

Summary
Chronic inflammation may drive development of cancer as observed in inflammation‐induced colorectal cancer (CRC). Though immune cells can infiltrate the tumour microenvironment, cancer cells seem to evade anti‐tumour responses, which is one of the established hallmarks of cancer. Targeting the programmed cell death protein‐1 (PD‐1)/PD‐L1 signalling pathway is currently at the forefront in the development of anti‐tumour immunity‐based therapies for multiple malignancies. By blocking the immune‐checkpoint of activated T‐cells, it is possible to rewire the adaptive resistance induced by the PD‐1 ligands expressed in the tumour microenvironment. However, adverse immunotherapy‐modulated events could complicate the treatment of individuals with preexisting chronic inflammatory conditions. In this study, we investigated the expression of different systemic and mucosal T‐cell subsets during the course of azoxymethane (AOM)/dextran sulphate sodium (DSS)‐induced colitis and colitis‐associated CRC. In addition, we examined the expression of PD‐1 and its ligands PD‐L1 and PD‐L2 as well as other molecular targets related to T‐cell exhaustion. We found a significant increase in PD‐1 expression on all examined mucosal T‐cell subsets of the colon and the ileum, which correlated with disease progression. We also observed an upregulation of PD‐L1 and PD‐L2 mRNA expression throughout the AOM/DSS regime. Blocking PD‐1 signalling with an anti‐PD1 antibody did not affect the tumour burden in the AOM/DSS‐treated mice, but did potentiate the weight loss in the third DSS cycle, indicating possible immune‐mediated toxicity. This raises a concern for patients with colitis‐associated CRCs and should be further investigated.
Keywords: AOM/DSS, colorectal cancer, inflammation, PD‐1
Introduction
Immune‐checkpoint molecules (ICMs) such as programmed cell death protein‐1 (PD‐1) are increasingly being recognized to play a pivotal role in the tumour‐mediated suppression of T‐cells that facilitates tumour escape from the immune system. Therapeutic antibodies against PD‐1 have recently been successfully implemented in the treatment of malignant melanoma,1 resulting in better survival rates, for example in treatment‐refractory malignant melanoma patients.2 In addition, PD‐1 blockade has also been approved for a variety of cancers, including non‐small‐cell lung carcinoma, bladder cancer and head and neck cancer. Also, PD‐1 blockade has been tested in colorectal cancer (CRC),3, 4 where a phase I study resulted in a complete response in one CRC patient,5 and recently the FDA approved the combination of nivolumab and ipilimumab (2018) in metastatic CRC with genetic features such as mismatch repair deficiency or high microsatellite instability. The reported complications of anti‐PD‐1‐induced colitis6, 7, 8 raise important concerns regarding dysimmune toxicity in relation to anti‐tumour efficacy. The risks of accelerating co‐existing inflammatory conditions have not been fully elucidated and could potentially be investigated in mouse models of inflammation‐induced cancer. Such studies could reveal the efficacy of anti‐PD‐1 treatment in addition to potential immune‐mediated toxicities in this colitis‐associated cancer (CAC) type.
Administration of the carcinogen azoxymethane (AOM) together with the inflammatory agent dextran sulphate sodium (DSS) is an established model of CAC (reviewed in Ref. 9). In this model, the association between ulcerative colitis and the increased development of colorectal dysplasia is reflected.
In most studies, PD‐1 expression on T‐cells has been investigated in cells from the spleen and regional lymph nodes, such as conventional CD4+ and CD8αβ TCRαβ cells. However, the expression of PD‐1 and other ICMs on non‐conventional T‐cells such as TCRγδ cells and the mesenteric lymph node (MLN) CD8αα TCRαβ cells is also of interest and has not been extensively investigated. Furthermore, the small and large intestines house a variety of additional T‐cell subsets, which in a cancer setting might be prone to tumour PD‐L1‐mediated suppression or suppression through other ICMs. This may be particularly true for intraepithelial lymphocytes (IELs) near the epithelial cells, from which colorectal adenocarcinoma originates. In mice, these include conventional CD4 or CD8αβ IELs that express TCRαβ, and unconventional IELs expressing CD8αβ and either TCRγδ or TCRαβ.10 While the role of classical TCRαβ T‐cells is well understood in relation to chronic inflammation and tumour immunology, the role of TCRγδ T‐cells and CD8αα TCRαβ T‐cells, as well as their expression of ICMs, is less understood.
Thus, a detailed analysis of both TCRαβ and TCRγδ subsets and changes in mucosal and systemic immune compartments may provide new insight into the immunopathology of this CAC model.
Although the role of PD‐1 in T‐cell suppression has been well described,11, 12 the discovery of new ICMs is ongoing. T‐cell immunoreceptor with Ig and ITIM domains (TIGIT) and Herpes virus entry mediator (HVEM) may be novel candidates for T‐cell suppression. TIGIT has been shown to be overexpressed on CD8+ tumour‐infiltrating lymphocytes (TILs).13 These TILs exhibited increased function after TIGIT and PD‐1 blockade,13 and were subsequently shown to elicit tumour rejection in mouse models.14 Similarly, HVEM has been shown to act as an ICM. For example, BTLA expressed on dendritic cells was recently shown to bind HVEM on T‐cells and upregulate forkhead box P3 (FOXP3),15 thereby inducing T‐cell tolerance. Thus, the involvement and regulation of ICMs such as PD‐1, TIGIT and HVEM in a more comprehensive panel of T‐cell subsets during the initiation of chronic colitis and in relation to inflammation‐induced CRC have not yet been described. Because PD‐1 signalling is usually thought to be the result of increased PD‐L1/PD‐L2 expression in tumours, it is also of interest to investigate if PD‐1 upregulation is the result of the chronic inflammation per se, which could be an additional mechanism that drives inflammation‐induced cancers.
Considering the clinical interest in anti‐PD‐1 treatment in different types of cancer, it is important to investigate whether this treatment is feasible in a highly inflammatory tumour environment. Anti‐PD‐1 therapy could potentially exacerbate the chronic mucosal inflammation seen after several cycles of DSS treatments. Moreover, to understand the regulation of PD‐1 in chronic inflammation and to comprehensively describe the mode of action of anti‐PD‐1 treatment, it is relevant to investigate PD‐1 expression in a larger repertoire of T‐cell subsets. Here, we investigated the changes in T‐cell subsets, such as CD4 and CD8αβ TCRαβ cells, TCRγδ cells and CD8αα TCRγδ or TCR αβ cells in the spleen, MLNs, and IELs in small and large intestines during the development of chronic colitis. We also analysed the expression of PD‐1, TIGIT and HVEM on these T‐cells. In addition, we investigated the effect of anti‐PD‐1‐antibody blockade in the AOM/DSS mouse model.
Materials and methods
Establishment of the AOM/DSS model
Forty C57BL/6 wild‐type age‐matched (10–12 weeks) female mice (Taconic, Lyngby, Denmark) were used for this study. Thirty mice were injected intraperitoneally (i.p.) with 7·4 mg/kg AOM, and 10 mice served as untreated healthy controls. The AOM‐injected mice were further subdivided into three groups of 10 mice.
The first group was given one cycle of 3% DSS in their drinking water in week 2 for 7 days and killed in week 4, 1 week after the end of the first DSS cycle.
The second group was given two cycles of 3% DSS for 7 days in weeks 2 and 5, and killed in week 7, 1 week after the end of the second DSS cycle.
The third group was given three cycles of 3% DSS for 7 days in weeks 2, 5 and 8, and killed in week 10, 2 weeks after the end of the third DSS cycle.
All mice were killed by cervical dislocation, and the number of macroscopic colonic tumours that had developed in the third group was analysed. All mice were housed in ventilated cages in a pathogen‐free animal facility (Panum Instituttet, University of Copenhagen) with ad libitum access to water and standard rodent diet. The mice were maintained under controlled temperature (22 ± 2°) and a 12‐hr light/dark cycle.
Colonoscopy
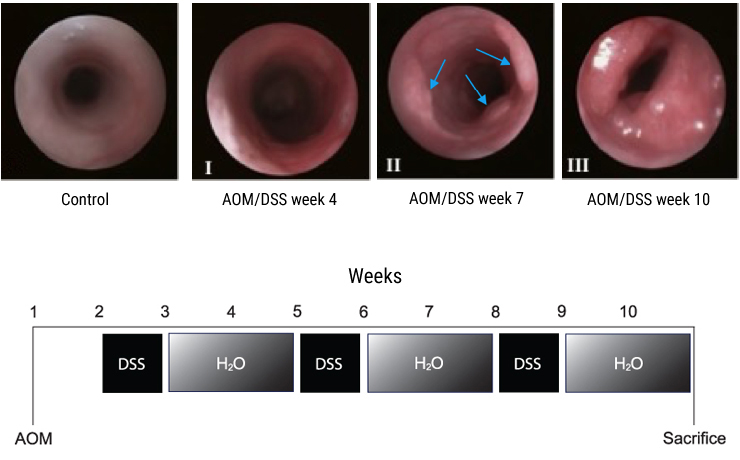
To follow the onset of tumour development, mice were anaesthetized with isoflurane prior to DSS administration, and after each DSS cycle during weeks 4, 7 and 10. In a separate cohort of mice, colonoscopies were performed using the Coloview mini‐endoscopic system (Karl Storz, Tuttlingen, Germany) in accordance with the protocol from Becker et al.16 The murine endoscopic index of colitis severity (MEICS) was used as described in the same protocol,16 with a maximal score of 15. Colonoscopy pictures are shown as Fig. S1 to demonstrate kinetics of tumour development, but the mice analysed in the analysis described in the manuscript did not undergo colonoscopy.
Anti‐PD‐1 treatment
Twenty age‐matched (10–12 weeks) female C57BL/6 mice (Taconic, Lyngby, Denmark) were injected i.p. with 7·4 mg/kg AOM, followed by three cycles of 3% DSS in their drinking water for 7 days in weeks 2, 5 and 8. The mice were divided into two groups of 10 and co‐housed to minimize any possible cage effects. Starting at week 6 and until death, the mice were injected i.p. twice a week with either anti‐mouse PD‐1 antibody (100 μg/200 μl; Bio X Cell, NH, USA) or a mouse IgG1 isotype control antibody (100 μg/200 μl; Bio X Cell, West Lebanon, NH) for a total of 10 injections. All mice were killed in week 10, 2 weeks after the end of the third DSS cycle, and macroscopic colonic tumours were analysed in a blinded setup.
Cell isolation and flow cytometry
Cells from MLNs and spleens were isolated by aseptically squeezing the fresh organs in phosphate‐buffered saline (PBS) through a 70‐μm cell strainer. IELs from the full‐length colon and terminal ileum were obtained from either AOM/DSS‐treated tumour‐bearing mice or control mice without tumours, whereas the number of obtained cells did not allow isolation from tumour versus non‐tumour tissue in the tumour‐bearing mice. Colon and terminal ileum were handled separately by cutting the intestine into small pieces, which were suspended in warm (37°C) PBS with 2 mm EDTA for 15 min. After incubation, the intestinal segments were vigorously shaken and then filtered through a 70‐μm cell strainer as described in Ref. 17 Cells were washed in PBS prior to a 30‐min staining with antibodies for flow cytometry. The analysis was performed using a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA) at the Core Facility for Flow Cytometry, Faculty of Health and Medical Sciences, University of Copenhagen. Data were analysed using the FlowJo software (Treestar, Woodburn, OR). For colon IELs we used the following gating strategy: first cells were gated on a lymphocyte gated based on FSC/SSC, and this was followed by a singlet gate. CD3‐positive cells were then gated on TCR‐β and TCR‐γδ cells. TCR‐γδ cells were further gated on CD8αα cells, and TCR‐β cells were gated on CD4 cells, CD8αβ and CD8αα cells. In each population of cells, surface markers were analysed as described. A similar approach was used for other organs.
Antibodies for flow cytometry
The following antibodies against mouse were used for the flow cytometry analysis: BV510‐A anti‐CD3, BV605‐A anti‐CD4, Alexa‐Fluor‐488‐A anti‐CD8α, BV650‐A anti‐CD8β, BV421‐A anti‐TCRγδ, BV‐711‐A anti‐TCRβ, PE‐Cy7‐A anti‐CD271 (PD‐1), APC‐A anti‐CD270 (HVEM) and PE‐A anti‐TIGIT (Vstm3). Viability dye 7‐AAD was used to exclude dead cells in the analyses. All antibodies are commercially available from BioLegend (San Diego, CA).
RNA extraction and quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR)
Colonic tissue harvested from wild‐type mice was stored in RNAlater prior to RNA extraction. Total RNA was extracted using a mirVana miRNA Isolation Kit (Ambion/Life Technologies, Waltham, MA) following the manufacturer's protocol. RNA quality was assessed with an Agilent Bioanalyzer (Agilent Technologies, Carpenteria, CA). For real‐time RT‐PCR, first‐strand cDNA was synthesized from 1 μg of total RNA in a total volume of 20 μl using a RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas GmbH, Leon‐Rot, Germany) in accordance with the manufacturer's protocol with minor modifications. A poly (dT) oligomer (Fermentas) was used as a primer. PCR primer pairs were selected using the PrimerBank resource: PDL‐1: ID11230798a1; PDL‐2: ID10946740a1 and PSMB6: ID26347247a1. Real‐time PCR was performed using a LightCycler 480 system (Roche, Basel, Switzerland) as described previously.18
Statistics
To determine the significance of difference from the untreated control group, Student's t‐test was performed. P‐values < 0·05 were considered statistically significant.
Ethical considerations
All experimental protocols were approved by the Danish Veterinary and Food Administration, Ministry of Environment and Food of Denmark, and performed according to the Danish guidelines.
Results
Tumour development can consistently be detected 6–8 weeks after AOM injection and administration of 2–3 cycles of 3% DSS. The development of endoscopic equipment for small animals has enabled the study of early tumour development time points as well as the monitoring of tumour development in individual animals over the course of an experiment without the need for killing the animals. In order to demonstrate kinetics of tumour development, we show colonoscopy images from a separate cohort of mice (Fig. S1). Most recently, methods for in vivo imaging have been applied to several other tumour models in rodents.19 However, for the AOM/DSS mouse model, reliable markers have not yet been reported.
Analysis of T‐cell subpopulations during chemically induced colitis
The major aim of the present study was to characterize in more detail T‐cell subsets during the course of chronic colitis and CAC. Thus, the frequencies of TCRαβ and TCRγδ CD3+ T‐cells, and the frequencies of CD4+, CD8αα+ and CD8αβ+ T‐cell subsets were analysed in the IELs of the colon and terminal ileum, and in the spleen and MLNs.
In colonic IELs, we found that the total CD3, TCRγδ and TCRαβ were increased at an early time point (week 4), and CD4 TCRαβ cells were massively increased, whereas CD8αβ TCRαβ, CD8αα TCRαβ and TCRγδ cells were only slightly decreased at certain time points (Fig. 1). In IELs of the ileum, CD8αα TCRγδ cells were transiently decreased but increased in the late phase of colitis (Fig. 2). CD8αα and CD8αβ TCRαβ cells were similarly increased in the late phase of colitis, whereas other subsets were unchanged during colitis (Fig. 2).
Figure 1.
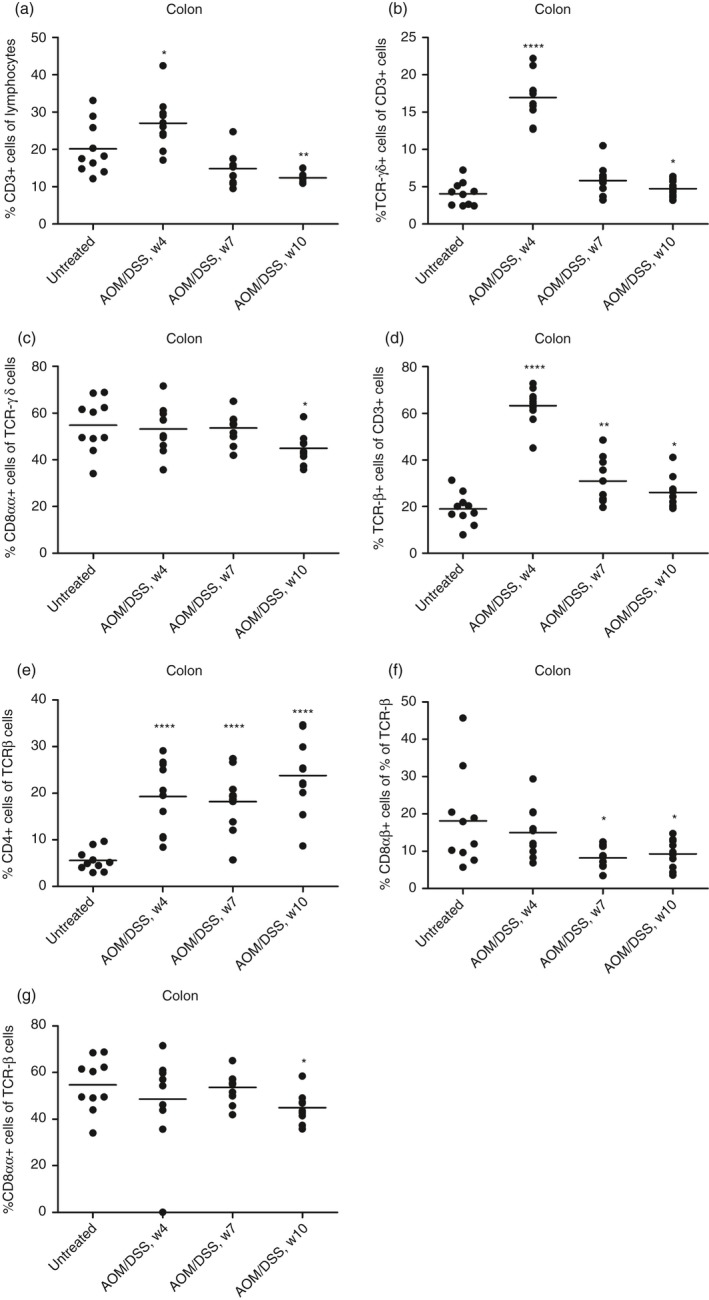
Flow cytometry analysis of isolated mouse colonic intraepithelial lymphocytes (IEL) subsets during dextran sulphate sodium (DSS)‐induced colitis. (a) % CD3+ cells of isolated colon IELs. (b) CD3+ cells were gated, and TCRγδ expression was measured. (c) TCRγδ+ cells were gated, and CD8αα expression was measured. (d) CD3+ cells were gated, and TCRαβ expression was measured. (e) TCRβ+ cells were gated, and CD4 expression was measured. (f) TCRβ+ cells were gated, and CD8αβ expression was measured. (g) TCRβ+ cells were gated, and CD8αα expression was measured. Each circle represents an individual mouse (n = 10), and the mean is shown. Statistical analysis was performed using Student's t‐test, *P < 0·05, **P < 0·01, ***P < 0·001.
Figure 2.
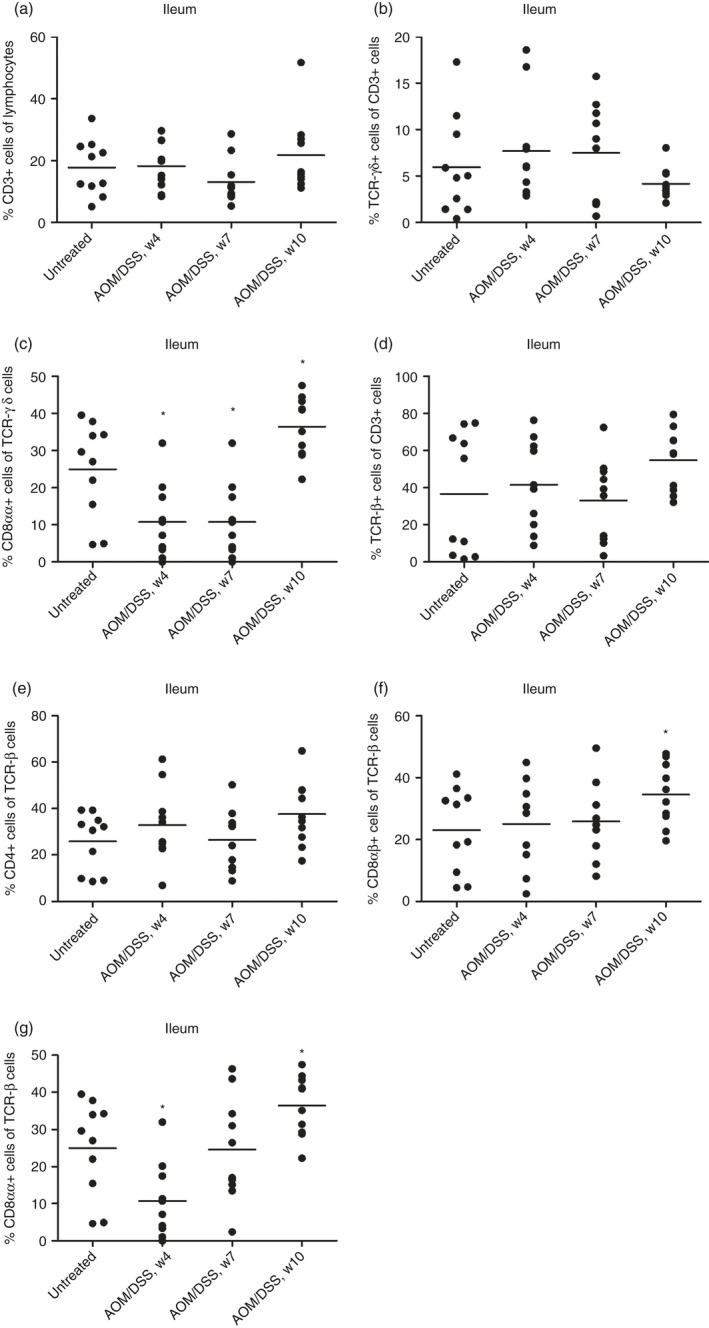
Flow cytometry analysis of isolated mouse ileal intraepithelial lymphocyte (IEL) subsets during dextran sulphate sodium (DSS)‐induced colitis. (a) % CD3+ cells of isolated ileal IELs. (b) CD3+ cells were gated, and TCRγδ expression was measured. (c) TCRγδ+ cells were gated, and CD8αα expression was measured. (d) CD3+ cells were gated, and TCRαβ expression was measured. (e) TCRβ+ cells were gated, and CD4 expression was measured. (f) TCRβ+ cells were gated, and CD8αβ expression was measured. (g) TCRβ+ cells were gated, and CD8αα expression was measured. Each circle represents an individual mouse (n = 10), and the mean is shown. Statistical analysis was performed using Student's t‐test, *P < 0·05, **P < 0·01, ***P < 0·001.
In the spleen, total CD3+ T‐cells declined during colitis development, accompanied by a decrease in CD8αβ and total TCRαβ T‐cells. By contrast, a transient increase in TCRγδ cells was observed, and a gradual increase in CD4+ TCRαβ T‐cells was evident (Fig. S2). In MLNs, no change in TCRγδ cells was observed, whereas the changes of the other subsets were the same as those observed for spleen cells (Fig. S3). In addition, an increase in CD8αα TCRαβ cells was noted in the late phase of chronic colitis (Fig. S3).
Upregulation of markers, including PD‐1
Our next aim was to study in detail the expression of ICMs on the various T‐cell subsets. For all T‐cell subsets analysed, we investigated the expression of PD‐1, TIGIT and HVEM over the course of AOM/DSS‐induced chronic colitis and CAC. However, TIGIT expression was generally not observed. In the spleen, the expression levels of PD‐1 were increased at a late time point in TCRγδ cells but less so in TCRαβ cells, whereas HVEM was increased at late time points in TCRαβ cells but not in TCRγδ cells (Fig. S4). In MLN cells, a similar pattern was observed, but HVEM was also upregulated in CD4 TCRαβ cells at late time points, and alterations in PD‐1 expression were minor (Fig. S5). In general, the expression levels of HVEM were higher than of PD‐1 in the spleen and MLNs.
By contrast, in colonic IELs, PD‐1 expression was greater than that of HVEM and significantly increased to reach its highest levels at week 10, at which point established colonic tumours were evident in all mice (Fig. 3). Conversely, changes in HVEM were minor. The same pattern was observed in ileal IELs for all subsets analysed (Fig. 4). However, PD‐1 expression declined after week 7 in all analysed IELs of the ileum (Fig. 4).
Figure 3.
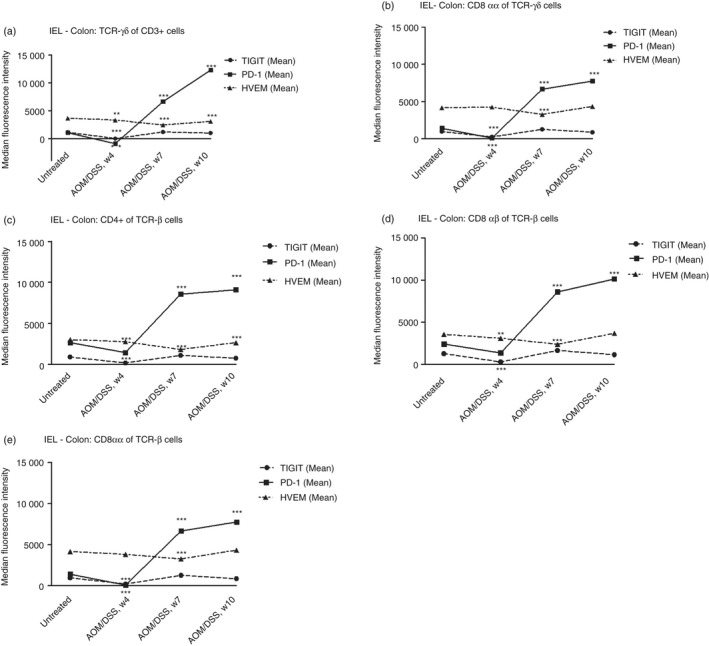
Flow cytometry analysis of programmed cell death protein‐1 (PD1), T‐cell immunoreceptor with Ig and ITIM domains (TIGIT) and Herpes virus entry mediator (HVEM) expression in different intraepithelial lymphocyte (IEL) subsets from mouse colons during dextran sulphate sodium (DSS)‐induced colitis. (a) PD1, TIGIT and HVEM expression in TCRγδ+ T‐cells, (b) CD8αα TCRγδ+ T‐cells, (c) CD4 TCRαβ+ T‐cells, (d) CD8αβ TCRαβ+ T‐cells and (e) CD8αα TCRαβ+ T‐cells. Each symbol represents the mean of 10 mice. Statistical analysis was performed using Student's t‐test, *P < 0·05, **P < 0·01, ***P < 0·001.
Figure 4.
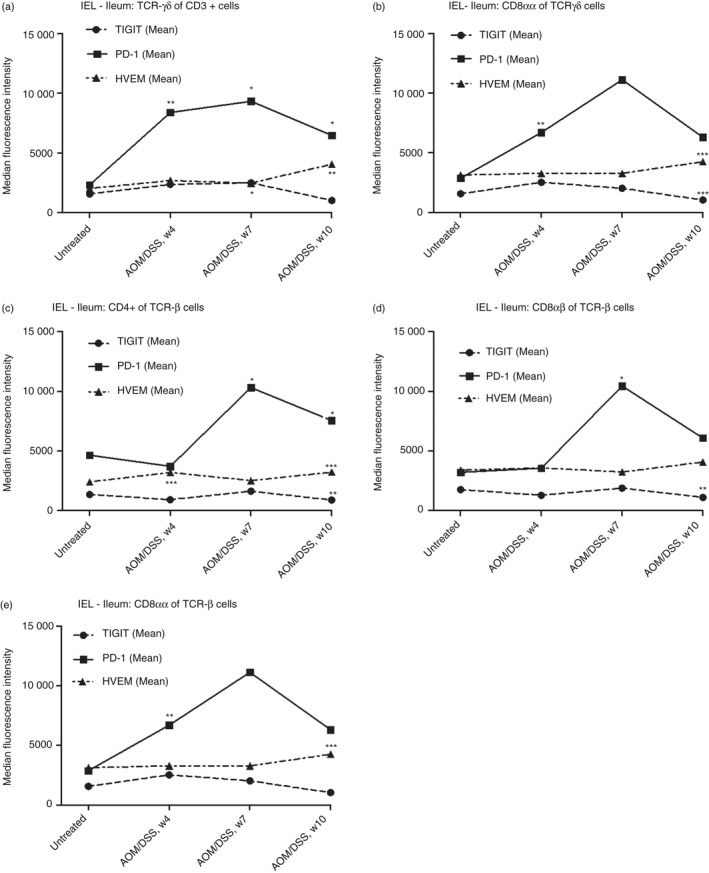
Flow cytometry analysis of programmed cell death protein‐1 (PD1), T‐cell immunoreceptor with Ig and ITIM domains (TIGIT) and Herpes virus entry mediator (HVEM) expression in different intraepithelial lymphocyte (IEL) subsets from mouse ileum during dextran sulphate sodium (DSS)‐induced colitis. (a) PD1, TIGIT and HVEM expression in TCRγδ+ T‐cells, (b) CD8αα TCRγδ+ T‐cells, (c) CD4 TCRαβ+ T‐cells, (d) CD8αβ TCRαβ+ T‐cells and (e) CD8αα TCRαβ+ T‐cells. Each symbol represents the mean of 10 mice. Statistical analysis was performed using Student's t‐test, *P < 0·05, **P < 0·01, ***P < 0·001.
Expression of PD‐1 ligands
The mRNA expression levels of PDL‐1 and PDL‐2 in the distal colon at different time points during the AOM/DSS treatment were investigated. Following normalization to the proteasome subunit beta type‐6 (PSMB6) housekeeping gene, it was found that PDL‐1 expression increased almost twofold in week 4 and week 7 (Fig. 5a). However, statistical significance was only evident in week 7. Furthermore, in week 10, when all mice developed chronic colitis and tumours, PDL‐1 expression in non‐tumour areas decreased to baseline levels. Interestingly, while PD‐L1 expression was lower at week 10 than at weeks 4 and 7, it tended to be expressed more in tumour tissues than non‐tumour areas from the same mice, although this was not statistically significant.
Figure 5.
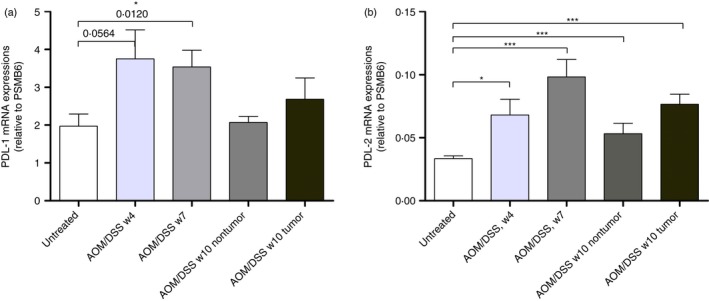
PD‐L1 and PD‐L2 mRNA expression levels in mouse colons during dextran sulphate sodium (DSS)‐induced colitis. (a) PD‐L1 mRNA expression in untreated and azoxymethane (AOM)/DSS‐treated colonic tissue from weeks 4, 7 and 10 (n = 10). (b) PD‐L2 mRNA expression in untreated and AOM/DSS‐treated colonic tissue from weeks 4, 7 and 10 (n = 10). The data are normalized to proteasome subunit beta type‐6 (PSMB6) and shown as the mean ± SEM. Statistical analysis was performed using Student's t‐test, and P‐values are shown. P‐values < 0·05 were considered statistically significant.
PDL‐2 showed the same tendency, with the highest significant increase observed in week 7 (Fig. 5b). Although the expression of PDL‐2 mRNA was much lower than that of PDL‐1 and typical for a low copy number transcript, colonic PDL‐2 expression remained significantly elevated at all stages during the AOM/DSS treatment. In contrast to PDL‐1 at the tumour stage, PDL‐2 expression remained significantly higher in non‐tumour areas than the untreated control mice. In addition, tumours showed higher PDL‐2 mRNA expression than non‐tumour areas.
The effect of PD‐1 blockade in the AOM/DSS tumour model
Finally, we investigated whether anti‐PD‐1 antibody administration at the onset of tumour development could potentially inhibit or abolish tumour progression. Mice were injected i.p. twice a week with 100 μg of an anti‐PD‐1 or isotype control antibody during the second half of the AOM/DSS 10 week model. Compared with the isotype control, anti‐PD‐1 antibody administration did not have any appreciable effect (Fig. 6a) on either total tumour numbers or tumour size (Fig. 6b). The anti‐PD‐1 treatment did, however, potentiate weight loss during the third DSS cycle (Fig. 6c). Interestingly, anti‐PD‐1 administration reduced the variation in tumour numbers between the mice compared with the control groups.
Figure 6.
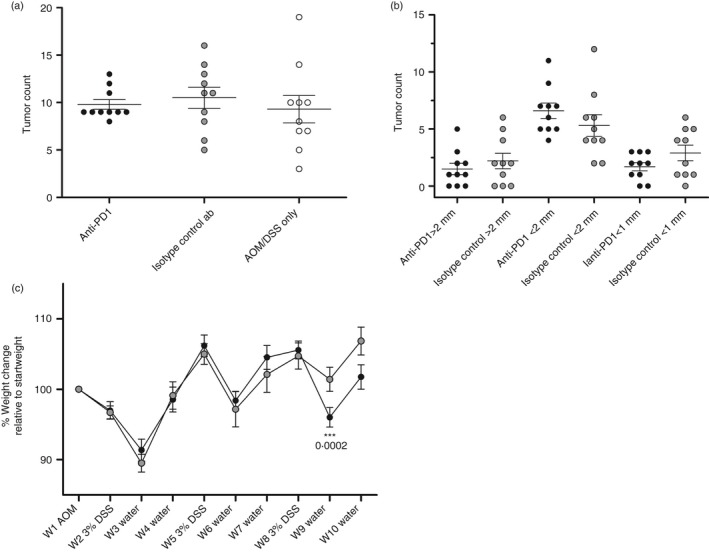
Tumorigenesis and weight development in azoxymethane (AOM)/dextran sulphate soidum (DSS)‐treated mice. (a) Comparison of the total numbers of macroscopic tumours in mice treated i.p. with an anti‐PD‐1 ab, isotype control ab, or AOM/DSS only. Each circle represents an individual mouse (n = 10), and the mean ± SEM is shown. Statistical analysis was performed using Student's t‐test, and no significant differences were found. (b) Comparison of the tumour size in mice treated i.p. with an anti‐PD‐1 ab or isotype control ab. (c) Percentage body weight change relative to the week 1 baseline during the course of three 3% DSS administrations. Weights are presented as the mean ± SEM (n = 10). Statistical analysis was performed using Student's t‐test. P‐values < 0·05 were considered statistically significant.
Discussion
The importance of T‐cells and immune‐checkpoints has been increasingly recognized in tumour immunology. Here, we investigated the mucosal and systemic changes in CD4+ and CD8+ T‐cell subsets, and analysed the expression of PD‐1, HVEM, TIGIT, PD‐L1 and PD‐L2 in the AOM/DSS model. Furthermore, we investigated the effects of PD‐1 antibody blockade in a highly inflamed setting during CAC development.
T‐cell subpopulations during DSS‐induced colitis
In the present work, we performed a detailed analysis of TCRαβ CD4+ and CD8+ T‐cell subsets and TCRγδ cells in various immune compartments. In colonic IELs, AOM/DSS‐induced inflammation resulted in an increase in total CD3+, TCRγδ and TCRαβ cells, including a massive increase in CD4+ TCRαβ cells, but moderate changes in CD8αβ TCRαβ and CD8αα TCRαβ/TCRγδ cells. This indicates a pivotal role for CD4+ T‐cell recruitment in regulating various parts of the mucosal immune response during acute and chronic DSS‐induced colitis. On the other hand, TCRγδ and CD8+ IELs of various subtypes are preexisting and present in sufficient numbers as part of the early innate immune system to maintain mucosal surveillance. Nonetheless, their function might potentially be regulated by the massive influx of CD4+ T‐cells. Our findings in ileum IELs confirmed that CD8+ cells of various subtypes were primarily recruited in the late chronic phase. At the systemic level, CD4+ T‐cells from the spleen and MLNs were also the major T‐cell population expanding in number at the early stage of inflammation.
Analysing ICMs may reveal novel insights into the functional role of CD8+ IELs during acute and chronic inflammation. Here, we analysed PD‐1, HVEM and TIGIT expression on all of the above T‐cell subsets. Whereas HVEM and TIGIT expressions were not markedly changed, PD‐1 expression increased dramatically in all IEL T‐cell subsets analysed. Importantly, this may indicate that the functional capacity of IELs in the colon is reduced as a result of the chronic inflammation, which may have implications for mucosal immune surveillance of gut infections and potentially local immune surveillance of developing cancer cells from the colonic epithelium.20, 21 It can be speculated that PD‐1 is upregulated on the T‐cells as a self‐protective mechanism induced by the emerging cancer cells, especially considering the early upregulation of both PD‐L1 and PD‐L2 mRNA levels in the colon. However, it cannot be excluded that the chronic inflammation per se affects the regulation of PD‐1 in an attempt to contain the excessive damage. PD‐1 was indeed initially discovered as a protective response from the host during chronic viral infections in order to avoid excessive immunopathology responses to the chronic antigen load.22 Our data clearly show that the chronic inflammation resulting in CAC development leads to massive PD‐1 upregulation in IELs. As such upregulation occurs on different CD4 and CD8 subsets altogether, this may reflect functional reduced capacity in both the early protective mechanisms of the innate lymphocyte repertoire (e.g. TCRγδ cells), as well as in the capacity to mount an anti‐tumour T‐cell response on the long term in the adaptive immune system (e.g. TCRαβ CD4+ and CD8+ T‐cells). Ultimately, this inhibits the immune capacity for pathogenic clearance and tumour immunosurveillance. Thus, PD‐1 upregulation of IELs might add to existing theories of inflammation‐induced cancers in the mucosal epithelium.
Anti‐PD‐1 intervention study
In our AOM/DSS model, the upregulation of PD‐1 in the examined T‐cells, particularly the strong upregulation in IELs of the colon, led us to examine therapeutic intervention with anti‐PD‐1 antibody. Along with the prospective anti‐tumour effect, we wanted to explore potential treatment‐related toxicities and determine whether blocking PD‐1 in a DSS‐induced colitis regime could exacerbate the inflammatory response. Administering anti‐PD‐1 twice a week throughout the last 5 weeks of the AOM/DSS model did not affect the total tumour count or tumour size. This lack of efficacy could be related to a low impact of PD1 blockade on gut immunotoxicity in mice in general, in contrast to what is observed in humans treated with various immune‐checkpoint inhibitors in the clinic. Another possible explanation could be the lack of any increase in infiltrating colonic CD8+ cytotoxic T lymphocytes at the late phase of inflammation. In fact, there was a significant decrease in all CD8+ subsets at the late phase compared with that in the untreated mice. Combinatorial therapies with immune‐modulating compounds such as immunogenic cell death inducers and vaccines might potentially solve this issue and enhance tumour immunosurveillance. Indeed, a recent study has demonstrated that the intra‐tumoral immunoscore, which is indicative of tumour‐infiltration, is a strong indicator of patient survival, exhibiting promising prognostic potential.23
One could argue for initiating early immune‐checkpoint intervention by blocking PD‐1 in the acute phase for durable anti‐tumour responses during DSS‐induced colitis. It is evident that the most dramatic increases in T lymphocytes occur after the first DSS cycle at the early phase of the intestinal inflammation. This also correlated with the expression of PD‐L1 and PD‐L2, for which the highest levels were observed in weeks 4 and 7, after the first and second DSS cycles, respectively. Indeed, a very interesting study in mice with DSS‐induced colitis demonstrated that blocking PD‐L1 also had gut‐protective effects, as anti‐PD‐L1 blockade clearly ameliorated colitis through a shift from a Th17 to a Th1 interferon‐γ response.24 On the other hand, one could argue that, in an acute and highly inflamed microenvironment, a PD1 blocking strategy is not without serious risks, and more explorative studies are required. Importantly and in contrast to the above gut‐protective effects of anti‐PD‐L1, another study in PD‐L1‐deficient mice has shown that parenchymal expression of PD‐L1, and not haematopoietic expression, promotes protection against gut injury. This mechanism was proposed to be dependent on innate immunity, more specifically the CD11c+ CD11b+ cells of the lamina propria, as PD‐L1 interaction with these cells was shown to suppress TNF‐α and promote IL‐22 production.25 Indeed, our finding of increased weight loss during PD1 blockade may suggest increased inflammation and be in line with these findings. In the clinic, unselected CRC patients have proved to be rather refractory to checkpoint‐blocking treatment. Still, a subset with mismatch repair‐deficient CRC appears to be the exception.26 The resulting microsatellite instability in the malignant cells in these patients increases the mutational load and hence the tumour immunogenicity, making these cells susceptible to PD‐1 targeting. This is evident in MSI‐high tumours that display a high density of tumour‐infiltrating immune cells together with an upregulation of PD‐L1.27 Therefore, it seems that a high level of somatic mutations correlates with a positive treatment outcome. Indeed, tumours with a higher mutational burden, such as melanoma and non‐small‐cell lung carcinoma, are the most responsive to such treatment.
Interestingly, during the third cycle of DSS, blocking PD‐1 did potentiate weight loss. This is most likely an immune‐mediated toxicity effect related to the antibody treatment. This is also evident in anti‐PD1‐induced colitis biopsies from humans, where CD8+ T‐cells were predominant in the intestinal epithelium and lamina propria.28 Thus, a clinical concern could be the potential risk of adverse events in patients with a preexisting autoimmune disorder, such as IBD patients receiving checkpoint‐blocking antibodies. This is especially relevant for patients with autoimmune conditions that require immune‐suppression and patients with organ transplants. Together, our results emphasize the challenges and major obstacles in making immunotherapy a cornerstone in CRC treatment. Currently, many clinical trials of gastrointestinal and CRC immunotherapies are ongoing (listed in Refs 29, 30 which could offer new successful combination strategies with more curative potential for CRC patients.
In conclusion, we found a significant upregulation of PD‐1 on all the examined IELs of the colon and the ileum. Especially for the IELs of the colon, PD‐1 upregulation showed a correlation with the severity of inflammation and tumour load, reaching its highest levels after the third cycle of DSS administration. Interestingly, we observed HVEM upregulation in the conventional T‐cell subsets of both the spleen and MLNs during chronic inflammation. This increase is suggested to be involved in the inhibition of the ability of gut‐homing effector CD8+ T‐cells to eradicate established cancer cells. Although PD‐L1 and PD‐L2 expression was elevated throughout the AOM/DSS regime, blocking PD‐1 signalling with an anti‐PD1 antibody did not affect the tumour burden in the antibody‐treated mice but did potentiate weight loss. Thus, more therapeutic interventions with anti‐PD‐1 antibodies are needed to rule out any practicalities affecting treatment efficacy, such as the concentration, optimal treatment onset and frequency of antibody administration.
Funding
This work was supported by research grants from ‘Sofus Friis and Wife's Foundation’, as well as The Faculty of Health and Medical Sciences, University of Copenhagen, Denmark.
Disclosures
AEP is currently affiliated with Merck A/S. MY is currently affiliated with AbbVie A/S. However, this work was performed prior to their employment, and not related to Merck A/S or AbbVie A/S. AEP and MY are still affiliated with the University of Copenhagen. The other authors declare no conflicts of interest.
Supporting information
Figure S1. In vivo colonoscopy images from AOM/DSS‐treated mice + AOM/DSS timeline.
{kind=link}
Figure S2. Flow cytometry analysis of isolated T cell subsets from mouse spleen during DSS‐induced colitis.
Figure S3. Flow cytometry analysis of isolated T cell subsets from mouse mesenteric lymph nodes during DSS‐induced colitis.
Figure S4. Flow cytometry analysis of PD1, TIGIT, HVEM expressions in isolated T cell subsets from mouse spleen during DSS‐induced colitis.
Figure S5. Flow cytometry analysis of PD1, TIGIT, HVEM expressions in isolated T cell subsets from mouse mesenteric lymph nodes during DSS‐induced colitis.
Acknowledgements
The authors would like to thank Elisabeth Veyhe Andersen and Lotte Laustsen for technical assistance.
References
- 1. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015; 14:561–84. [DOI] [PubMed] [Google Scholar]
- 2. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH et al Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH et al Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28:3167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA et al Durable cancer regression off‐treatment and effective reinduction therapy with an anti‐PD‐1 antibody. Clin Cancer Res 2013; 19:462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen JH, Pezhouh MK, Lauwers GY, Masia R. Histopathologic features of colitis due to immunotherapy with anti‐PD‐1 antibodies. Am J Surg Pathol 2017; 41:643–54. [DOI] [PubMed] [Google Scholar]
- 7. Baroudjian B, Lourenco N, Pages C, Chami I, Maillet M, Bertheau P et al Anti‐PD1‐induced collagenous colitis in a melanoma patient. Melanoma Res 2016; 26:308–11. [DOI] [PubMed] [Google Scholar]
- 8. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel‐Vinay S et al Immune‐related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer 2016; 54:139–48. [DOI] [PubMed] [Google Scholar]
- 9. De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L et al The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J Carcinog 2011; 10:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014; 14:667–85. [DOI] [PubMed] [Google Scholar]
- 11. Francisco LM, Sage PT, Sharpe AH. The PD‐1 pathway in tolerance and autoimmunity. Immunol Rev 2010; 236:219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C et al TIGIT and PD‐1 impair tumor antigen‐specific CD8(+) T cells in melanoma patients. J Clin Invest 2015; 125:2046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johnston RJ, Comps‐Agrar L, Hackney J, Yu X, Huseni M, Yang Y et al The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014; 26:923–37. [DOI] [PubMed] [Google Scholar]
- 15. Jones A, Bourque J, Kuehm L, Opejin A, Teague RM, Gross C et al Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity 2016; 45:1066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Becker C, Fantini MC, Neurath MF. High resolution colonoscopy in live mice. Nat Protoc 2006; 1:2900–4. [DOI] [PubMed] [Google Scholar]
- 17. Tougaard P, Skov S, Pedersen AE, Krych L, Nielsen DS, Bahl MI et al TL1A regulates TCRgammadelta+ intraepithelial lymphocytes and gut microbial composition. Eur J Immunol 2015; 45:865–75. [DOI] [PubMed] [Google Scholar]
- 18. Bjerrum JT, Nyberg C, Olsen J, Nielsen OH. Assessment of the validity of a multigene analysis in the diagnostics of inflammatory bowel disease. J Intern Med 2014; 275:484–93. [DOI] [PubMed] [Google Scholar]
- 19. Ntziachristos V. Fluorescence molecular imaging. Annu Rev Biomed Eng 2006; 8:1–33. [DOI] [PubMed] [Google Scholar]
- 20. Roberts AI, O'Connell SM, Ebert EC. Intestinal intraepithelial lymphocytes bind to colon cancer cells by HML‐1 and CD11a. Cancer Res 1993; 53:1608–11. [PubMed] [Google Scholar]
- 21. Golby SJ, Chinyama C, Spencer J. Proliferation of T‐cell subsets that contact tumour cells in colorectal cancer. Clin Exp Immunol 2002; 127:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S et al PD‐1 expression on HIV‐specific T cells is associated with T‐cell exhaustion and disease progression. Nature 2006; 443:350–4. [DOI] [PubMed] [Google Scholar]
- 23. Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D et al Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016; 44:698–711. [DOI] [PubMed] [Google Scholar]
- 24. Song MY, Hong CP, Park SJ, Kim JH, Yang BG, Park Y et al Protective effects of Fc‐fused PD‐L1 on two different animal models of colitis. Gut 2015; 64:260–71. [DOI] [PubMed] [Google Scholar]
- 25. Scandiuzzi L, Ghosh K, Hofmeyer KA, Abadi YM, Lazar‐Molnar E, Lin EY et al Tissue‐expressed B7‐H1 critically controls intestinal inflammation. Cell Rep 2014; 6:625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD et al PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015; 372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM et al The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter‐inhibitory checkpoints. Cancer Discov 2015; 5:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coutzac C, Adam J, Soularue E, Collins M, Racine A, Mussini C et al Colon immune‐related adverse events: anti‐CTLA‐4 and anti‐PD‐1 blockade induce distinct immunopathological entities. J Crohns Colitis 2017; 11:1238–46. [DOI] [PubMed] [Google Scholar]
- 29. Lote H, Cafferkey C, Chau I. PD‐1 and PD‐L1 blockade in gastrointestinal malignancies. Cancer Treat Rev 2015; 41:893–903. [DOI] [PubMed] [Google Scholar]
- 30. Bilgin B, Sendur MA, Bulent Akinci M, Sener Dede D, Yalcin B. Targeting the PD‐1 pathway: a new hope for gastrointestinal cancers. Curr Med Res Opin 2017; 33:749–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. In vivo colonoscopy images from AOM/DSS‐treated mice + AOM/DSS timeline.
Figure S2. Flow cytometry analysis of isolated T cell subsets from mouse spleen during DSS‐induced colitis.
Figure S3. Flow cytometry analysis of isolated T cell subsets from mouse mesenteric lymph nodes during DSS‐induced colitis.
Figure S4. Flow cytometry analysis of PD1, TIGIT, HVEM expressions in isolated T cell subsets from mouse spleen during DSS‐induced colitis.
Figure S5. Flow cytometry analysis of PD1, TIGIT, HVEM expressions in isolated T cell subsets from mouse mesenteric lymph nodes during DSS‐induced colitis.