

Abstract
We present a 44-year-old female with an initial presentation with distal renal tubular acidosis (RTA) after she presented with hypokalaemia and normal anion gap acidosis. Three years following the diagnosis, she presented with progressive renal impairment. In the absence of any clinical, biochemical and radiological clues, she underwent a renal biopsy which showed severe tubulitis secondary to lymphocytic infiltration. Serological investigations subsequently revealed positive anti-nuclear, anti-Sjögren’s syndrome related antigen A (SS-A), and anti-Sjögren’s syndrome related antigen B (SS-B) antibodies, supporting the diagnosis of Sjögren’s syndrome. This case is unique in that distal RTA was the presenting clinical manifestation of Sjögren’s syndrome. We hope that a consideration for Sjögren’s syndrome is made in patients with seemingly idiopathic RTA.
Keywords: renal system, acute renal failure, sjogren’s syndrome
Background
Sjögren’s syndrome is a rare autoimmune condition typically involving chronic inflammation of exocrine organs such as lacrimal and salivary glands, typically manifesting as dry eyes and mouth.1 However, extra-glandular renal involvement is not uncommon. Pathologically, the disease is characterised by lymphocytic infiltration with tissue damage in affected organs. Based on biopsy reports in the available literature, tubulointerstitial nephritis (TIN) is the most common histological abnormality, followed by glomerulonephritis as a distant second.2 While TIN is the most common histological finding, the biochemical presentation of hypokalaemia suggestive of distal renal tubular acidosis has been infrequently reported.3 In this case report, we present a middle-aged Caucasian female who initially presented with hypokalaemia and acidosis and subsequently was diagnosed as having Sjögren’s syndrome.
Case presentation
A 44-year-old female was initially seen in the nephrology outpatient clinic for evaluation of hypokalaemia. She reported feeling tired and listless for 4 months prior to her review. She denied any fever, night sweats, lymphadenopathy or weight loss. There was no history of diabetes mellitus, hypertension or rheumatological illnesses, and in particular, she denied a history of dry eyes and mouth. She had no history of nephrolithiasis or pre-existing renal disease. Systems review did not reveal abdominal pain, emesis, diarrhoea, painful swollen stiff joints or the use of non-steroidal anti-inflammatories (NSAIDs). Her past medical history was significant for pernicious anaemia and hypothyroidism for which she was prescribed vitamin B12 1200 mcg daily and L-thyroxine 75 mcg daily.
There was no personal or family history of autoimmune disease, but she appeared to recollect that her aunt and cousin were also hypokalaemic on supplements. She quit smoking 15 years ago, reported minimal alcohol intake, worked as a receptionist and was single. Her vital signs were unremarkable and no abnormal finding was identified on physical examination. Initial lab investigations revealed sodium 136 mmol/L, potassium 2.8 mmol/L, chloride 116 mmol/L and bicarbonate 16 mmol/L (normal anion gap acidosis). The pH on venous gas was 7.29. Her serum potassium, chloride and bicarbonate were within the normal limits 6 months prior to her review. Urine sodium was 61 mmol/L, urine potassium 38 mmol/L and urine chloride 55 mmol/L (+ve urine anion gap: 44 mmol/L).
Without a history of gastrointestinal losses or administration of intravenous fluids, a diagnosis of renal tubular acidosis (RTA) was contemplated. The presence of metabolic acidosis and a urine pH of 6.5 support a diagnosis of distal (type I) RTA. A 24 hours urine for calcium was 3.7 mmol/day (normal for her dietary intake). Incidentally, her total protein level was elevated at 88 g/L (60 to 80 g/L). Serum IgG 30.10 g/L (5.5 to 17.2 g/L), IgA <0.25 g/L (0.87 to 3.94 g/L) and IgM g/L 1.08 (0.44 to 2.47 g/L). As myeloma is associated with renal tubular anomalies including RTA, she was investigated further with serum protein electrophoresis, which revealed polyclonal elevation of IgG. There was no monoclonal band identified on serum immunofixation. Urine for immunofixation did not reveal any Bence Jones protein. Urinalysis did not reveal any blood or protein. Her serum albumin was 39 g/L and serum creatinine was 116 μmol/L. Renal ultrasound revealed normal sized kidneys with preserved cortical thickness and corticomedullary distinction. Specifically, there was no evidence of nephrocalcinosis or calculus. Potassium citrate (K-Lyte) was initiated at a dose of 45 mEq twice daily and she felt better on the medication. She was followed for a year and discharged back to primary care.
However, 3 years later, she was referred by her family physician for a progressive increase in her serum creatinine (elevated at 172 μmol (initially 116 μmol)). She denied any recent illnesses. There had been no change in her medications and she claimed adherence with K-Lyte. Her potassium (4.1 mmol/L) and bicarbonate (24 mmol/L) had been maintained within a normal range for the last 2 years. Specifically, she denied any recent exposure to NSAIDs, over-the-counter medications or chemotherapeutic agents. She denied swollen joints, early morning joint stiffness, rash, haemoptysis or bloody sinus discharge. Her urea was 6.4 mmol/L, creatinine 172 μmol, potassium 4.1 mmol/L and bicarbonate 24 mmol/L. Urinalysis was negative for blood and protein and the albumin-to-creatinine ratio was 0.3 mg/mmol (normal range: <2.8 mg/mmol). A 24 hours urine did not reveal any protein. Ultrasound scan of the kidneys revealed no evidence of obstruction, with preserved cortex bilaterally.
In the absence of any clinical or biochemical cause to account for the acute rise in creatinine, a renal biopsy was performed. The primary pathological alteration was evident in the tubulointerstitial compartment with a diffuse multifocal dense plasma cell rich interstitial inflammation and many foci of lymphocytic tubulitis (figures 1–3). There was multifocal acute tubular injury accompanying the interstitial inflammation. In-situ hybridisation for kappa/lambda light chains highlighted the polyclonality of the infiltrating plasma cells (figure 4). There were only rare IgG4-expressing plasma cells among the infiltrate. The less pronounced lymphocytic population was composed of both T cells and B cells with the predominance of T lymphocytes. B cells did not express aberrant surface markers and had low proliferation rate. Immunofluorescent microscopy was unremarkable and electron microscopy showed unremarkable glomerular architecture and no electron-dense deposits. Many tubuloreticular inclusions were seen inside the endothelial cytoplasm. The case was reviewed by a haematologist to rule out the possibility of a haematological neoplasm, who concurred with the reactive nature of the plasma cell and lymphoid population.
Figure 1.

Sections of paraffin-embedded tissue stained with haematoxylin and eosin shows core needle biopsy of kidney cortex with multifocal dense interstitial inflammation (arrows) - 100x magnification.
Figure 2.

Higher magnification of the inflamed tissue reveals plasma-cell rich infiltrate (inside black arrows) surrounding one tubule affected by lymphocytic tubulitis (white arrow) - 400x magnification; haematoxylin and eosin stain.
Figure 3.

Sections of paraffin-embedded tissue stained with periodic acid-Schiff shows one tubule (arrow) attacked by many lymphocytes invading its epithelial lining (400x magnification).
Figure 4.
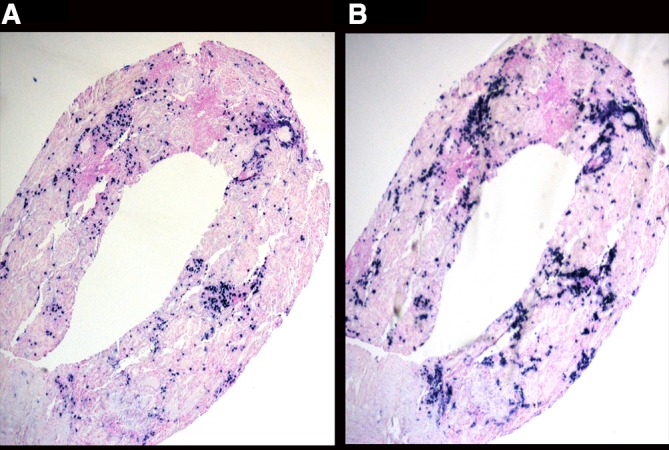
In-situ hybridisation for kappa (A) and lambda (B) light chain reveals the expression of both light chains in the infiltrating plasma cells.
On the quantitative immunoglobulin panel, IgA levels were low at 0.27 g/L (0.87 to 3.94 g/L). IgG was elevated at 34.80 g/L (5.5 to 17.2 g/L) and IgM levels were normal. On serum immunofixation, there was a very faint IgG kappa restriction band of undetermined significance. A 24 hours urine for protein failed to detect Bence-Jones protein. Serum free light chains revealed that both kappa and lambda were elevated but the ratio was 2.22. Due to diffuse plasma cell infiltration, she underwent a CT scan of the chest, abdomen and pelvis and there was no evidence of enlarged lymph node or lymphoma. Based on the findings of TIN on histology, and the absence of enlarged lymph nodes and lymphoma on imaging, an autoimmune workup was performed. Serological biochemical markers are listed in table 1.
Table 1.
Results of serological investigations
| ANA | Positive, speckled pattern, 1:2560 dilution |
| Rheumatoid factor | 192 IU/mL (0–0.29 IU/mL) |
| Anti-SS-A | Positive |
| Anti-SS-B | Positive |
| Anti-Smith | Negative |
| Anti-RNP | Negative |
| Anti-Jo | Negative |
| Anti-SCL | Negative |
| C3 (g/L) | 0.97 (0.90–1.80 g/L) |
| C4 (g/L) | 0.09 (0.10–0.40 g/L) |
| Anti-MPO | Negative |
| Anti-PR3 | Negative |
| Anti-GBM | Negative |
| Anti-thyroid peroxidase | Negative |
ANA, anti-nuclear antibody; anti-GBM, anti-glomerular basement membrane antibody; anti-MPO, anti-myeloperoxidase antibody; anti-PR3, anti-proteinase 3 antibody; anti-RNP, anti-ribonucleoprotein; anti-SCL, anti-topoisomerase; anti-SS-A, anti-Sjögren’s syndrome related antigen A; anti-SS-B, anti-Sjögren’s syndrome related antigen B; C3, Complement 3; C4, Complement 4.
Despite the presence of positive rheumatoid factor, in the absence of small joint stiffness, arthritis and other clinical manifestations, rheumatoid arthritis was not considered as a potential diagnosis. However, on further questioning, she revealed having dry eyes and mouth for 2 years and attributed the symptoms to side effects of her existing medications. Based on positive anti-Sjögren’s syndrome related antigen A (anti-SS-A) and anti-Sjögren’s syndrome related antigen B (anti-SS-B) antibodies in the presence of sicca symptoms, a presumed diagnosis of Sjögren’s syndrome was made.
Differential diagnosis
Common causes of distal RTA include: autoimmune disorders (Sjögren’s syndrome, systemic lupus erythematosus (SLE) and rheumatoid arthritis), medications (ifosfamide, amphotericin B, lithium carbonate and ibuprofen), hypercalciuric conditions (hyperparathyroidism, vitamin D intoxication and sarcoidosis) and others, including familial causes (medullary sponge kidney and Wilson’s disease).
Common causes of TIN include: medications (NSAIDs, penicillins and cephalosporins, sulfonamides, diuretics, allopurinol, proton pump inhibitors), infections (Legionella, Corynebacterium diphtheriae, Cytomegalovirus, Epstein-Barr virus, Leptospira, mycobacterium, streptococcus), tubulointerstitial nephritis and uveitis syndrome and autoimmune disorders (sarcoidosis, Sjögren’s syndrome, SLE).
Treatment
The patient was initiated on prednisolone at a dose of 1 mg/kg body weight, but within a month developed cushingoid features. Steroids were rapidly tapered and she was eventually initiated on mycophenolate mofetil at a dose of 750 mg three times per day.
Outcome and follow-up
Creatinine levels measured 3 months post initiation of therapy had improved to 123 μmol. Her dryness of eyes responded to artificial tears. No specific therapy was needed for her dry mouth, which improved significantly in the 3 months since treatment was initiated. She continues to work full-time and does not report any noticeable changes in her health, with the understanding that the progression of renal disease can often be asymptomatic.
Discussion
Sjögren’s syndrome is an autoimmune condition which typically involves lymphocytic infiltration of the salivary, parotid and lacrimal glands, resulting in the characteristic symptoms of xerosis (dry eyes) and xerostomia (dry mouth). This immune process can also affect non-exocrine organs, such as the skin, lungs, gastrointestinal tract and the kidneys.
Although Sjögren’s syndrome is often characterised by sicca symptoms, they are not always present. A case series by Shioji et al 4 described four cases of Sjögren’s syndrome complicated by RTA, in which three of the four patients presented with arthralgia or muscle weakness. Only two reported dry mouth and none reported any ocular abnormality.4 A number of case reports have also reported muscle weakness,5 pathological fractures6 and hypokalaemic paralysis7 as the presenting symptoms of Sjögren’s syndrome secondary to distal RTA. Therefore, symptoms related to RTA, even in the absence of sicca symptoms, may be a clue that triggers the discovery of Sjögren’s syndrome.
The diagnosis of Sjögren’s presents a challenge to clinicians, particularly when the initial presentation differs from the exocrine manifestation of dry eyes and mouth. This is highlighted by the emphasis diagnostic criteria have placed on ocular and oral findings. To make a diagnosis of primary Sjögren’s, the American-European Consensus Classification Criteria requires four of six criteria, including: ocular or oral symptoms, objective ocular or oral signs, histopathology from a lip biopsy and the presence of autoantibodies.1 Our patient only met three of the criteria (dry eyes and mouth for more than 3 months and positive anti-SS-A and anti-SS-B antibodies), which led to a presumed diagnosis of primary Sjögren’s.
Although findings on renal biopsy are not a part of the diagnostic criteria of Sjögren’s, they can help support the diagnosis. The incidence of renal involvement in Sjögren’s syndrome varies in the literature from 0.3% to 27%, based on either biopsy findings or biochemical impairment.8 9 Tubulointerstitial inflammation in the form of TIN is the most common renal manifestation of Sjögren’s syndrome.8 10 Depending on the segment of the nephron impacted by lymphocytic infiltration, TIN has been shown to manifest as hypokalaemia, acidosis, RTA (predominantly distal and rarely proximal), Gitelman syndrome, Fanconi syndrome and diabetes insipidus.11 Similarly, it can also lead to nephrocalcinosis and renal calculi.12 Histological findings in RTA secondary to Sjögren’s are typically described as diffuse lymphocytic infiltration in the renal interstitial tissues, interspersed with plasma cells.4 In cases of Sjögren’s syndrome with RTA, similarities in histological findings in the lacrimal glands, salivary glands and kidneys suggest that tubular dysfunction is likely a result of the same immunologic process underlying Sjögren’s syndrome.4
IgG-4 related disease is often confused with Sjögren’s syndrome, as the former can similarly affect the lacrimal and salivary glands, and present with TIN. Although there are some epidemiological differences between the two entities (with Sjögren’s more commonly affecting women between the ages of 30 to 50 and IgG-4 related diseases mainly affecting men older than 60), differentiation is made largely based on serological and histopathological findings.13 Patients with IgG-4 related diseases generally test seronegative for anti-SS-A and SS-B antibodies, and have a significantly elevated serum level of IgG4.13 Demonstration of IgG4-positive plasma cells (with a ratio of IgG4-positive/IgG-positive plasma cells over 40%) is characteristic of IgG-4 related disease, while lymphocytes with a T-cell dominance are found in Sjögren’s-related TIN.14 In contrast to in Sjögren’s syndrome, severe tubulitis is extremely rare in IgG-4 related disease,15 where storiform fibrosis are the characteristic histopathological features.16
Our patient presented with renal tubular acidosis 2 years prior to the onset of sicca symptoms. Her creatinine at the time was elevated (but unfortunately not pursued), raising the possibility that an infiltrative process may have already been taking place. By the time a renal biopsy was performed, the extent of inflammation was already quite severe. This case emphasises the need to consider a wide set of differential diagnoses, including Sjögren’s syndrome, in cases of unexplained renal tubular acidosis. Diagnosis at an early stage may allow for better control and prevent the progression of disease.
Learning points.
Although Sjögren’s syndrome is characteristically associated with symptoms of dry eyes and mouth, extra-glandular renal manifestations are not uncommon presentations.
The most common renal manifestation of Sjögren’s syndrome is tubulointerstitial nephritis, which can manifest as renal tubular acidosis.
Individuals with seemingly idiopathic distal renal tubular acidosis should be screened for Sjögren’s syndrome.
Acknowledgments
The authors would like to acknowledge Dr Elan Paluck and her entire team at the Research and Performance Support at the Regina General Hospital.
Footnotes
Contributors: KH wrote the initial draft. PD contributed to the pathology images, MM assisted with the drafts and BP wrote the final version. All the authors have read the final version.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Vitali C, Bombardieri S, Jonsson R, et al. . Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8. 10.1136/ard.61.6.554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maripuri S, Grande JP, Osborn TG, et al. . Renal involvement in primary Sjögren’s syndrome: a clinicopathologic study. Clin J Am Soc Nephrol 2009;4:1423–31. 10.2215/CJN.00980209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siamopoulos KC, Elisaf M, Drosos AA, et al. . Renal tubular acidosis in primary Sjögren’s syndrome. Clin Rheumatol 1992;11:226–30. 10.1007/BF02207962 [DOI] [PubMed] [Google Scholar]
- 4. Shioji R, Furuyama T, Onodera S, et al. . Sjögren’s syndrome and renal tubular acidosis. Am J Med 1970;48:456–63. 10.1016/0002-9343(70)90045-8 [DOI] [PubMed] [Google Scholar]
- 5. Jung SW, Park EJ, Kim JS, et al. . Renal Tubular Acidosis in Patients with Primary Sjögren’s Syndrome. Electrolyte Blood Press 2017;15:17–22. 10.5049/EBP.2017.15.1.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abdulla MC, Zuhara S, Parambil AAK, et al. . Pathological fracture in Sjögren’s syndrome due to distal renal tubular acidosis. Int J Rheum Dis 2017;20:2162–4. 10.1111/1756-185X.13193 [DOI] [PubMed] [Google Scholar]
- 7. Sedhain A, Acharya K, Sharma A, et al. . Renal Tubular Acidosis and Hypokalemic Paralysis as a First Presentation of Primary Sjögren’s Syndrome. Case Rep Nephrol 2018;2018:9847826 10.1155/2018/9847826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goules AV, Tatouli IP, Moutsopoulos HM, et al. . Clinically significant renal involvement in primary Sjögren’s syndrome: clinical presentation and outcome. Arthritis Rheum 2013;65:2945–53. 10.1002/art.38100 [DOI] [PubMed] [Google Scholar]
- 9. Kaufman I, Schwartz D, Caspi D, et al. . Sjögren’s syndrome - not just Sicca: renal involvement in Sjögren’s syndrome. Scand J Rheumatol 2008;37:213–8. 10.1080/03009740701867323 [DOI] [PubMed] [Google Scholar]
- 10. Yang HX, Wang J, Wen YB, et al. . Renal involvement in primary Sjögren’s syndrome: a retrospective study of 103 biopsy-proven cases from a single center in China. Int J Rheum Dis 2018;21:223–9. 10.1111/1756-185X.13182 [DOI] [PubMed] [Google Scholar]
- 11. Jasiek M, Karras A, Le Guern V, et al. . A multicentre study of 95 biopsy-proven cases of renal disease in primary Sjögren’s syndrome. Rheumatology 2017;56:362–70. 10.1093/rheumatology/kew376 [DOI] [PubMed] [Google Scholar]
- 12. Moutsopoulos HM, Cledes J, Skopouli FN, et al. . Nephrocalcinosis in Sjögren’s syndrome: a late sequela of renal tubular acidosis. J Intern Med 1991;230:187–91. 10.1111/j.1365-2796.1991.tb00429.x [DOI] [PubMed] [Google Scholar]
- 13. Maślińska M, Przygodzka M, Kwiatkowska B, et al. . Sjögren’s syndrome: still not fully understood disease. Rheumatol Int 2015;35:233–41. 10.1007/s00296-014-3072-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeong HJ, Shin SJ, Lim BJ. Overview of IgG4-related tubulointerstitial nephritis and its mimickers. J Pathol Transl Med 2016;50:26–36. 10.4132/jptm.2015.11.09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshita K, Kawano M, Mizushima I, et al. . Light-microscopic characteristics of IgG4-related tubulointerstitial nephritis: distinction from non-IgG4-related tubulointerstitial nephritis. Nephrol Dial Transplant 2012;27:2755–61. 10.1093/ndt/gfr761 [DOI] [PubMed] [Google Scholar]
- 16. Saeki T, Kawano M. IgG4-related kidney disease. Kidney Int 2014;85:251–7. 10.1038/ki.2013.393 [DOI] [PubMed] [Google Scholar]