

Abstract
Metaphyseal dysplasias are a heterogeneous group of skeletal dysplasias characterised by metaphyseal irregularities. Due to the presence of metaphyseal changes accompanied with bowing deformity of lower limb, they are likely to be mistaken for rickets. We present a case of a 7-year-old boy, finally diagnosed with metaphyseal dysplasia, Spahr type (MDST) (OMIM # 250400) after his exome sequencing revealed novel variations in the MMP13 gene (OMIM * 600108). This is a rare skeletal dysplasia with only a few cases reported in literature. A compilation of the presentation of the reported cases is given to help the reader understand this rare disorder. To the best of our knowledge, this case of MDST is the first to be reported from India.
Keywords: orthopaedics, genetics
Background
Metaphyseal dysplasias are a heterogeneous group of skeletal dysplasias characterised by metaphyseal irregularities. Due to the presence of metaphyseal changes accompanied with bowing deformity of lower limb, they are likely to be mistaken for rickets. We present a case of a 7-year-old boy finally diagnosed with metaphyseal dysplasia, Spahr type (MDST) (OMIM # 250400) after his exome sequencing revealed novel variations in the MMP13 gene (OMIM * 600108). This is a rare skeletal dysplasia with only a few cases reported in literature. A compilation of the presentation of the reported cases is given to help the reader understand this rare disorder.
Case presentation
The case presented here is that of a 7-year-old boy who was symptomatic since 3 years of age in the form of increasing deformity of the lower limbs (figure 1). His medical history was not suggestive of any chronic illness such as chronic diarrhoea or recurrent fractures. Several physicians were consulted for this complaint and every time he was diagnosed to be having rickets, and vitamin D and calcium were given to him on several occasions but with no improvement. On physical examination, the child had preserved anthropometry with height 129.3 cm (90th centile; as per Indian Academy of Pediatrics (IAP) growth charts), weight 30.7 kg (72nd centile), upper:lower segment ratio of 1.24:1. He had bowing of both lower limbs with coxa vara. However, the other systemic signs of rickets such as frontal bossing, rachitic rosary double malleoli were absent. The radiographs of lower limbs showed widening and irregularity of metaphyses with sclerosis of the lower end of femur and upper end of tibia along with bowing of bilateral femora (figures 2, 3). No major abnormality in spine and the upper limbs was noted(figures 4, 5). Osteopenia was not observed in any of the child’s radiographs. Laboratory investigations showed a serum calcium of 9.4 mg/dL (8.8–10.8 mg/dL), phosphate of 6.8 mg/dL (3.7–5.6 mg/dL) and alkaline phosphatase of 243 IU/L (145–420 U/L). Vitamin D level was found to be 125.74 nmol/L (deficiency <50 nmol/L). A possibility of metaphyseal skeletal dysplasia was considered and exome sequencing was done which showed compound heterozygous state of the MMP13 gene. A non-sense variation NM_002427:c.889C>T (p.Arg297*) was found on one allele and the other allele showed a missense variation NM_002427:c.797A>G (p.Tyr266Cys). These variants in the proband were confirmed by sanger sequencing. The non-sense variation was classified as likely pathogenic according to ACMG guidelines,1 whereas the missense variation seen was classified as variant of uncertain significance. Targeted parental testing by sanger sequencing was done which showed the father to be having heterozygous carrier state of c.797A>G (p.Tyr266Cys) and mother being carrier of heterozygous variant c.889C>T (p.Arg297*). After careful clinical correlation of the phenotype with the molecular pathology, a final diagnosis of autosomal recessive MDST was made. The parents were explained the prognosis and genetic counselling was offered.
Figure 1.
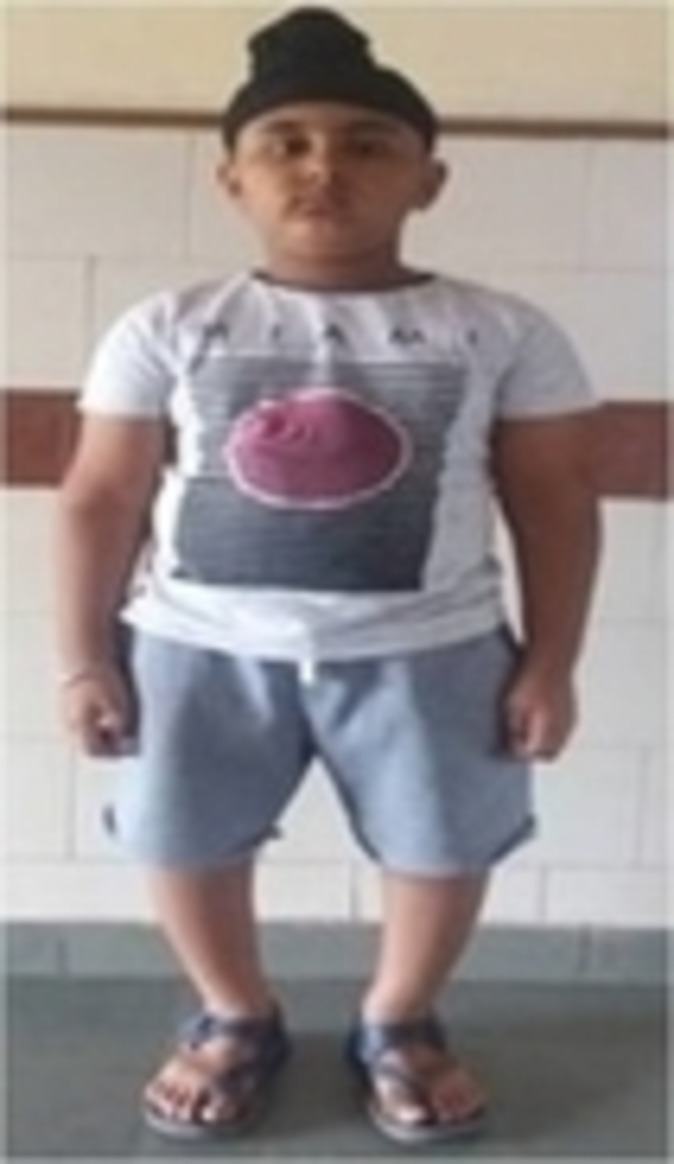
Clinical photograph of the child showing bowing of the lower limbs.
Figure 2.

Radiograph showing bowing of right femur with metaphyseal widening, irregularity and sclerosis of the lower end of femur.
Figure 3.

Radiograph of the pelvis showing metaphyseal irregularity of the femoral head along with irregular acetabular margins.
Figure 4.
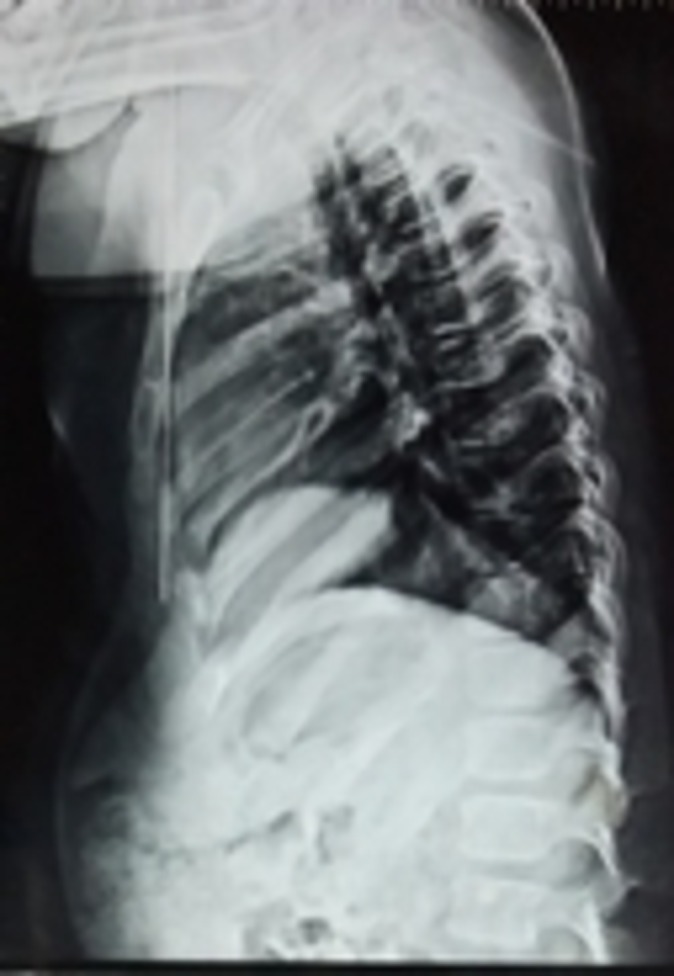
Radiograph showing normal spine.
Figure 5.
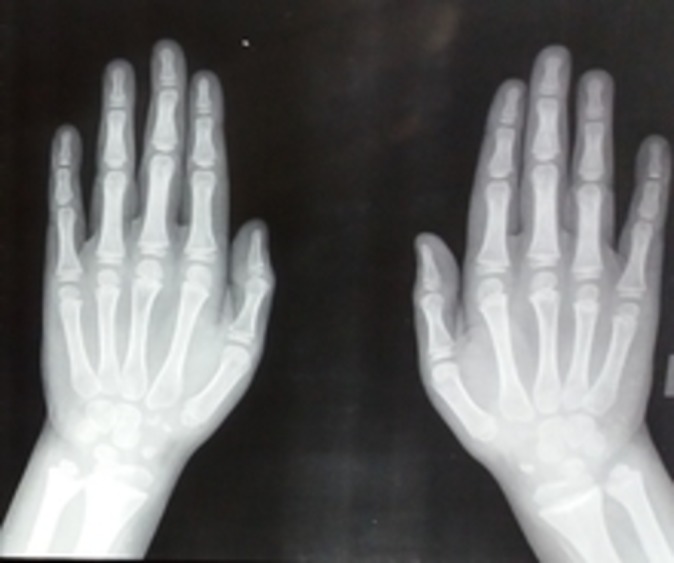
Radiograph showing normal hands.
Outcome and follow-up
The child was followed up after 1 year and 4 months of the initial examination date. He is currently 8 years with a height of 137.5 cm (90th centile). The bowing of the lower limbs is less noticeable and the child is doing well with his daily activities. His parents planned next pregnancy and after explaining the relatively benign nature of the skeletal dysplasia diagnosed in their child, they opted for prenatal diagnosis for the same. The results of fetal targeted mutation analysis showed fetus to be carrier of c.889C>T (p.Arg297*) variation found in the mother. The parents were counselled that their fetus was unaffected and the pregnancy is ongoing at the time of drafting this manuscript.
Discussion
Metaphyseal dysplasias belong to group 11 of the 2015 revised nosological classification of skeletal dysplasias.2 They are characterised by metaphyseal changes in long bones of the lower limbs causing short stature and varus deformity. There are subtle differences between the major types, that is, metaphyseal chondrodysplasia, Schimd type (MCDS) (OMIM # 156500), MDST (OMIM # 250400) and metaphyseal anadysplasia (MAND) (OMIM # 602111). While the age of onset is relatively earlier (usually in infancy) with more severe metaphyseal changes in MAND, the radiological changes typically improve by 6 years and the adult height is normal. MCDS and MDST are slightly late to present, the changes persist and short stature though mild is seen in adulthood. The varus deformity is considered to be more severe in MCDS than in MDST. Knee pain has been reported more commonly in MDST as compared with other metaphyseal dysplasias. Although we initially kept the possibility of MCDS in our case, after knowing the genetic diagnosis retrospectively we think that perhaps the varus deformity in our case is relatively mild as is seen in MDST.
MDST (OMIM # 250400) was recognised as early as in 1961; however, its molecular basis has been discovered relatively recently in 2014.3 Only a few cases of this disorder have been described in literature. The clinical and molecular findings in these cases have been summarised in table 1.
Table 1.
Summary of the important clinical and molecular findings of the reported cases of metaphyseal dysplasia, Spahr type
| Authors | Year | No. of patients | Consanguinity | Stature at specified age | Mutation reported in MMP13 gene |
| Andre and Ines Spahr3 | 1961 | 4 siblings | Yes | 47 years/F-138 cm M-167 cm M-154 cm |
c.619T>G/C.619T>G (in 3 out of 4 siblings) p.Trp207Gly (sequenced in 2014) |
| Farag and Teebi7 | 1990 | 4 siblings | Yes | 12 years /F-127 cm (<3rd centile) | − |
| Mé garbané et al 8 | 2008 | 2 siblings | Yes | 7 years 8 months-3rd centile 1 year-between 25th and 50th centile |
Excluded RMRP and COL10A1 gene mutations and suggested resemblance to MDST |
| Lausch et al 5 | 2009 | 1 | 3 years 2 months/M-1 SD | c.722C>A p.H213N |
|
| Bonafé et al 3 | 2014 | 1 patient related to family at S.No. 1 | Yes | 7 years/F-2.5 SD | c.619T>G/c.619T>G p.Trp207Gly |
| Li et al 9 | 2015 | 2 siblings | Denied | 15 years/M-2.6 SD and 7 years 5 months/M-1.9 SD | c.325C>T/c.325C>T p.Arg109Ter |
| Tadros et al 10 | 2017 | 2 siblings | Yes | 11 years/F-0.4th centile 6y/M-9th centile |
c.619T>G/c.619T>G, p.Trp207Gly |
| Presented case | 2019 | 1 | Denied | 7 years/M-0.82 z score | c.797A>G/c.889C>T p.Tyr266Cys/p.Arg297* |
The 11-year-old girl described by Tadros et al appears to have more severe bowing of the lower limb when compared with the case described by us. She also had chronic knee pain and even underwent surgical intervention in the form of epiphyseodesis at the knee joints. The presentation of her brother aged 6 years seems to resemble more like our case with height being less affected and only mild bowing of the lower limbs.
To summarise, the presentation of MDST in majority of the cases has been the observation of postnatal bowing of limbs causing disproportionate short stature with metaphyseal abnormalities. The metaphyseal abnormalities include metaphyseal irregularities and areas of sclerosis. In the absence of spine changes and any other systemic signs, these group of skeletal dysplasias very often mimic rickets and most of the children receive high doses of vitamin D prior to the genetic diagnosis.4 It is not expected to cause a significant short stature, though knee pain and abnormal gait can be expected.
MDST is caused by mutations in MMP13 gene. Matrix metalloproteinases are a family of proteases which are involved in extracellular matrix turnover and remodelling of the cartilage of growth plates. Mouse models have shown that the variations in MMP13 and MMP9 (OMIM * 120361) (which work in synergism) cause inactivation of these enzymes resulting in disordered cartilage matrix degradation and chondrocyte proliferation.5 Song et al have reviewed the pathogenic variations in MMP13 reported in literature until. According to this study, it appears that the missense pathogenic variations affecting the prodomain of MMP13 protein are associated with dominant metaphyseal anadysplasia 1 while the non-sense and missense variants in the catalytic domain are associated with recessive MDST.6 The two variations in the MMP13 gene found in our case were identified by exome sequencing and later confirmed by sanger sequencing. The c.889C>T is a stop gain mutation located in exon 6, qualifying as likely pathogenic according to ACMG guidelines.1 The c.797A>G variation is a missense variation located in exon 5 and was predicted to be deleterious by in-silico softwares (SIFT, PolyPhen, Mutation taster). To the best of our knowledge, this case is the first case of MDST to be reported from India.
Learning points.
Metaphyseal dysplasia, Spahr type (MDST) (OMIM # 250400) is a close mimicker of rickets and in any child presenting with skeletal features of rickets in whom the biochemistry is normal, the possibility of a metaphyseal dysplasia must be considered.
The investigation of choice for MDST is next generation sequencing.
The long-term prognosis of MDST is good.
Acknowledgments
We thank Dr Girisha KM for giving his expert opinion on our findings and supporting our diagnosis of this case.
Footnotes
Contributors: AK: substantial contributions to the conception or design of the work; acquisition, analysis or interpretation of data for the work. MB: drafting the work. AS: substantial contribution in radiograph analysis and final approval of the version to be published. NRG: substantial contribution in acquisition of data, radiograph analysis and final approval of the version to be published. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Parental/guardian consent obtained.
References
- 1. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–23. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015;167A:2869–92. 10.1002/ajmg.a.37365 [DOI] [PubMed] [Google Scholar]
- 3. Bonafé L, Liang J, Gorna MW, et al. MMP13 mutations are the cause of recessive metaphyseal dysplasia, Spahr type. Am J Med Genet A 2014;164A:1175–9. 10.1002/ajmg.a.36431 [DOI] [PubMed] [Google Scholar]
- 4. Dahl M, Birkebaek NH. [Metaphyseal chondrodysplasia as differential diagnosis to rickets]. Ugeskr Laeger 1996;158:1683–4. [PubMed] [Google Scholar]
- 5. Lausch E, Keppler R, Hilbert K, et al. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphyseal anadysplasia. Am J Hum Genet 2009;85:168–78. 10.1016/j.ajhg.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song C, Li N, Hu X, et al. A de novo variant in MMP13 identified in a patient with dominant metaphyseal anadysplasia. Eur J Med Genet 2018:30150–2. 10.1016/j.ejmg.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 7. Farag TI, Teebi AS. The second family with Spahr-type metaphyseal chondrodysplasia: autosomal recessive inheritance confirmed. Clin Genet 1990;38:237–9. [PubMed] [Google Scholar]
- 8. Mégarbané A, Chouery E, Ghanem I. A multiplex family with possible metaphyseal Spahr-type dysplasia and exclusion of RMRP and COL10A1 as candidate genes. Am J Med Genet A 2008;146A:1865–70. 10.1002/ajmg.a.32390 [DOI] [PubMed] [Google Scholar]
- 9. Li D, Weber DR, Deardorff MA, et al. Exome sequencing reveals a nonsense mutation in MMP13 as a new cause of autosomal recessive metaphyseal anadysplasia. Eur J Hum Genet 2015;23:264–6. 10.1038/ejhg.2014.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tadros S, Scott RH, Calder AD, et al. Metaphyseal dysplasia, Spahr type; missense MMP13 mutations in two Iraqi siblings. Clin Dysmorphol 2017;26:13–17. 10.1097/MCD.0000000000000151 [DOI] [PubMed] [Google Scholar]