763C > T-p.Arg255* (47 patients), c.806delG-p.G269fs (13 patients), c.808C > T-p.Arg270* (31 patients), c.880C > T-p.Arg294* (21 patients) and c.916C > T-p.Arg306Cys (25 patients); and 2 patients with a large deletion in MECP2.
Samples of blood genomic DNA (gDNA) were obtained from peripheral blood leukocytes. Samples of brain gDNA were obtained postmortem from several brain regions (frontal, occipital, temporal and parietal cortex; white matter, brain stem, striatum and cerebellum) of two patients with c.763C > T mutation. RNA was also obtained from the frontal and occipital cortices of such patients. DNA samples were isolated using the saline extraction kit PUREGENE® DNA Isolation Kit of Gentra Systems®, and brain RNA samples were extracted using TRIzol™ Reagent from Invitrogen™.
Ethical approval and informed consent
The study was approved by the ethical committees of Hospital Sant Joan de Déu, CEIC: Comitè d’Ètica d’Investigació Clínica- Fundació Sant Joan de Déu (internal code: PIC-101-15). Written informed consent from the legal guardians of the patients was obtained in accordance with the corresponding ethical protocols to perform the genetic studies, and tissue samples from patients and controls were obtained according to the Helsinki Declaration of 1964, as revised in 200134.
HpaII and HinfI digestion
Digestion of gDNA samples was performed with one of the methylation-sensitive restriction enzymes HpaII or HinfI (New England BioLabs® Inc.), depending on the presence of the relevant enzyme target sequences near the studied loci. In the AR, c.455C > G, c.473C > T, c.502C > T, c.674C > G, c.763C > T, c.806delG, c.808C > T, c.880C > T, c.916C > T and deletion 2 (NM_004992.3: c.887_10015 + 18460del) loci assays HpaII was used, while in the deletion 1 (NM_004992.3: c.27-10677_1192del) locus assay HinfI was used. A total volume of 500 ng of gDNA was digested with 0.5 μL of enzyme in a 25 μL reaction volume in CutSmart 1x Buffer (New England Biolabs® Inc.). Digestions were incubated at 37 °C for 20 minutes followed by another 20 minutes at 80 °C for enzyme inactivation, as established in the enzyme protocol.
PCR amplification and fragment analysis
A pair of primers with the sequences described in Allen, et al.35 was used to amplify the AR polymorphic locus. Allele-specific primers were designed for each MECP2 recurrent mutation included in the study. Primer design was carried out following the recommendations in Liu, et al.36. For the deletion assays, a forward primer was designed inside the deletion locus and another primer immediately after the deletion; they were both amplified with a reverse primer outside the deleted region. All primers used were designed using Primer3web version 4.1.037,38, and they are shown together with PCR conditions for each pair in Supplementary Tables S2, S3 and S4. One primer of each pair was FAM-labeled at the 5′ end.
PCR amplification was performed using the resulting DNA after the digestion and nondigestion of each sample. PCR products were analyzed on a 3500 Genetic Analyzer (Applied Biosystems®) using GeneScan™ – 500 LIZ® Size Standard of Applied Biosystems® as an internal size standard and Peak Scanner Software v1.0. The X chromosome inactivation ratios were calculated as described elsewhere35.
Brain RNA analysis
RT-PCR was performed with frontal and occipital cortex RNA of two patients with the c.763C > T mutation, following the recommendations provided with the SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR from Invitrogen™. Subsequently, Sanger sequencing of the cDNA obtained in the RT-PCR reaction was performed. qPCR was performed in a QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems™) with TaqMan™ Gene Expression Master Mix (Applied Biosystems™) and specific TaqMan™ MGB probes to amplify the mutant and the wild-type alleles. qPCR data were analyzed using the comparative Ct method. Primers and probes were designed using Primer3web version 4.1.036,37, and they are listed in Supplementary Table S5.
Patient phenotype evaluation and correlation analysis
When clinical data were available (181/221 patients), the RTT phenotype was evaluated, and a score was assigned following the scale of evaluation of the RTT phenotype published by Monrós, et al.27.
The linear correlation between the inactivation patterns of the WT allele and the global score of each patient was evaluated using statistical methods that are based on Ordinary Least Squares (OLS) regression models, grouping patients with the same mutation.
Supplementary information
X chromosome inactivation does not necessarily determine the severity of the phenotype in Rett syndrome patients
Acknowledgements
We thank all patients and their families who contributed to this study. The work was supported by grants from the Spanish Ministry of Health (Instituto de Salud Carlos III/FEDER, PI15/01159); Crowdfunding program PRECIPITA, from the Spanish Ministry of Health (Fundación Española para la Ciencia y la Tecnología); the Catalan Association for Rett Syndrome; Fondobiorett and Mi Princesa Rett.
Author Contributions
J.A., S.V., C.X. and M.P. conceived and supervised the study. C.X., S.V., P.P., N.B., A.P., E.G., M.O. and L.B. performed the experiments and collected the data. C.X., J.A. and S.V., analyzed the results. J.A., M.O. and M.P. provided samples and patients’ clinical and genetic information. C.X., J.A. and M.P. wrote the manuscript. All the authors reviewed the article critically for intellectual content.
Data Availability
All data from this article is available in the Supplementary Data.
Competing Interests
The authors declare no competing interests.
Footnotes
A comprehensive list of consortium members appears at the end of the paper
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Judith Armstrong, Email: jarmstrong@sjdhospitalbarcelona.org.
Rett Working Group:
Francisco Javier Aguirre, Montserrat Aleu, Xènia Alonso, Mercè Alsius, Maria Inmaculada Amorós, Guillermo Antiñolo, Lourdes Aquino, Carmen Arellano, Gema Arriola, Rosa Arteaga, Neus Baena, Montserrat Barcos, Nuria Belzunces, Susana Boronat, Tomás Camacho, Jaume Campistol, Miguel del Campo, Andrea Campo, Ramon Cancho, Ramon Candau, Ignacio Canós, María del Carmen Carrascosa, Francisco Carratalá-Marco, Jovaní Casano, Pedro Castro, Ana Cobo, Jaime Colomer, David Conejo, Maria José Corrales, Rocío Cortés, Gabriel Cruz, Gábor Csányi, María Teresa de Santos, María de Toledo, Miguel Del Campo, Mireia Del Toro, Rosario Domingo, Anna Duat, Rosario Duque, Ana María Esparza, Rosa Fernández, Maria Carme Fons, Ana Fontalba, Enrique Galán, Pia Gallano, María José Gamundi, Pedro Luis García, María del Mar García, María García-Barcina, María Jesús Garcia-Catalan, Ángels García-Cazorla, Sixto García-Miñaur, Juan Jose Garcia-Peñas, María Teresa García-Silva, Rosa Gassio, Esther Geán, Belén Gil, Sarenur Gökben, Luis Gonzalez, Veronica Gonzalez, Julieta Gonzalez, Gloria González, Encarna Guillén, Miriam Guitart, Montserrat Guitet, Juan Manuel Gutierrez, Eva Gutiérrez, Jose Luís Herranz, Gemma Iglesias, Iva Karacic, Carlos H. Lahoz, José Ignacio Lao, Pablo Lapunzina, María Jesús Lautre-Ecenarro, María Dolores Lluch, Laura López, Asunción López-Ariztegui, Alfons Macaya, Rosario Marín, Charles M. Lourenço Marquez, Elena Martín, Beatriz Martínez, Eduardo Martínez-Salcedo, María José Mas, Gonzalo Mateo, Pilar Mendez, Amparo Morant Jimenez, Sira Moreno, Fernando Mulas, Juan Narbona, Andrés Nascimento, Manuel Nieto, Tania Fabiola Nunes, Núria Núñez, María Obón, Ignacio Onsurbe, Carlos Ignacio Ortez, Emilio Orts, Francisco Martinez, Rafael Parrilla, Samuel Ignacio Pascual, Ana Patiño, Maria Pérez-Poyato, Belén Pérez-Dueñas, Pilar Póo, Eliodoro Puche, Feliciano Ramos, Miquel Raspall, Ana Roche, Susana Roldan, Jordi Rosell, Cesar Ruiz, María Luz Ruiz-Falcó, Maria Eugenia Russi, Jordi Samarra, Victoria San Antonio, Ivan Sanchez, Xavier Sanmartin, Ana Sans, Alfredo Santacana, Sabine Scholl-Bürgi, Nuria Serrano, Mercedes Serrano, Pilar Martin-Tamayo, Adrián Tendero, Jaime Torrents, Diego Tortosa, Emma Triviño, Ledia Troncoso, Eulàlia Turón, Pilar Vázquez, Carlos Vázquez, Ramón Velázquez, Clara Ventura, Alfonso Verdú, Anna Vernet, M. Tomás Vila, and Cristina Villar
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48385-w.
References
- 1.Neul JL, et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaving LS, Ellaway CJ, Gécz J, Christodoulou J. Rett syndrome: clinical review and genetic update. J Med Genet. 2005;42:1–7. doi: 10.1136/jmg.2004.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ip JPK, Mellios N, Sur M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018;19:368–382. doi: 10.1038/s41583-018-0006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liyanage VRB, Rastegar M. Rett syndrome and MeCP2. NeuroMolecular Med. 2014;16:231–264. doi: 10.1007/s12017-014-8295-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landucci E, et al. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018;368:225–235. doi: 10.1016/j.yexcr.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidal S, et al. The utility of Next Generation Sequencing for molecular diagnostics in Rett syndrome. Sci. Rep. 2017;7:1–11. doi: 10.1038/s41598-016-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weaving LS, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004;75:1079–93. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mencarelli MA, et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J. Med. Genet. 2010;47:49–53. doi: 10.1136/jmg.2009.067884. [DOI] [PubMed] [Google Scholar]
- 9.Percy AK, et al. Rett syndrome diagnostic criteria: Lessons from the Natural History Study. Ann. Neurol. 2010;68:951–955. doi: 10.1002/ana.22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrhart F, et al. Rett syndrome - Biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016;11:1–13. doi: 10.1186/s13023-016-0545-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzales ML, LaSalle JM. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr. Psychiatry Rep. 2010;12:127–134. doi: 10.1007/s11920-010-0097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffbuhr KC, Moses LM, Jerdonek MA, Naidu S, Hoffman EP. Associations between MeCP2 mutations, X-chromosome inactivation, and phenotype. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:99–105. doi: 10.1002/mrdd.10026. [DOI] [PubMed] [Google Scholar]
- 13.Amir RE, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann. Neurol. 2000;47:670–679. doi: 10.1002/1531-8249(200005)47:5<670::AID-ANA20>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 14.Vacca M, Della Ragione F, Scalabrì F, D’Esposito M. X inactivation and reactivation in X-linked diseases. Semin. Cell Dev. Biol. 2016;56:78–87. doi: 10.1016/j.semcdb.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Gartler, S. M. & Goldman, M. A. X-Chromosome Inactivation. Encycl. Life Sci. 1–6, 10.1038/npg.els.0004172 (2001).
- 16.Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Ret Syndrome: Confirmation of X-Linked Dominant Inheritance, and Localization of the Gene to Xq28. Am. J. Hum. Genet. 1998;63:1552–1558. doi: 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, et al. Familial cases and male cases with MECP2 mutations. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017;174:451–457. doi: 10.1002/ajmg.b.32534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wan M, et al. Rett Syndrome and Beyond: Recurrent Spontaneous and Familial MECP2 Mutations at CpG Hotspots. Am. J. Hum. Genet. 1999;65:1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ørstavik KH. X chromosome inactivation in clinical practice. Hum. Genet. 2009;126:363–373. doi: 10.1007/s00439-009-0670-5. [DOI] [PubMed] [Google Scholar]
- 20.Van Den Veyver IB, Zoghbi HY. Mutations in the gene encoding methyl-CpG-binding protein 2 cause Rett syndrome. Brain Dev. 2001;23:147–151. doi: 10.1016/S0387-7604(01)00376-X. [DOI] [PubMed] [Google Scholar]
- 21.Bertelsen B, Tümer Z, Ravn K. Three new loci for determining X chromosome inactivation patterns. J. Mol. Diagnostics. 2011;13:537–540. doi: 10.1016/j.jmoldx.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Hoon B, Monkhorst K, Riegman P, Laven JSE, Gribnau J. Buccal swab as a reliable predictor for X inactivation ratio in inaccessible tissues. J. Med. Genet. 2015;52:784–790. doi: 10.1136/jmedgenet-2015-103194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weaving LS, et al. Effects ofMECP2 mutation type, location and X-inactivation in modulating Rett syndrome phenotype. Am. J. Med. Genet. 2003;118A:103–114. doi: 10.1002/ajmg.a.10053. [DOI] [PubMed] [Google Scholar]
- 24.Clerc P, Avner P. Random X-chromosome inactivation: skewing lessons for mice and men. Curr. Opin. Genet. Dev. 2006;16:246–253. doi: 10.1016/j.gde.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Peeters SB, Yang C, Brown CJ. Have humans lost control: The elusive X-controlling element. Semin. Cell Dev. Biol. 2016;56:71–77. doi: 10.1016/j.semcdb.2016.01.044. [DOI] [PubMed] [Google Scholar]
- 26.Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, Willard HF. Report Skewed X-Chromosome Inactivation Is a Common Feature of X-Linked Mental Retardation Disorders. Am. J. Hum. Genet. 2002;71:168–173. doi: 10.1086/341123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monrós E, et al. Rett syndrome in Spain: mutation analysis and clinical correlations. Brain Dev. 2001;23(Suppl 1):S251–S253. doi: 10.1016/S0387-7604(01)00374-6. [DOI] [PubMed] [Google Scholar]
- 28.Gibson JH, Williamson SL, Arbuckle S, Christodoulou J. X chromosome inactivation patterns in brain in Rett syndrome: Implications for the disease phenotype. Brain Dev. 2005;27:266–270. doi: 10.1016/j.braindev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Gale RE, Wheadon H, Boulos P, Linch DC. Tissue specificity of X-chromosome inactivation patterns. Blood. 1994;83:2899–2905. [PubMed] [Google Scholar]
- 30.Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2016;13:37–51. doi: 10.1038/nrneurol.2016.186. [DOI] [PubMed] [Google Scholar]
- 31.Shahbazian MD, Sun Y, Zoghbi HY. Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am. J. Med. Genet. 2002;111:164–168. doi: 10.1002/ajmg.10557. [DOI] [PubMed] [Google Scholar]
- 32.Swierczek SI, et al. Methylation of AR locus does not always reflect X chromosome inactivation state. Blood. 2012;119:e100–e109. doi: 10.1182/blood-2011-11-390351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lake B. B., Ai R., Kaeser G. E., Salathia N. S., Yung Y. C., Liu R., Wildberg A., Gao D., Fung H.-L., Chen S., Vijayaraghavan R., Wong J., Chen A., Sheng X., Kaper F., Shen R., Ronaghi M., Fan J.-B., Wang W., Chun J., Zhang K. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352(6293):1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlson, R. V., Boyd, K. M. & Webb, D. J. The revision of the Declaration of Helsinki: Past, present and future. Vol. 57, British Journal of Clinical Pharmacology. p. 695–713 (2004). [DOI] [PMC free article] [PubMed]
- 35.Cutler Allen R, Zoghbi HY, Annemarie Moseley IB, Rosenblatt HM, Belmont JW. Methylation of Hpall and Hhal Sites Near the Polymorphic CAG Repeat in the Human Androgen-Receptor Gene Correlates with X Chromosome Inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, et al. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012;8:1. doi: 10.1186/1746-4811-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Untergasser A, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40:1–12. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
X chromosome inactivation does not necessarily determine the severity of the phenotype in Rett syndrome patients
Data Availability Statement
All data from this article is available in the Supplementary Data.